

Introduction
Alveolar soft part sarcoma (ASPS), also called alveolar soft-tissue sarcoma (ASTS), is a rare malignant soft-tissue neoplasm, which was first described by Christopherson and colleagues[1] in 1952. Its prevalence is less than 1% of all primary soft-tissue sarcomas.[2] The histogenesis of ASPS is not clear; however, a myogenic phenotype and skeletal muscle differentiation are favored.[3,4] It is more common in adolescents and young adults, especially in those who are 15-35 years old; occurs more commonly in women; and is rarely seen in children.[5]
ASPS most commonly originates from the deep tissues of the proximal parts of the extremities, especially the lower limbs,[5] and shows right-sided laterality, as described by Fassbender.[6] In children the primary site is in the head and neck. It usually appears as a painless mass in the thigh or leg, but it may initially appear from other sites of the body. Regardless of its indolent growth pattern, the ultimate prognosis remains poor because of early metastatic spread to the lungs, bone, brain, and lymph nodes in order of frequency.[7]
In many cases, the first manifestations are due to distant metastases. Surgery remains the mainstay of therapy.[8] Little is known about other therapies, ie, chemotherapy, radiation therapy, and biologic therapy, and the data are still inconclusive.
Readers are encouraged to respond to George Lundberg, MD, Editor of MedGenMed, for the editor's eyes only or for possible publication via email: glundberg@medscape.net
Case Report
The patient is a 20-year-old woman who has recently immigrated to Iran from Afghanistan. She was taken to the emergency department due to respiratory distress. She had a left subclavicular mass for 3 years that had been growing slowly. The mass gradually became painful and restricted the movements of her left arm. It was associated with fever, anorexia, and weight loss (11 kg during the last 5 months). Over time, she has also developed dyspnea, productive cough, and generalized bone pain – especially around the hip and sacroiliac joints. She has never had any other disease requiring hospital admission, and has never smoked or abused substances. The patient was ill-appearing and cachectic with bitemporal atrophy. A prominent tortuous venous pattern over the left hemithorax was obvious, and an edema extending from the upper intercostals spaces to the midcervical region was present. A soft, warm, and tender mass of 20 × 15 cm size was noted over the anterior aspect of the upper left hemithorax with a bruit heard over the mass. Another mass of 3 × 5 size was notable at the left posterior chest wall, just opposite to the first one. No mass was detected in the breasts. Pulmonary auscultation revealed diffuse bilateral coarse crackles and wheezing. A III/VI holosystolic murmur was detected over the left sternal border. Abdomen was not distended, and there were no clinically detectable ascites. The liver was palpable 2-3 cm below the right costal margin with a span of 15 cm; no splenomegaly was detected clinically. Both active and passive movements of the left arm were limited, and a tense edema was present over the upper parts of the left arm. There was no evidence of focal neurologic deficit, optic atrophy, or papilledema. Lab tests, chest x-ray, and ultrasonography (US) of the mass, liver, and peritoneal cavity were performed (see Figures 1 and 2).
Figure 1.
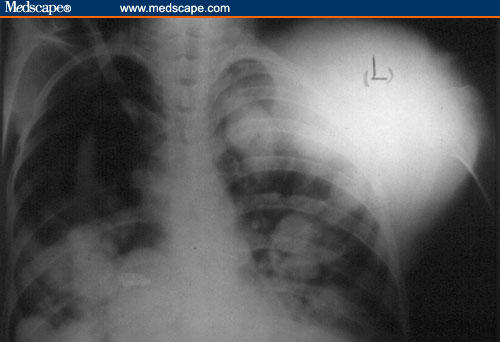
Chest x-ray: indicative of a large mass over the left anterior chest wall, and multiple well-defined, round-shaped bilateral pulmonary opacities, due to metastatic involvement of the lungs.
Figure 2.
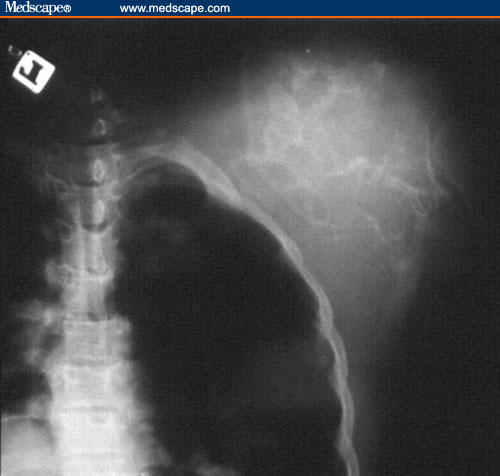
X-ray of the mass, showing a tumor of soft-tissue density with a highly vascular context.
