

Abstract
The P4 helix is an essential element of ribonuclease P (RNase P) that is believed to bind catalytically important metals. Here, we applied a combination of NMR residual dipolar couplings (RDCs) and a recently introduced domain-elongation strategy for measuring “motionally decoupled” relaxation data to characterize the structural dynamics of the P4 helix from Bacillus subtilis RNase P. In the absence of divalent ions, the two P4 helical domains undergo small amplitude (~13°) collective motions about an average interhelical angle of 10°. The highly conserved U7 bulge and helical residue C8, which are proposed to be important for substrate recognition and metal binding, are locally mobile at pico- to nanosecond timescales and together form the pivot point for the collective domain motions. Chemical shift mapping reveals significant association of Mg2+ ions at the P4 major groove near the flexible pivot point at residues (A5, G22, G23) previously identified to bind catalytically important metals. The Mg2+ ions do not, however, significantly alter the structure or dynamics of P4. Analysis of results in the context of available X-ray structures of the RNA component of RNase P and structural models that include the pre-tRNA substrate suggest that the internal motions observed in P4 likely facilitate adaptive changes in conformation that take place during folding and substrate recognition, possibly aided by interactions with Mg2+ ions. Our results add to a growing view supporting the existence of functionally important internal motions in RNA occurring at nanosecond timescales.
Keywords: RNA dynamics, electrostatics, collective motions, catalysis, RNP folding
INTRODUCTION
Ribonuclease P (RNase P) is a ubiquitous ribonucleoprotein (RNP) enzyme that catalyzes hydrolysis of phosphodiester bonds in precursors of transfer RNA (pre-tRNA) to form mature tRNA. In bacteria, RNase P consists of a large (350–400 nt), essential RNA component and one or more protein components that are dispensable for catalysis at high salt and metal concentrations (Guerrier-Takada et al. 1983; Kurz and Fierke 2000). In both the presence and absence of protein, divalent cations are required to activate catalysis by RNase P, Mg2+ being the preferred metal in vivo (Guerrier-Takada et al. 1986; Smith and Pace 1993; Kurz and Fierke 2000). The divalent ions can play at least three roles: they can aid in folding of the enzyme, participate in substrate binding, and be directly involved in phosphodiester bond hydrolysis (Guerrier-Takada et al. 1986; Pan 1995; Beebe et al. 1996).
A large body of work has established the importance of the universally conserved P4 helical element located at the junction of the RNase P catalytic and specificity domains (Fig. 1). Phosphorothioate substitution studies (Hardt et al. 1995; Harris and Pace 1995; Crary et al. 2002) together with nucleotide analog interference mapping (NAIM) (Hardt et al. 1996; Kazantsev and Pace 1998; Heide et al. 1999; Siew et al. 1999; Kaye et al. 2002a) have shown that residues below the bulge are important for both tRNA binding (Fig. 1A, blue symbols) and catalysis (Fig. 1A, red symbols). In many cases, the phosphorothioate substitution effects could be rescued by Mn2+, indicating the involvement of metal cations in both substrate binding (Fig. 1A, black filled blue arrows; Hardt et al. 1995) and catalysis (Fig. 1A, black filled red arrows; Harris and Pace 1995; Christian et al. 2000; Crary et al. 2002). The rescue of catalysis at the A50 position raised the possibility that the water-activating catalytic metal binds to this site in P4 (Harris and Pace 1995). However, recent studies have called into question the direct involvement of P4 and its bound metals in the RNase P catalytic step. Two crystal structures of the RNA component of A- and B-type RNase P (Torres-Larios et al. 2005; Kazantsev et al. 2005) show a large distance between P4 and the sites in the catalytic domain that cross-link to the cleavage site in pre-tRNA (Fig. 1B, blue and green, respectively; Christian et al. 1998). Thus, any direct involvement of P4 in catalysis would require a conformational change following substrate recognition. This is not unprecedented given that conformational changes in the RNA component of RNase P have been observed via cross-linking studies to pre-tRNA and mature tRNA (Pomeranz Krummel and Altman 1999). A recent study involving cross-linking data and Cd2+ rescue at the active site suggests that the metals bound to P4 are important for positioning the pre-tRNA substrate relative to the active site rather than directly catalyzing hydrolysis (Christian et al. 2006), thereby reconciling the biochemical and crystallographic data.
FIGURE 1.

The P4 element of RNase P. (A) Secondary structure of P4 derived from the crystal structure of B. stearothermophilus RNase P (Kazantsev et al. 2005). Green circles indicate Mg2+ binding sites identified in E. coli P4 by NMR (Schmitz and Tinoco 2000). Black open circle indicates a site of mutation that disrupts catalysis (Frank and Pace 1997); arrows indicate sites where phosphorothioate substitutions interfere with catalysis (red) and substrate binding (blue), or both (black; Hardt et al. 1995; Harris and Pace 1995; Christian et al. 2000; Crary et al. 2002); black filled arrows indicate sites where interference is rescued with Mn2+ (Hardt et al. 1995; Harris and Pace 1995; Christian et al. 2000; Crary et al. 2002); filled circles indicate sites where nucleotide analog substitutions interfere with catalysis (red; Kazantsev and Pace 1998; Kaye et al. 2002a), substrate binding (blue; Heide et al. 1999; Siew et al. 1999), or both (black; Kazantsev and Pace 1998; Heide et al. 1999; Kaye et al. 2002a); blue stars indicate 2′OH sites that are important for substrate binding (Hardt et al. 1996). Uppercase letters are universally conserved residues in RNase P (Chen and Pace 1997). (B) Crystal structure of B. stearothermophilus RNase P (Kazantsev et al. 2005). Highlighted in color are P4 (blue), the P4 bulge (yellow), L8 (pink), the specificity domain (white), the catalytic domain (orange), and tRNA cross-link sites (green; Christian et al. 1998). (C) The P4 construct from B. subtilis used in our NMR studies with hydrogen bond alignments derived from the JNN-COSY experiment (Dingley and Grzesiek 1998; Pervushin et al. 1998). For comparison, the P4 construct from E. coli used in previous NMR studies is also shown (Schmitz and Tinoco 2000).
Although its precise role in catalysis remains to be elucidated, P4 is an integral element of RNase P whose function is intimately linked to metal binding. NMR structures and metal binding properties of the Escherichia coli P4 helix have previously been reported (Schmitz and Tinoco 2000). However, little is known about the intrinsic structural plasticity of P4 and its potential role in RNase P function. Given its central location near the substrate binding site, there is little doubt that the RNase P catalytic cycle, beginning with folding and assembly of the ribonucleoprotein followed by recognition of substrate, catalysis, and finally product release, will require changes in the P4 conformation that allow the structural requirements of these distinct steps to be dynamically satisfied by a single enzyme. Mounting studies indicate that the dynamical properties of RNA structures play an important role in such adaptive conformational changes (Williamson 2000; Leulliot and Varani 2001; Al-Hashimi 2005). Recent advances in NMR methodology, including the measurement of residual dipolar couplings (RDCs) (Tolman et al. 1995; Tjandra and Bax 1997; Prestegard et al. 2000; Al-Hashimi et al. 2002b; Latham et al. 2005; Leeper and Varani 2005; Richards et al. 2006) and 13C/15N relaxation data (Duchardt and Schwalbe 2005; Shajani and Varani 2005; Showalter and Hall 2005; Vallurupalli and Kay 2005; Zhang et al. 2006), are allowing the dynamical properties to be visualized at atomic resolution.