CBC: White blood cell count, 7100/mcL (polymorphonuclear, 83%; lymph, 17%); red blood cell count, 3.55 × 106/mcL; hemoglobin, 11.7 g/dL; platelets: 271 × 103/mcL.
Biochemistry: Na, 136 mEq/L; K, 4.2 mEq/L; bilirubin, 1.09 (T), 0.6 (D) mg/dL; Ca, 8.8 mg/dL; P, 4.5 mg/dL; erythrocyte sedimentation rate (ESR), 25 mm/hour; C-reactive protein (CRP), ++(1/100); aspartate aminotransferase (AST), 67 IU/L; alanine aminotransferase (ALT), 33 IU/L; alkaline phosphate, 359 IU/L; creatine kinase, 127 IU/L; lactic dehydrogenase, 1229 IU/L; BUN, 13 mg/dL; Cr, 0.7 mg/dL; PT, 15; PTT, 33 seconds; total Pr, 5.2 g/L; serum albumin, 3.1 g/L; BS, 121 mg/dL.
Urinanalysis: Normal.
Urine culture: Negative.
Stool exam: Normal.
Blood culture (x3): Negative.
Arterial blood gas: pH, 7.45; PaCO2, 32.1 mm Hg; HCO3, 25.4 mEq/L; PaO2, 84 mm Hg; O2 saturation, 90%.
US revealed a highly vascular mass of the left upper hemithorax. Abdominal US was normal. Computerized tomography (CT) of the chest was requested on the basis of the chest x-ray findings. As the patient's level of consciousness began to fall following admission, a brain CT scan was performed to assess the possible cerebral involvement (see Figures 3 and 4).
Figure 3.
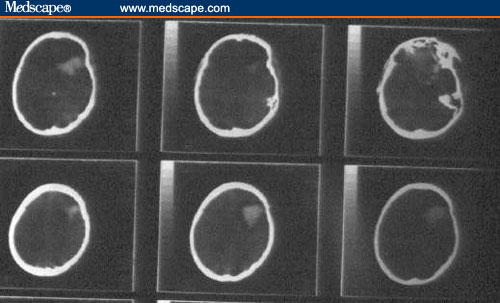
Computerized tomographic scan of the brain: a hyperdense space occupying lesion is noted in the left frontal lobe, indicating a metastatic focus.
Figure 4.
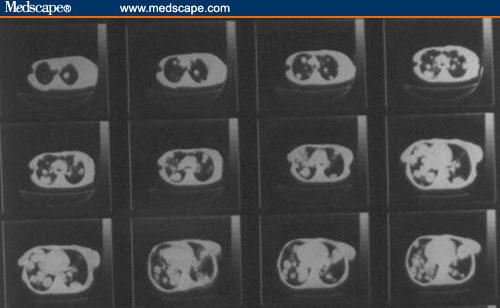
Chest computerized tomographic scan, showing multiple pulmonary metastases on both sides.
Diagnosis of a soft-tissue malignant neoplasm with pulmonary and cerebral metastases was made on the basis of clinical and imaging findings. Unfortunately, the patient died due to respiratory distress and apnea that were not responsive to supportive care. Necropsy of the chest wall mass and liver lesions revealed soft, pale gray poorly circumscribed masses. Microscopic examination revealed clusters of tumor cells separated from each other by dense fibrous trabeculae, each cluster further divided into groups of cells with central degeneration and loss of cohesion resulting in an alveolar or pseudoglandular pattern, occasionally surrounded by thin vasculature. The tumor cells were round to polygonal in shape with large, finely granular cytoplasm and well-defined borders. They had large, round-to-oval eccentrically placed nuclei, with prominent single-to-multiple nucleoli. Few binucleate and occasional multinucleate cells were present (Figures 5–8).
Figure 5.
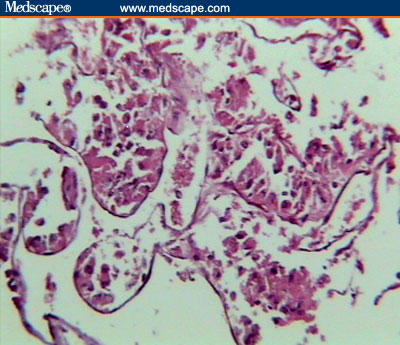
Light microscopic view, showing pseudoalveolar arrangement of the neoplastic cells (H&E stain, medium power).
Figure 8.
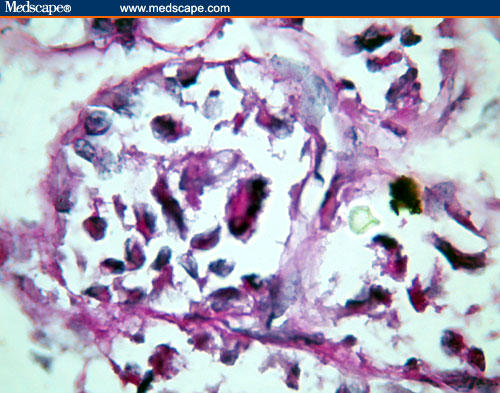
A tumor cell nest shows a bizarre tumor cell with periodic acid-Schiff-positive reaction in the cytoplasm.
The periodic acid-Schiff (PAS) reparation revealed varying amounts of extracellular glycogen and PAS-positive, diastase-resistant intracytoplasmic crystalline inclusions. On the basis of these cytoarchitectural findings, a diagnosis of ASPS was offered. The immunohistochemistry study results were in favor of a diagnosis of ASPS – strongly positive for muscle-specific actin; weakly positive for desmin; and negative for S 100, cytokeratin, and vimentin.
Discussion
The exact cause of ASPS is not entirely understood; however, studies indicate that genetic alterations may play a role. Certain inherited conditions are associated with an increased risk of developing soft-tissue sarcomas, including Li-Fraumeni syndrome (alterations in the p53 gene) and neurofibromatosis (alterations in the NF1 gene).
ASPS shows a characteristic histopathologic pattern and distinctive cytoarchitectural features.[5,9] It is composed of cells with granular cytoplasm arranged along delicate fibrous septa, giving rise to an alveolar or organoid pattern.[7]
The most striking feature is the presence of needle-shaped crystalline intracytoplasmic inclusions and a granule material in the Giemsa-stained smears.[4,7] The crystals are strongly positive with PAS stain and are diastase-resistant.[4,7] The typical crystals are considered diagnostic of ASPS.
These structures are a characteristic, if not pathognomonic, feature of this enigmatic neoplasm. The histogenesis of this tumor has been subject to extensive research, and different hypotheses varying from skeletal muscle to neural origin have been proposed, but none of them found acceptance.[10,11] Ronald and Heal[12] hypothesized that ASPS arise from displaced paraganglionic mesoderm and have a close homology with paragangliomas of the carotid body type. Most of the other studies indicate a muscle origin.[13,14] Implied in the question is our naive belief that all tumors must necessarily relate to some normal tissue type; not all sarcomas may follow this paradigm and ASPS may be the prime example.[10]
The histologic differential diagnosis of ASPS on cytology includes metastatic renal cell carcinoma, granular cell tumor, metastatic seminoma, bronchoalveolar carcinoma, clear-cell sarcoma, and epithelioid sarcoma.[4] The tumor contains numerous vascular channels around the clusters of tumor cells, reminiscent of a paraganglioma or an endocrine neoplasm.[4,7] Most often they can be excluded on the basis of clinical presentation and a definitive cytomorphologic picture.[4]
Although ASPS has distinctive histologic features, it may cause diagnostic problems when it arises in unusual locations.[15]
Studies have shown that the typical crystals are immunoreactive to MCT1 (monocarboxylate transporter) and its chaperone protein CD 147.[10,11,16,17]
MCT1 is normally located at the cell membrane, but in ASPS patients it has a cytoplasmic localization, in addition to cell-surface expression, that is distinguished from the exclusive cell-membrane expression in normal tissues and other neoplasms.[16,17]
Recently, ASPS was found to have a nonreciprocal translocation, der (17)t(x;17)(p11.2;q 25).[10,17,18] This translocation fuses the TFE3 transcription factor gene at Xp11 to a novel gene at 17q25, designated ASPL.[10,15,18,19] The transcription in ASPS differs from that in most other sarcomas in that it is unbalanced.[10] Because females have an extra X chromosome, their likelihood of developing an X autosome translocation is theoretically double that of males, and this may be an explanation for female predominance in ASPS.[18] The presence of the ASPL-TFE3 fusion appears highly specific and sensitive for ASPS among sarcomas; the ASPL-TFE3 fusion transcripts can serve as a useful marker in the diagnosis of ASPS and may also be helpful in elucidating the underlying pathogenesis of ASPS.[19]
The tumor generally shows slow growth and late occurrence of metastases. Because ASPS affects tissue that is elastic and easily moved, a tumor may exist for a long time before being discovered, growing large and pushing aside surrounding tissue. The following are the most common clinical manifestations of ASPS: a painless swelling or lump, pain or soreness caused by compressed nerves or muscles, limping or other difficulty using the limb, and a diminished range of motion in the affected area.