Here, we applied a combination of RDCs and motionally decoupled relaxation data together with chemical shift mapping experiments to characterize at atomic resolution the structural plasticity and Mg2+ binding properties of P4 from Bacillus subtilis. Our results show the power of combining these NMR techniques in characterizing RNA dynamics and identify local and global structural plasticity and possible Mg2+ binding sites in P4 that may play a role in dynamical steps of RNase P catalysis.
RESULTS
Resonance assignments, hydrogen bond alignments, and RDC measurements in P4 in the absence of divalent ions
The B. subtilis P4 construct used in our study differs slightly from the E. coli construct used in previous NMR studies (Fig. 1C; Schmitz and Tinoco 2000). While the bulge and flanking Watson–Crick base pairs are identical (as well as the UUCG loop added for stability), our P4 construct contains an additional A3–U27 base pair that serves to conserve the stretch of three consecutive adenines below the bulge. Other differences in the Watson–Crick identity of residues in domain II reflect differences between the E. coli and B. subtilis P4 sequences (Fig. 1C; Kazantsev et al. 2005). Our initial NMR characterization focused on P4 under low ionic strength conditions in the absence of divalent ions (buffer conditions: 10 mM sodium chloride, 10 mM sodium phosphate, pH ~6.2). This was followed by studies examining Mg2+ binding and its effects on the P4 structural dynamics.
The 1H, 13C, and 15N resonances in B. subtilis P4 in the absence of divalent ions were assigned using standard NMR experiments employing uniformly 13C/15N labeled or unlabeled P4. The nonexchangeable NOE walk (Wüthrich 1986) was uninterrupted from G18 to U25, indicating a continuous helical conformation. While the NOE walk was interrupted at the U7–C8 junction on the opposite strand, observation of a weak NOE cross-peak between H1′ of G6 and H6 of C8 is consistent with a pseudo-continuous helical conformation involving an unstacked looped out uridine bulge (Fig. 2A). No or weak NOE connectivities were observed between terminal residues G1/G2 and C28/C29, likely due to end-fraying effects. In addition, the NOE cross-peaks between C8 and C9 and between G23 and C24 were comparatively weak, suggesting that the C8–G23 base pair is perturbed from ideality. The latter was confirmed using the JNN-COSY experiment (Fig. 2B; Dingley and Grzesiek 1998; Pervushin et al. 1998). The intensity of the auto- and cross-peaks for G23 are attenuated by ~44% and ~50%, respectively, relative to mean guanine values (excluding the end-fraying G2). The corresponding attenuation was less significant for G6 below the bulge (~24% and ~20%, respectively). Thus, C8–G23 and to a lesser extent G6–C24 experience considerable exchange broadening. In addition, the 2hJNN coupling constant obtained for C8–G23 (4.8 Hz) and to a lesser extent G6–C24 (5.2 Hz) are smaller than typical values expected for G–C base pairs (~5.5 Hz) (Furtig et al. 2003). These results suggest that the bulge introduces deviations from ideality in its flanking G–C base pairs, with the deviations being greater for the base pair above the bulge.
FIGURE 2.

NMR spectroscopy of P4 in the absence of Mg2+. (A) Expanded region of the nonexchangable NOESY of P4 showing the NOE walk for the A3–C13 strand. (B) Detection of hydrogen bond alignments using the JNN-COSY experiment (Dingley and Grzesiek 1998; Pervushin et al. 1998). The measured 2hJNN coupling constants (in hertz) are shown adjacent to each cross peak (Dingley et al. 1999). (C) One bond C–H RDCs measured in P4 as a function of residue.
Comparison of one bond C–H RDCs [C1′H1′, C2H2, C5H5, C6H6, and C8H8] measured in P4 in the absence of divalent ions using 15 mg/mL of Pf1-phage (Tjandra and Bax 1997; Clore et al. 1998; Hansen et al. 1998; Prestegard et al. 2000) provided qualitative insight into the structural dynamics of P4. As expected from a pseudo-continuous helical conformation, RDCs measured for a given type of vector fall within a relatively narrow range (Fig. 2C). The small RDCs observed for G1 likely reflect dynamical averaging due to end fraying. Much smaller RDCs (near zero values) were observed in base and sugar moieties of the U7 bulge (Fig. 2C). Though an unusual static conformation could not be ruled out, these attenuated RDCs suggest a high degree of local mobility at U7 occurring at submillisecond timescales. The comparatively smaller RDCs in the nucleobases of C8 and G23 are also indicative of an unusual conformation and/or flexibility that may explain their perturbed hydrogen bond alignment (Fig. 2C).
Conformation of P4 in the absence of divalent ions using RDCs
We determined the relative orientation and dynamics of the two P4 helical domains in the absence of divalent ions by subjecting RDCs measured in nonterminal residues to an order tensor analysis (Losonczi et al. 1999; Tolman et al. 2001; Al-Hashimi et al. 2002b). Watson–Crick regions of the two domains were modeled using an idealized A-form helix geometry, whereas the UUCG loop in domain II was modeled using an X-ray structure (Ennifar et al. 2000) as previously described for TAR (Al-Hashimi et al. 2002a). The idealized A-form geometry has been validated for nonterminal Watson–Crick base pairs using RDCs in a number of different contexts (Mollova et al. 2000; Sibille et al. 2001; Al-Hashimi et al. 2002b; Pitt et al. 2004, 2005; Hansen and Al-Hashimi 2006; Musselman et al. 2006).
Shown in Figure 3A is the correlation between measured and back-calculated RDCs (16 and 17 for domains I and II, respectively) when independently fitting each domain to an order tensor. Owing to potential deviations from ideality, RDCs from G1–C29, G2—C28, and C8–G23 were not included in this analysis. A very good fit is observed with the root-mean-square deviation (RMSD = 2.5 and 2.9 Hz for domains I and II/loop, respectively) approaching the RDC measurement uncertainty (2.4 Hz) estimated from duplicate measurements (Pitt et al. 2004). This indicates that the targeted Watson–Crick residues in the two domains adopt the expected local A-form helix geometry. Importantly, significant deviations (~9.3 Hz) were observed between the sugar and base RDCs measured in residue C8 and values back-calculated assuming the idealized A-form helix and the above order tensor obtained from a fit that excludes the G23 and C8 RDCs (Fig. 3A, open symbols). The better agreement observed for the base RDCs in G23 suggests that its base moiety behaves as expected for an ideal Watson–Crick base pair. Thus, local structural perturbations in C8, which is adjacent to the flexible U7 bulge (see below), may explain the anomalous hydrogen bonding observed in the C8–G23 base pair (Fig. 2B). Such RDC deviations were not observed in the G6–C24 base pair below the bulge (Fig. 3A).
FIGURE 3.

Order tensor analysis of RDCs measured in P4 in the absence of Mg2+. (A) Correlation plot between measured RDCs and back-calculated values when independently fitting order tensors to an idealized A-form geometry for domains I (red) and II (blue). Error bars represent the RDC measurement uncertainty (2.4 Hz). RDCs from residues C8 and C24 (open squares) and G23 and G6 (open diamonds) are shown but C8 and G23 were not included in the singular value decomposition fit. (B) The RDC derived average interdomain orientation obtained from superimposing order tensor frames (left). For comparison, the X-ray structure of P4 in B. stearothermophilus (Kazantsev et al. 2005) is also shown (right). (C) Correlation plot between measured and back-calculated RDCs when fitting RDCs against the X-ray structure of P4 (Kazantsev et al. 2005). RDCs from domains I and II are shown in red and blue, respectively. Inset shows residues common to both the NMR construct and crystal structure (Kazantsev et al. 2005). (D) The best-fit GDOs for domains I (red) and II (blue) as a function of input RDCs over 25 iterations (circles) upon the removal of two random RDCs. Best-fit values when using all RDCs in each domain are shown as horizontal lines.
The order tensor analysis of RDCs yields an order tensor frame for each helical domain (Sxx, Syy, Szz), which describes the average domain orientation relative to the applied magnetic field. The average orientation of helical domains relative to one another can be determined by superimposing their respective order tensor frames (Losonczi et al. 1999; Tolman et al. 2001; Al-Hashimi et al. 2002b). The latter amounts to insisting that the two domains have, on average, an equivalent view of the magnetic field direction when assembled into a proper average conformation. The relative orientation of the two P4 helical domains obtained by superimposing their respective RDC-derived order tensor frames is shown in Figure 3B. Consistent with the NOE data, the interhelical conformation only slightly deviates from coaxial alignment. The average interhelical bend and twist angles are 10° ± 5° and 14° ± 30°, respectively (Fig. 3B). This P4 conformation falls within the distribution of conformations reported in the previous NOE-based NMR structures of E. coli P4 (interhelical bend and twist angles ranging between 12° and 34° and −42° and 43°, respectively) (Schmitz and Tinoco 2000). The P4 interhelical angle of 10° is also in excellent agreement with that determined using transient electric birefringence for double helices containing a single uridine bulge (10° ± 5°) (Zacharias and Hagerman 1995).
Comparison of the isolated P4 conformation derived by RDCs with that observed in the context of the 3.3-Å X-ray structure of the 417-nt RNA component of RNase P from the related bacteria Bacillis stearothermophilus allowed us to evaluate the relevance of our P4 construct as well as assess possible differences arising from interactions present in the larger RNA context (Fig. 3B; Kazantsev et al. 2005). While the P4 interhelical twist observed in the X-ray structure (~15°) is in excellent agreement with that obtained by RDCs (~14°), the X-ray structure has a slightly more bent interhelical angle (~17° compared to ~10°) (Fig. 3B). Due to the latter difference and the presence of interdomain motions (see below), slightly less favorable agreement is observed when directly fitting the RDCs to matching regions on the P4 X-ray structure (RMSD = 3.3 Hz) (Fig. 3C). This difference cannot be attributed to the presence of divalent ions in the crystallization conditions since our results indicate that Mg2+ binding does not significantly alter the P4 interhelical alignment (see below). Small differences in the interhelical angle may arise from tertiary contacts between the upper stem of P4 and L8 that are present in the X-ray structure (Fig. 1B) but not in the isolated P4 construct (Kazantsev et al. 2005). Another point of agreement between the X-ray structure (Fig. 1B) and NMR data is the looped out U7 bulge conformation in which the nucleobase is not sterically forbidden from freely rotating around the χ angle (Fig. 3B; Kazantsev et al. 2005). Thus, the internal motions observed in U7 by RDCs (Fig. 2C) and motional narrowing (see below) also likely occur in the larger RNA context.
Conformational dynamics of P4 using RDCs
While the mean P4 interhelical angle derived by RDCs in the absence of divalent ions differs slightly from the X-ray structure, the RDCs provide evidence for dynamical excursions about the mean angle that would allow access to the conformation observed in the X-ray structure. The order tensor analysis of RDCs also yields a generalized degree of order (GDO, ϑ) (Tolman et al. 2001; Al-Hashimi et al. 2002b), which describes the degree of alignment experienced by each domain. Rigidly held domains will experience a common degree of alignment and therefore report identical GDOs (Tolman et al. 2001; Al-Hashimi et al. 2002b). However, interdomain motions can result in different levels of domain alignment and hence different domain GDOs. In the latter case, the ratio of the domain GDOs, referred to as the internal generalized degree of order (GDOint, ϑint) can be used to assess the amplitude of interdomain motions varying between 1 for interdomain rigidity and 0 for maximum interdomain motions (Tolman et al. 2001; Al-Hashimi et al. 2002b). In P4, the GDO value obtained for domain II (1.25 × 10−3 ± 8%) was smaller than that obtained for domain I (1.35 × 10−3 ± 3%) (Fig. 3D, horizontal lines) yielding a GDOint value of ~0.93 ± 9%. The GDO uncertainties reflect both RDC measurement uncertainty and structural noise in the A-form geometry as implemented in the program Aform-RDC (Musselman et al. 2006). Although the difference in the domain GDOs is comparable to their experimental uncertainty, it was consistently observed regardless of the input RDCs used in the order tensor determination (Fig. 3D). More importantly, independent evidence for domain motions was obtained using motionally decoupled spin relaxation data (see below). The smaller level of order observed for domain II may be attributed to the location of the disordered C8–G23 base pair, and therefore the hinge point is closer to domain II as compared to domain I. Assuming isotropic motions in a cone (Tolman et al. 1997; Al-Hashimi et al. 2002b), the observed GDOint value translates into domain motional amplitudes of 13°, yielding a P4 conformation in which the interhelical angle varies dynamically between −3° and 23°. The P4 conformation observed in the X-ray structure of B. stearothermophilus (Fig. 3B) falls within this dynamical envelope. Thus, it is possible that tertiary contacts between the upper helix of P4 and L8 capture one of many possible P4 interhelical conformations during folding of RNase P.