The most conclusive diagnostic procedure is a biopsy, taken by a simple surgical procedure. Various imaging studies may be performed: Simple x-rays that are usually normal unless the bone adjacent to the tumor is eroded; magnetic resonance imaging (MRI) outlines the extent of the tumor and its relation to other soft-tissue structures; a CT scan is used primarily to assess the chest and lung for metastatic tumors; and a bone scan cannot distinguish between tumor, infection, or fractures.
Once ASPS has been diagnosed, the tumor should be staged. The stage of a tumor suggests which form of treatment is most appropriate, and gives some indication of prognosis.
Primary soft-tissue sarcomas of the chest wall are uncommon, and data concerning treatment and results are sparse. Most studies have categorized these tumors as truncal sarcomas and inferred a poor prognosis. In a review of 149 cases of chest wall sarcomas from 1948 to 1988, only 3 patients (3%) had ASPS.[20]
To our knowledge, only a few cases of primary chest wall ASPS have been reported – less than 20 cases in the last half-century.[20–22] Reports on primary presentation of ASPS with intracranial metastases are even more rare, and to our knowledge, only 3 cases have been reported.[23] Because of a low socioeconomic state, our patient did not seek medical care despite the obvious mass over her chest wall, and came with symptoms of pulmonary and cerebral metastases, a finding that is usually expected in patients with the tumor in the extremities, not the chest wall.
Surgical excision remains the mainstay of therapy for most soft-tissue sarcomas, particularly for localized ASPS, which tends to be slow-growing and unresponsive to chemotherapy.[11,20] Surgical excision of the pulmonary metastases has resulted in prolonged survival in some patients, but adjuvant radiation and/or chemotherapy are generally thought to be ineffective.[11,24] ASPS patients should not be treated with chemotherapy outside of controlled clinical trials.[25]
If surgery to remove the entire tumor isn't possible or the tumor has metastasized, surgery may be combined with radiation therapy. Supportive medical care and continuous long-term follow-up care are vital to the successful treatment of ASPS.[1,11]
As with any cancer, prognosis and long-term survival can vary greatly between individuals. ASPS is a relatively slow-growing tumor and seldom recurs locally after complete resection, but is highly metastatic. Metastasis can occur early in the course of the disease, sometimes prior to the detection of the primary lesion or much later, even decades, after resection of the primary tumor despite the absence of local recurrence.
The influence of tumor size and site on the prognosis are subject to controversy; whereas in some series tumor size correlated with metastatic disease at onset,[8] and is considered as an important prognostic factor[8] and ASPS arising from atypical sites is thought to have a less favorable prognosis,[5] some other authorities believe that they have no significant bearing on prognosis.[11,20] In general, tumor size greater than 5 cm indicates a poorer prognosis. Regardless of the last 2 factors, tumor respectability and metastatic disease at presentation are generally agreed on as the crucial prognostic markers.[11,20,25] ASPS that involves the orbit or head and neck or that occurs in younger patients seems to confer a better outcome than its adult counterpart. Pediatric ASPS is known as one of the most common types of nonrhabdomyosarcomatous soft-tissue sarcomas in children.[26]
In a study of 91 patients who were diagnosed and followed up between 1923 and 1986, among the patients without metastases at the time of diagnosis, 2-year survival of 77%, 5-year survival of 60%, 10-year survival of 38%, and 20-year survival of 15% were detected.[27]
Numerous research studies are focused on this puzzling soft-tissue sarcoma. Although many novel findings have been noted in recent years, they do not shed light on the histogenesis and differentiation of ASPS.
Finding new targets for specific biologically directed therapies, such as interferon-alpha and angiogenesis inhibitors, is another hot point of research.
Figure 6.