Collective and librational motions in P4 in the absence of divalent ions from motionally decoupled 15N relaxation
To obtain further insight into both librational motions and collective dynamics, we used a recently described domain-elongation strategy to decouple internal motions in P4 from overall reorientation (Zhang et al. 2006). The elongation is accomplished by extending the size of the terminal domain I using a stretch of 22 Watson–Crick base pairs (Fig. 4A). This slows down overall rotational diffusion, allowing its separation from internal motions occurring at timescales comparable to overall molecular tumbling of non-elongated P4 (Lipari and Szabo 1982). To avoid increasing NMR spectral overlap, two RNA constructs were prepared that are elongated using stretches of either unlabeled A–U (E-AU-P4) or G–C (E-GC-P4) base pairs in a background of uniformly 13C/15N-labeled G–C or A–U nucleotides, respectively (Fig. 4A, colored residues).
FIGURE 4.

Measurement and extended model-free analysis of E-P4 15N relaxation data in the absence of Mg2+. (A) Secondary structure of E-GC-P4 and E-AU-P4. 13C/15N labeled and unlabeled residues are shown in color and black, respectively. (B) 2D 1H–13C HSQC spectra of aromatic region of E-GC-P4+E-AU-P4 (red) overlaid on non-elongated P4 (black). Natural abundance signals from elongation residues are indicated with an asterisk. (C) Examples of 15N R 1 and R 2(CPMG) monoexponential decays in E-P4. (D) The measured R 2/R 1 values for guanine (filled circles) and uridine (filled diamonds) residues in domains I (red) and II (blue) of P4 (left) and E-P4 (right). Values predicted using hydrodynamic calculations are shown as open symbols. (E) Dynamical parameters for E-P4 derived from extended model-free analysis (Lipari and Szabo 1982; Clore et al. 1990). Shown are the time constants/amplitudes for fast (τf/S2 f) and slow (τs/S2 s) internal motions in the absence of divalent ions (filled symbols) and in the presence of 15 mM MgCl2 (open symbols).
We obtained high quality NMR spectra for E-P4 that exhibit excellent overlap with spectra of non-elongated P4 with minor chemical shift differences observed near the site of elongation (Fig. 4B). Folding of elongation residues into their predicted helical structure was confirmed based on a number of factors, including observation of intense imino proton peaks at the expected Watson–Crick chemical shift region (data not shown). The successful elongation of P4 supports the general applicability of this technique in studies of RNA dynamics.
Imino nitrogen (15N) relaxation data were measured for guanine and uridine residues in both non-elongated and elongated P4 in the absence of divalent ions (Fig. 4C; Zhang et al. 2006). As shown in Figure 4D, the transverse (R 2) to longitudinal (R 1) relaxation rate ratios (R 2/R 1) measured in the two domains of P4 are very similar. Given the expected isotropic overall tumbling of non-elongated P4, these data would normally be interpreted as evidence that the two domains are held rigid relative to one another. However, these data do not rule out the presence of domain motions that cannot be separated from overall molecular tumbling because they occur at similar timescales. Indeed, the corresponding data measured in E-P4 provides evidence for domain motions. As shown in Figure 4D, significantly smaller R 2/R 1 values are observed for the smaller domain II as compared to the elongated domain I (Fig. 4D). This is despite the fact that for a rigid conformation, near minimum R 2/R 1 values are expected for domain I given that its N–H bonds are oriented perpendicular to the long principal axis of anisotropic rotational diffusion (Zhang et al. 2006). Rather, the uniformly smaller domain II R 2/R 1 values are exactly as would be expected if domain II reoriented semi-independently at timescales faster than overall E-P4 tumbling. The excellent agreement between the domain I R 2/R 1 values and those predicted based on the RDC-derived E-P4 conformation and hydrodynamic calculations provides further support that elongation residues adopt the expected helical conformation (Fig. 4D, open symbols; Garcia de la Torre et al. 2000).
We subjected the E-P4 15N relaxation data to an extended model-free analysis (Lipari and Szabo 1982; Clore et al. 1990; Mandel et al. 1995; Zhang et al. 2006). This yielded a time constant for overall E-P4 rotational diffusion in the absence of divalent ions (τM = 19.7 ± 0.2 and 19.1 ± 0.2 nsec for E-AU-P4 and E-GC-P4, respectively) that is in very good agreement with hydrodynamic predictions (18.5 ns at 298 K) (Garcia de la Torre et al. 2000). Similar N–H librations were observed throughout the two helices with time constants (τf) in the 23–27 psec range and amplitudes ranging between S 2 f = 0.83–0.89, where S 2 f is the Lipari–Szabo (Lipari and Szabo 1982) spin relaxation order parameter, which varies between 0 and 1 for maximum and minimum motions (Fig. 4E, filled symbols). The N–H sites in domain II experience an additional slower motional mode characterized by uniform amplitudes (S 2 s ~ 0.90) and time constants in the near nanosecond range (τs = 0.9–1.3 nsec) (Fig. 4E, filled symbols). This is as expected if all of the N–H sites moved together as part of a common collective dynamical process. Remarkably, the amplitude of domain motions observed by relaxation (Ss = 0.91/2 = 0.95) is in very good agreement with that obtained independently by RDCs (ϑint = 0.93). This suggests that the domain motions sensed by RDCs occur at nanosecond timescales and that little to no motions occur at micro- to millisecond timescales.
Local motions from motional narrowing of resonances in E-P4 in the absence of divalent ions
The E-P4 resonance intensities exposed local motions that evade detection in non-elongated P4 because they occur at timescales approaching overall molecular tumbling. Ignoring chemical exchange, the resonance intensity for a given C–H bond reports on the net bond vector reorientational dynamics relative to the applied magnetic field. In P4, very similar intensities were observed for various C–H sites (Fig. 5A). In contrast, significantly higher intensities indicative of fast internal motions were observed for a number of sites in E-P4, the most notable being the U7 bulge and UUCG loop (Fig. 5B). These motions escape detection in non-elongated P4 likely because they occur at timescales slower than overall tumbling (~5.6 nsec). The motional narrowing at U7 confirms that the near zero RDCs measured at U7 reflect extensive dynamical averaging (Fig. 2C), painting a picture for the U7 bulge, which counter to previous NMR studies of P4 (Schmitz and Tinoco 2000) is essentially unstructured. Interestingly, slightly higher than average intensities were also observed for the neighboring C8, indicating that its unusual RDCs and hydrogen bond alignment may arise from dynamical disorder. Together, the locally mobile U7 and C8 provide a flexible hinge and thus a molecular basis for the observed domain motions. The virtually identical chemical shifts observed for U7 and C8 in E-P4 and P4 (Fig. 4B) underscore the point that the observed dynamics in E-P4 are not induced by elongation, but rather escape detection in non-elongated P4. As expected, the relative intensities for the UUCG loop are in excellent agreement with those reported for the UUCG loop in E-TAR (Zhang et al. 2006), which are in turn in excellent agreement with motional amplitudes reported previously by 2H relaxation (Vallurupalli and Kay 2005).
FIGURE 5.

Local motions in E-P4 in the absence of Mg2+ from motional narrowing of resonances. Shown are the normalized resonance intensities as a function of residue in (A) P4 and (B) E-P4 obtained from nonconstant time 1H–13C HSQC experiments. Intensities for a given type of C-H vector are normalized relative to a minimum value of 0.1 indicated by a horizontal line (Zhang et al. 2006).
Mapping Mg2+ association by chemical shift mapping and electrostatic calculations
To characterize the Mg2+ binding properties of P4, we recorded 2D HSQC spectra following incremental addition of Mg2+ up to 15 mM. Relative to previously published NMR chemical shift mapping data on E. coli P4 (Schmitz and Tinoco 2000), we employed a lower background of monovalent ions (10 mM compared to 100 mM) and used a combination of N–H and C–H chemical shift probes. Relatively small chemical shift perturbations were observed throughout P4 that are indicative of nonspecific interactions with diffusive counterions. Some of the largest perturbations are observed at residue A5 (Fig. 6A), which based on phosphorothioate rescue experiments is the site that binds a catalytically important metal ion (Harris and Pace 1995). Association of Mg2+ ions at this site may have escaped detection in the previous NMR study, which relied on imino proton probes that are absent in adenine residues. In agreement with previous NMR Mg2+ mapping data (Schmitz and Tinoco 2000), we also observe Mg2+ association within a region above the bulge around the perturbed C8–G23 base pair (Fig. 6A).
FIGURE 6.

Mg2+ association with P4. (A) Selected regions of 2D 13C/15N/1H HSQC spectra of P4 in the presence (red) and absence (black) of 15 mM MgCl2. Significant chemical shifts (>0.08 ppm) are highlighted. Mg2+-induced chemical shift perturbations in P4 (>0.08 ppm) observed for C6H6/C8H8 (circles), C5H5 (squares), C1′H1′ (stars), and N1H1/N3H3 (“x”) with the highest shifts across all sites shown in red. (B) Representative titration curves as a function of total Mg2+ concentration with Kd values highlighted on the P4 secondary structure (terminal residues not shown): red <1.0 mM, orange 1–2 mM, green 2–4 mM, and blue >4 mM. Chemical shift mapping data were available in certain cases without corresponding Kd values because titration spectra were recorded with reduced resolution in the indirect dimension. (C) Electrostatic surface potential of P4 computed using the B-type X-ray structure (Glemarec et al. 1996; Kazantsev et al. 2005) with negative and positive potentials indicated in red and blue, respectively.
To obtain further insight into metal binding, we measured apparent Kd values by fitting the chemical shift titration data to a two-state binding model (Fig. 6B). The apparent Kd values ranged between 0.7 and 7.3 mM, with the highest affinity observed for metal binding at G22. High affinity is also observed at A5 (Kd of ~2.0 mM). To aid interpretation of these results, we used the X-ray structure of P4 from the 417-nt RNA component to compute the electrostatic potential surface employing the Delphi (Accelrys) module of Insight II (Molecular Simulations, Inc). Results revealed a strong surface accessible electrostatic potential precisely around residues exhibiting the largest Mg2+-induced chemical shift perturbations and tight Mg2+ association, namely, residues A5, G22, and G23 above and below the bulge that cluster together in the major groove of P4 (Fig. 6C). The electrostatic potential in this region is weaker for an idealized coaxial A-form helix containing the P4 sequence without the bulge (data not shown). Thus, the distortion in the interhelical alignment and the C8–G23 base pair induced by the U7 bulge appears to promote formation of strong electrostatic potentials in regions where Mg2+ association is observed.
How does Mg2+ affect the structure and dynamics of P4?
To examine if and how Mg2+ affects the P4 structural dynamics, we repeated the NMR characterization in the presence of 15–20 mM Mg2+. Assuming a simple two state model, these Mg2+ concentrations are expected to lead to up to 89% of P4 being in the Mg2+ associated state. Results are summarized in Figure 7. Consistent with the relatively small Mg2+-induced chemical shift perturbations, our results indicate that Mg2+ binding does not significantly alter the structure or dynamics of P4. This is in good agreement with previous studies on E. coli P4, in which NOESY data also argued that neither Mg2+ nor cobalt (III) hexamine binding led to detectable changes in the P4 conformation (Schmitz and Tinoco 2000). The JNN-COSY spectrum recorded in the presence of Mg2+ shows hydrogen bond alignments very similar to those observed in free P4 (Fig. 2B), including diminished auto- and cross-peak intensities as well as 2hJNN coupling constants for the C8–G23 base pair above the bulge (Fig. 7A). Interestingly, however, Mg2+ binding does result in an increase in all of the measured 2hJNN coupling constants, consistent with greater stabilization of the helical structure relative to the low ionic strength conditions.
FIGURE 7.

Structural dynamics of P4 in the presence of Mg2+. (A) JNN-COSY spectrum of P4 in Mg2+. The 2hJNN coupling constants (in hertz) are shown next to each cross peak (Dingley et al. 1999). (B) Correlation plot between P4 RDCs measured in 20 mM Mg2+ and values back-calculated when independently fitting order tensors to an idealized A-form geometry for domains I (red) and II (blue). Error bars represent measurement uncertainty (2.4 Hz as for free, Fig. 3A). RDCs from residues C8 (open squares) and G23 (open diamonds) are shown but were not included in the analysis. (C) Orientation of order tensor frames (Sxx, Syy, and Szz) for domains I (in red) and II (in blue) determined in the presence of Mg2+. Results are shown relative to the free P4 interhelical conformation determined using RDCs in the absence of Mg2+. (D) 15N R 2/R 1 values for guanine (circles) and uridine (diamonds) residues measured in P4 in the presence of 15 mM Mg2+. Average values for each domain are shown as dotted lines. (E) One-bond C–H RDCs in P4 (left) and normalized intensities in E-P4 (right) in the bulge and flanking residues measured in the absence (filled symbols) and presence (open symbols) of MgCl2. (F) Schematic diagram of local and global motions in P4 and Mg2+ association (yellow residues).
Order tensor analysis of P4 RDCs measured in the presence of Mg2+ also showed very little change in the local conformation of domains or their average relative orientation. The two domains exhibit an equally good fit to the idealized A-form geometry (Fig. 7B) as observed for the RDCs measured in the absence of Mg2+ (Fig. 3A). The RDCs also show that deviations of C8 from ideality persist in the presence of Mg2+ (Fig. 7B). The average interhelical alignment obtained from the RDCs measured in Mg2+ (interhelical bend and twist angles of 11° ± 5° and 15° ± 30°) is virtually identical to that obtained in the absence of Mg2+ (interhelical bend and twist angles of 10° ± 5° and 14° ± 30°, respectively). This result is illustrated in Figure 7C, where the domain I and II Mg2+ order tensor frames are depicted relative to the free P4 interhelical alignment derived using RDCs. The domain I and II order tensor frames overlap one another about all three principal directions (Sxx, Syy, and Szz), indicating that Mg2+ binding does not alter the average P4 interhelical alignment. Interestingly, the same order tensor analysis yields a ratio for the domain order (ϑint = 0.80 ± 8%), which is smaller than that observed in the absence of Mg2+ (ϑint = 0.93 ± 9%). Thus, Mg2+ binding does not arrest domain motions in P4.