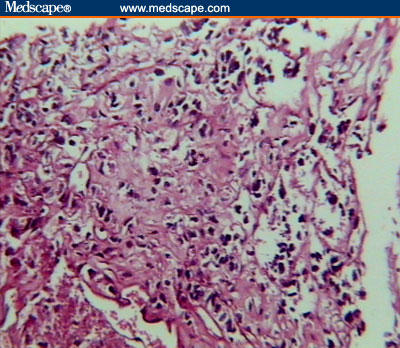
Clusters of tumor cells separated from each other by dense fibrous trabeculae, each cluster further divided into groups of cells with central degeneration and loss of cohesion resulting in an alveolar or pseudoglandular pattern (H&E stain, low power).
Figure 7.
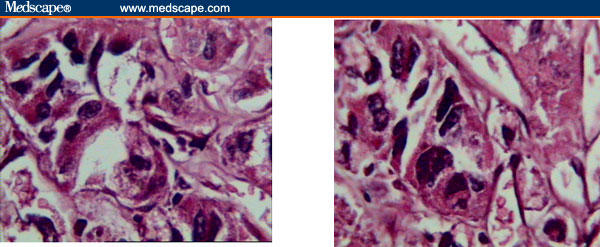
Neoplastic cells, round to polygonal in shape, with large finely granular cytoplasm and well-defined borders. Large, round-to-oval eccentrically placed nuclei, with prominent single-to-multiple nucleoli, are sometimes seen.
Contributor Information
Farhad Zamani, GI and Liver Disease Research Center, Iran University of Medical Sciences, Tehran, Iran.
Mosadegh Jabbari, Department of Nephrology, Iran University of Medical Sciences, Tehran, Iran.
Seyed Maysam Alimohamadi, Digestive Disease Research Center, Tehran University of Medical Sciences, Tehran, Iran.
Ramin Shakeri, Digestive Disease Research Center, Tehran University of Medical Sciences, Tehran, Iran. Email: rshakeri@gmail.com.
Zohreh Rostami, Department of Nephrology, Iran University of Medical Sciences, Tehran, Iran.
Behnoush Abedi, Digestive Disease Research Center, Tehran University of Medical Sciences, Tehran, Iran.
Mahshid Hormazdi, Department of Pathology, Iran University of Medical Sciences, Tehran, Iran.
Reza Malekzadeh, Digestive Disease Research Center, Tehran University of Medical Sciences, Tehran, Iran.
References
- 1.Christopherson WM, Foote FW, Jr, Stewart FW. Alveolar soft-part sarcomas: structurally characteristic tumors of uncertain histogenesis. Cancer. 1952;5:100–111. doi: 10.1002/1097-0142(195201)5:1<100::aid-cncr2820050112>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 2.Sreedevi SRBR. Alveolar soft part sarcoma of knee joint. Indian J Radiol Imag. 1991;(suppl):628–631. [Google Scholar]
- 3.Ordonez NG, Mackay B. Alveolar soft-part sarcoma: a review of the pathology and histogenesis. Ultrastruct Pathol. 1998;22:275–292. doi: 10.3109/01913129809103349. [DOI] [PubMed] [Google Scholar]
- 4.Gupta S, Jain S, Sodhani P. Alveolar soft part sarcoma: a rare entity with unique cytomorphological features. Cytopathology. 2003;14:40–41. doi: 10.1046/j.1365-2303.2003.12051.x. [DOI] [PubMed] [Google Scholar]
- 5.Poroshin KK, Krylov LM, Kudriavtsev BN. Alveolar soft tissue sarcoma [in Russian] Arkh Patol. 1989;51:51–58. [PubMed] [Google Scholar]
- 6.Fassbender HG. Alveolar myoblastic sarcoma of the skeletal musculature [in German] Oncologia. 1960;13:184–191. [PubMed] [Google Scholar]
- 7.Park YK, Unni KK, Kim YW, et al. Primary alveolar soft part sarcoma of bone. Histopathology. 1999;35:411–417. doi: 10.1046/j.1365-2559.1999.035005411.x. [DOI] [PubMed] [Google Scholar]
- 8.Casanova M, Ferrari A, Bisogno G, et al. Alveolar soft part sarcoma in children and adolescents: a report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol. 2000;11:1445–1449. doi: 10.1023/a:1026579623136. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda T, Saito M, Nakajima T. Giemsa staining for alveolar soft part sarcoma. Acta Cytol. 1999;43:519–521. [PubMed] [Google Scholar]
- 10.Weiss SW. Alveolar soft part sarcoma: are we at the end or just the beginning of our quest? Am J Pathol. 2002;160:1197–1199. doi: 10.1016/S0002-9440(10)62545-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nair A, Pai DR, Jagdish S, Krishnan R. Alveolar soft part sarcoma: a unique tumor with disputed histogenesis. Indian J Cancer. 2003;40:82–83. [PubMed] [Google Scholar]