The persistence of domain motions in the presence of Mg2+ was also confirmed by the 15N relaxation data measured in E-P4 in 20 mM Mg2+. As shown in Figure 7D, the R 2/R 1 values measured in domain II exhibit a similar level of attenuation (~12%) relative to values measured in domain I as is observed in free E-P4 (~15%) (Fig. 4D). Model-free analysis of the Mg2+ relaxation data yielded dynamical parameters that are very similar to those obtained in free E-P4 (Fig. 4E, open symbols). It is important to note that the Mg2+-induced chemical shift perturbations observed in E-P4 were virtually identical to those observed in non-elongated P4, further confirming that elongation does not disturb the integrity of P4 (data not shown). Slightly higher Mg2+ concentrations were required to induce the chemical shift perturbations observed in small P4, likely due to having competing sites in the elongation residues of E-P4. Interestingly however, unlike for free E-P4, we observe systematically higher R 2/R 1 values for E-GC-P4 compared to E-AU-P4 in the presence of Mg2+ (Fig. 7D). This systematic difference reflects the slightly reduced overall tumbling rate for E-GC-P4 (τM = 20.2 nsec) compared to E-AU-P4 (τM = 19.2 nsec) in the presence of Mg2+. This may arise from greater association of Mg2+ ions with long stretches of alternating GCs in the E-GC-P4 construct, which may modify the overall hydrodynamic properties of the RNA.
Finally, results also indicate the persistence of significant local motions in the U7 bulge in the presence of Mg2+. As shown in Figure 7E, the U7 RDCs remain near zero and are hardly affected by Mg2+ binding. Although a small reduction in the U7 resonance intensities in E-P4 is observed relative to the free form, they remain significantly elevated relative to the reference value of 0.1. The observed decrease in resonance intensities may also reflect exchange broadening due to nonspecific Mg2+ association and not necessarily a reduction in motional amplitudes.
DISCUSSION
The P4 helix is universally conserved across all three kingdoms of life, has a central location in the RNase P catalytic domain, and has been the subject of many biochemical studies that show it plays an essential role in both substrate binding and catalysis. Our study illuminates dynamical aspects and metal binding properties of P4 that may be relevant to its function. It also provides new insight into the dynamical nature of RNA and how it can be studied using a combination of NMR techniques.
The combined application of RDCs and motionally decoupled spin relaxation data allowed us to better define the overall dynamics of P4 both in providing complementary information as well as in affording a unique opportunity to cross-validate results. The domain elongation proved to be absolutely critical for the spin relaxation studies (Zhang et al. 2006), without which little to no information would have been obtained about the dynamics of P4. Our ability to observe small amplitude domain motions in P4 in the presence and absence of Mg2+ ions was greatly aided by independent observation of the motions using RDCs and spin relaxation. This also afforded an opportunity to probe the amplitude of motions at two different timescale regimes, namely, pico- to nanosecond and pico- to millisecond. The similar domain motional amplitudes obtained by RDCs and spin relaxation in the absence of Mg2+ suggest that the domain motions occur at nanosecond timescales and that few to no domain motions occur at micro- to millisecond timescales. In contrast, the presence of slower domain motions at micro- to millisecond timescales cannot be ruled out in the presence of Mg2+, given that the RDCs report slightly larger domain motional amplitudes compared to the 15N relaxation data. There is precedence for such small amplitude domain motions across the single uridine bulge in P4. The P4 domain motional amplitudes in the absence of Mg2+ are roughly one-third those observed across the trinucleotide TAR bulge under nearly identical salt conditions (Zhang et al. 2006) and about half those observed across dinucleotide symmetric uridine bulges by electric birefringence (Zacharias and Hagerman 1996). The combination of RDCs and spin relaxation also proved critical in deciphering the local dynamics of P4. While the near zero RDCs measured in the U7 bulge in the presence and absence of Mg2+ provided evidence for extensive local mobility, an unusual conformation with bond vectors oriented along the magic angle could not be ruled out. The motional narrowing of U7 observed in both the presence and absence of Mg2+ provided independent support for local mobility at this site, allowing interpretation of the RDCs in terms of an unstructured U7 conformation. It is remarkable that the mobility at U7 seems to entirely escape detection in non-elongated P4. This is likely because the motions occur at timescales approaching overall molecular tumbling. Taken together, our results reinforce the existence of a universe of nanosecond motions that has so far been difficult to probe using conventional NMR relaxation methods because motions occur at timescales slower than overall tumbling of the non-elongated RNA (Zhang et al. 2006).
What, if any, is the functional role of the newly discovered P4 flexibility? It is increasingly recognized that RNA residues involved in intermolecular recognition are often flexible in the unbound state (Williamson 2000; Leulliot and Varani 2001; Al-Hashimi 2005). Such structural plasticity allows for dynamic optimization of induced fit intermolecular interactions with diverse targets. In this context, the interdomain mobility observed in P4 may play a role in facilitating changes in domain orientation that lead to tertiary interactions between the upper helix of P4 and L8 during RNase P folding. It is also possible that the P4 interhelical alignment undergoes further changes during the course of catalysis especially to allow recognition and cleavage of the diverse set of pre-tRNA molecules. In this regard, our results uncover a high degree of local mobility in two highly conserved residues, U7 and C8, which have been shown to be important in binding pre-tRNA (Frank and Pace 1997; Kaye et al. 2002b). In addition to phosphorothioate substitution studies (Fig. 1A; Hardt et al. 1995), cross-linking data suggest that U7 directly interacts with the tRNA acceptor stem five residues from the cleavage site (Christian et al. 2006). Available models show this interaction with the P4 helix running parallel to the acceptor stem of the pre-tRNA substrate (Tsai et al. 2003; Kazantsev et al. 2005; Torres-Larios et al. 2005, 2006). Dynamics at U7 is consistent with its looped out conformation observed in the larger RNase P context (Fig. 3B). These local motions, together with fluctuations at C8 and interdomain flexibility, may allow for adaptive conformational changes that allow induced-fit interaction with diverse pre-tRNA substrates.
It is instructive to try to explain the motions observed in P4 in terms of its sequence. The NOE, JNN-COSY, RDC, and resonance intensity data all suggest that the presence of the uridine bulge leads to greater perturbation of the C8–G23 base pair, and specifically C8 above the bulge compared to the the G6–C24 base pair below the bulge (Fig. 7F). This is likely due to the more flexible nature of the U7–C8 versus G6–U7 stack. Thus, the P4 sequence appears to code for a flexible U7/C8 pivot point and concomitant interdomain flexibility. These are subtle features determined by the P4 sequence that are difficult to visualize on the basis of static structures (Fig. 7F). In addition to changing the relative orientation of the two helical domains, movement of the bulge, which has been shown to have deleterious consequences on RNase P activity (Kaye et al. 2002b), will likely disrupt this dynamical network.
Most biochemical studies have shown that catalytically important metals bind to P4 in the adenine stretch below the bulge (Harris and Pace 1995; Christian et al. 2000; Crary et al. 2002). In contrast, NMR studies of E. coli P4 revealed a primary Mg2+ binding site above the bulge that contains many residues in domain II (Fig. 1A; Schmitz and Tinoco 2000). Our study reconciles this apparent discrepancy by providing evidence for association of Mg2+ ions in an electrostatic hot spot composed of residues both above (G22 and G23) and below (A5) the bulge in the major groove of P4 (Fig. 6C). The association between Mg2+ and A5 may have escaped detection in previous NMR studies due to reliance on imino proton chemical shift probes (Schmitz and Tinoco 2000). The role of these Mg2+ ion(s) and relevance in the overall RNase P context remains to be determined. Our results show that Mg2+ binding does not significantly alter the structure and/or dynamics of P4 and thus does not seem to play the role of changing the isolated P4 conformation. Rather, the association of Mg2+ ions near a site of flexibility (U7 and C8) that is known to interact with the pre-tRNA substrate suggests a possible role in substrate binding. It is possible that the P4-associated metals form nonspecific intermolecular backbone bridging interactions with the tRNA substrate. Such Mg2+ intermolecular bridges have been observed between 16S and 30S ribosomal subunits (Ghosh and Joseph 2005) and between the thiamine pyrophosphate riboswitch and its cognate ligand TPP (Serganov et al. 2006). These interactions could aid in positioning the pre-tRNA substrate relative to the active site (Christian et al. 2006).
MATERIALS AND METHODS
Preparation and purification of 13C/15N labeled RNA
P4, E-AU-P4, and E-GC-P4 RNA samples (Figs. 1C, 4A) for NMR studies were prepared by in vitro transcription reactions using T7 RNA polymerase (Milligan and Uhlenbeck 1989), uniformly 13C/15N labeled (Spectra Stable Isotopes, Inc.) or unlabeled (Sigma) nucleotide triphosphates, and synthetic DNA templates (Integrated DNA Technologies, Inc.) containing the T7 promoter and sequence of interest. A second P4 NMR sample for measurement of RDCs was prepared containing 15 mg/mL of Pf1 phage (Asla Biotech; Hansen et al. 1998). RNA transcripts were purified on 15% (w/v) denaturing polyacrylamide gels containing 8 M urea/TBE. RNA was recovered from gels by passive elution followed by ethanol precipitation. The RNA pellet was dissolved and repeatedly exchanged into NMR buffer (10 mM sodium chloride, 10 mM sodium phosphate at pH ~6.2) using a Centricon Ultracel YM-3 concentrator (Millipore Corp). The elongated samples were annealed at 95°C for 2 min in H2O to ensure proper folding, then exchanged into NMR buffer. For nonexchangeable NOE assignment, precipitated RNA was dissolved in D2O and repeatedly exchanged into the above NMR buffer made with 99.96% D2O (Cambridge Isotope Laboratories, Inc.). The final RNA concentration in the NMR samples was 0.1 mM for MgCl2 titrations and ranged between 0.6 and 1 mM for all other experiments.
NMR spectroscopy
Resonance assignments and relaxation experiments were carried out on an Avance Bruker 600 MHz NMR spectrometer equipped with a triple-resonance 5-mm cryogenic probe at 298 K (288 K for nonexchangeable NOESY). The RDCs for P4 and P4-Mg were measured on an Avance Bruker 600 MHz NMR spectrometer equipped with a triple-resonance 5-mm cryogenic probe and an 800 MHz Varian Inova spectrometer equipped with a triple-resonance Z-gradient probe, respectively. Sample volumes of ~300 μL in Shigemi tubes were used in all experiments. NMR spectra were processed using NMRPipe/NMRDraw (Delaglio et al. 1995) or Felix (Accelrys, Inc. 2002), analyzed using NMRView (Johnson and Blevins 1994), and overlaid using SPARKY 3 (Goddard and Kneller 2004). The 1H–13C splittings were measured in the divalent ion-free P4 sample as the difference in the upfield and downfield components of the 13C–1H doublet along the 1H and 13C dimension, which were measured in separate subspectra using 2D 13C–1H S3CT HSQC experiments (Meissner et al. 1997; Meissner and Sorensen 1999; Pitt et al. 2005). The two sets of RDCs computed from the differences in splittings measured in the presence and absence of phage (15 mg/mL) yielded an RMSD of 2.4 Hz (data not shown). The average value from the two independent measurements was used whenever possible. The RDCs for P4 in the presence of 20 mM Mg2+ were obtained using splittings measured in the 13C dimension. In all cases, spectra of P4 in the presence and absence of the phage medium were nearly identical (barring the expected small perturbations from residual chemical shift anisotropies), indicating that phage does not directly or indirectly (e.g., by sequestering Mg2+) affect the P4 conformation. In all cases, RDCs from base pairs G1–C29, G2–C28, and C8–G23, which based on the JNN-COSY experiment have partial or no hydrogen bonding, were excluded from analysis. All resonance intensities were measured using 2D 1H–13C HSQC experiments as previously described (Zhang et al. 2006). The intensity for each type of C–H spin was normalized to a minimum value of 0.1 independently for G–C and A–U residues (Fig. 5A,B).
All of the 15N relaxation data were measured in free (P4 and E-P4) and Mg2+ (E-P4) samples closely following the procedure described previously (Zhang et al. 2006). Briefly, imino 15N longitudinal (R 1) and transverse (R 2(CPMG)) relaxation rates and 1H–15N NOEs were measured using well-established 2D experiments (Palmer 2004). The R 1 experiment incorporated a recently described heat compensation element (Yip and Zuiderweg 2005). The relaxation delays for free P4 were 0.02, 0.06, 0.12, 0.24, 0.48, 0.64, and 0.80 sec and for E-P4, and E-P4-Mg were 0.02, 0.06, 0.12, 0.24, 0.48, 0.64, 0.80, and 1.2 sec. The R 2(CPMG) experiment employed a recently described [0013]N phase cycle in the Carr–Purcell–Meiboom–Gill (CPMG) pulse train for suppressing artifacts arising from off-resonance effects (Yip and Zuiderweg 2004). The following relaxation delays were used for P4, E-P4, and E-P4-Mg: 0.0062, 0.0124, 0.0248, 0.0372, 0.0496, 0.0620, and 0.0744 sec. The final R 2 values were corrected to account for off-resonance effects as previously described (Yip and Zuiderweg 2004). The uncertainties were also verified based on duplicate measurements of relaxation data. The 1H–15N NOEs were measured from pairs of spectra recorded with and without proton saturation during the recycle delay. The NOE uncertainty was estimated from the standard deviation of the baseline in the spectra.