- 12.Ronald AW, Heal DMF. Histopathogenesis of alveolar soft part sarcoma. Cancer. 1972;29:191–204. doi: 10.1002/1097-0142(197201)29:1<191::aid-cncr2820290129>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 13.Miettinen M, Ekfors T. Alveolar soft part sarcoma Immunohistochemical evidence for muscle cell differentiation. Am J Clin Pathol. 1990;93:32–38. doi: 10.1093/ajcp/93.1.32. [DOI] [PubMed] [Google Scholar]
- 14.Mukai M, Heal TCI. Histogenesis of alveolar soft part sarcoma. Am J Surg Pathol. 1986;10:218. doi: 10.1097/00000478-198603000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Wu J, Brinker DA, Haas M, Montgomery EA, Argani P. Primary alveolar soft part sarcoma (ASPS) of the breast: report of a deceptive case with xanthomatous features confirmed by TFE3 immunohistochemistry and electron microscopy. Int J Surg Pathol. 2005;13:81–85. doi: 10.1177/106689690501300112. [DOI] [PubMed] [Google Scholar]
- 16.Ladanyi M, Antonescu CR, Drobnjak M, et al. The precrystalline cytoplasmic granules of alveolar soft part sarcoma contain monocarboxylate transporter 1 and CD147. Am J Pathol. 2002;160:1215–1221. doi: 10.1016/S0002-9440(10)62548-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spagnolo DV. Alveolar soft part sarcoma: a new piece in the puzzle [news In brief] Adv Anatomic Pathol. 2003;10:48. [Google Scholar]
- 18.Bu X, Bernstein L. A proposed explanation for female predominance in alveolar soft part sarcoma. Noninactivation of X: autosome translocation fusion gene? Cancer. 2005;103:1245–1253. doi: 10.1002/cncr.20899. [DOI] [PubMed] [Google Scholar]
- 19.Pang LJ, Li F, Chang B, et al. Detection of ASPL-TFE3 fusion gene by reverse transcriptase polymerase chain reaction in paraffin-embedded tumor tissues of alveolar soft part sarcoma [in Chinese] Zhonghua Bing Li Xue Za Zhi. 2004;33:508–512. [PubMed] [Google Scholar]
- 20.Gordon MS, Hajdu SI, Bains MS, Burt ME. Soft tissue sarcomas of the chest wall. Results of surgical resection. J Thorac Cardiovasc Surg. 1991;101:843–854. [PubMed] [Google Scholar]
- 21.Portera CA, Jr, Ho V, Patel SR, et al. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001;91:585–591. doi: 10.1002/1097-0142(20010201)91:3<585::aid-cncr1038>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 22.Heller DS, Frydman CP, Gordon RE, Jagirdar J, Schwartz IS. An unusual organoid tumor. Alveolar soft part sarcoma or paraganglioma? Cancer. 1991;67:1894–1899. doi: 10.1002/1097-0142(19910401)67:7<1894::aid-cncr2820670713>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 23.Sujit Kumar GS, Chacko G, Chacko AG, Rajshekhar V. Alveolar soft-part sarcoma presenting with multiple intracranial metastases. Neurol India. 2004;52:257–258. [PubMed] [Google Scholar]
- 24.Nickerson HJ, Silberman T, Jacobsen FS, Krawisz BR, Maki HS, Arndt CA. Alveolar soft-part sarcoma responsive to intensive chemotherapy. J Pediatr Hematol Oncol. 2004;26:233–235. doi: 10.1097/00043426-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Reichardt P, Lindner T, Pink D, Thuss-Patience PC, Kretzschmar A, Dorken B. Chemotherapy in alveolar soft part sarcomas. What do we know? Eur J Cancer. 2003;39:1511–1516. doi: 10.1016/s0959-8049(03)00264-8. [DOI] [PubMed] [Google Scholar]
- 26.Pappo AS, Rao BN, Jenkins JJ, et al. Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol. 1999;33:76–82. doi: 10.1002/(sici)1096-911x(199908)33:2<76::aid-mpo3>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 27.Lieberman PH, Brennan MF, Kimmel M, Erlandson RA, Garin-Chesa P, Flehinger BY. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer. 1989;63:1–13. doi: 10.1002/1097-0142(19890101)63:1<1::aid-cncr2820630102>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]