Order tensor analysis of RDCs
Watson–Crick residues in domains I and II were modeled using idealized A-form helices constructed using the program Insight II (Molecular Simulations, Inc), noting that the propeller twist angles had to be corrected from +15° to the standard A-form value of −15°. An X-ray structure of the UUCG loop (Ennifar et al. 2000) was appended to domain II by superimposing the common closing base pairs as previously described (Al-Hashimi et al. 2002a). RDCs from the highly flexible U15 and C16 loop residues were also excluded from analysis (Vallurupalli and Kay 2005). The 16/17 and 15/15 RDCs measured in domains I/II in the absence and presence of Mg2+, respectively, together with the above domain structures were used to obtain best-fit order tensors for each domain using singular value decomposition implemented in the program RAMAH (an in-house modified version of ORDERTEN_SVD; Losonczi et al. 1999). The overall structure of P4 was assembled by rotation of each domain into the principal axis system of its best-fit order tensor and fixing the translational disposition of the two domains by setting the distance between C24(P) and G23(O3′) equal to ~1.59 Å (Fig. 3B). Three of four degenerate domain–domain orientations involving rotation of 180° about the three order tensor principal axes were omitted: two solutions (180° Sxx and 180° Syy) because they yielded anti-parallel helical alignments and a third (180° Szz) because it yielded a G6(O3′)–C8(P) distance (~28 Å) that could not be linked using a single U7 residue. The program PALES (http://spin.niddk.nih.gov/bax/; Zweckstetter et al. 2004) was used to compare the experimental order tensor frame for P4 with its counterpart predicted based on its overall molecular shape (difference in the Szz orientation was ~4° and ~3° for the free and Mg2+ bound P4, respectively). Interhelical angles were calculated using in-house written software (Zhang et al. 2003). In all cases, the uncertainty in the order tensor parameters was computed using the program Aform-RDC, which takes into account both RDC uncertainty and parameterized deviations in the assumed idealized A-form helix geometry (Musselman et al. 2006). An order tensor fit was also carried out against the X-ray structure of P4 from B. stearothermophilus (PDB ID #2A64, Fig. 3C; Kazantsev et al. 2005) using 16 [A5 (1DC1′H1′, 1DC2H2, 1DC8H8), G6 (1DC8H8), C9 (1DC5H5, 1DC6H6), A10 (1DC2H2, 1DC8H8), G12 (1DC8H8), A20 (1DC2H2, 1DC8H8), G22 (1DC8H8), and C24 (1DC6H6), U25 (1DC1′H1′, 1DC5H5, 1DC6H6)] and 14 [A5 (1DC1′H1′, 1DC2H2, 1DC8H8), G6 (1DC8H8), C9 (1DC6H6), A10 (1DC8H8), U11 (1DC1′H1′, 1DC5H5), G12 (1DC8H8), A20 (1DC8H8), U21(1DC5H5), G22 (1DC8H8), U25 (1DC5H5, 1DC6H6)] RDCs measured in free and Mg2+-bound P4, respectively, that correspond to bond vectors in common sequence regions in the two domains (i.e., residues 50, 51, 54, 55, 57, 392, 394, 396, and 397).
Model-free analysis of relaxation data
Insight II (Molecular Simulations, Inc.) was used to in silico elongate domain I of the RDC-derived P4 structure using a stretch of 22 base pairs. The E-P4 and P4 structures were subjected to hydrodynamic calculations using the program HYDRONMR (Garcia de la Torre et al. 2000) as previously described (Zhang et al. 2006). The HYDRONMR computed diffusion tensor parameters and P4/E-P4 structure were then used to compute 15N R 2/R 1 values using in-house-written software.
The R 1 and R 2 values and their uncertainties measured from 15N relaxation data were determined by nonlinear least-squares fitting of experimental data to the monoexponential function, I(t) = I(0)e (−R×t) based on the Levenberg–Marquardt (LM) algorithm implemented in the software Origin (Origin Lab Corp.; Fig. 4C). The final R 2 values were corrected to account for off-resonance effects as previously described (Yip and Zuiderweg 2004). The R 1, R 2, and NOE values measured in E-P4 and E-P4-Mg were subjected to an extended model-free analysis (Fig. 4E; Lipari and Szabo 1982; Clore et al. 1990) using the software Modelfree (Version 4.16 for Linux) provided by Palmer and coworkers (Mandel et al. 1995) closely following the procedure described previously (Zhang et al. 2006). This analysis assumed (1) the decoupling approximation (Lipari and Szabo 1982; Clore et al. 1990), (2) an axially symmetric overall diffusion tensor, and (3) a diffusion tensor with Dratio (D ///D ⊥ = 5.3) and orientation computed using HYDRONMR (Garcia de la Torre et al. 2000) and the RDC derived average P4 interhelical conformation. Model selection of the 15N relaxation data was carried out following the procedure recommended by Palmer and coworkers (Mandel et al. 1995) as recently described in detail (Zhang et al. 2006). The final model selections comprised models 2 and 5 for residues in domains I and II, respectively, in E-P4. With the exception of residue U25 in domain I (model 1), the model selections for E-P4-Mg were also models 2 and 5 for residues in domains I and II, respectively.
Detection of Mg2+ association by chemical shift mapping
Changes in chemical shift upon addition of Mg2+ were quantified using the equation 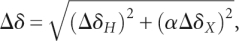 , where ΔδH and ΔδX are the changes in proton and carbon/nitrogen chemical shift and α is the ratio of the H and C/N gyromagnetic ratio. The most significant changes (>0.08 ppm, using the above equation) for each vector type are highlighted on the P4 secondary structure in Figure 6A. The terminal residues were excluded from the analysis. To measure site-specific binding affinities for P4, 2D HSQC spectra were recorded following incremental addition of MgCl2 (0.5, 1, 3, 5, 10, and 15 mM). The apparent Kd values were obtained using the following equation (Gonzalez and Tinoco 1999):
, where ΔδH and ΔδX are the changes in proton and carbon/nitrogen chemical shift and α is the ratio of the H and C/N gyromagnetic ratio. The most significant changes (>0.08 ppm, using the above equation) for each vector type are highlighted on the P4 secondary structure in Figure 6A. The terminal residues were excluded from the analysis. To measure site-specific binding affinities for P4, 2D HSQC spectra were recorded following incremental addition of MgCl2 (0.5, 1, 3, 5, 10, and 15 mM). The apparent Kd values were obtained using the following equation (Gonzalez and Tinoco 1999):
 |
where δobs is the observed chemical shift, ΔδT is the chemical shift difference between the free and fully bound state, [Mg2+]T is the amount of added MgCl2, and [RNA]T is the RNA concentration obtained from its absorbance at 260 nm. The data were fitted using Origin (Origin Lab Corp.) in which the unknown values ΔδT and Kd are optimized during the fit (Fig. 6B).
Structure-based electrostatic calculations
Residues 48–57 and 391–398 from the crystal structure of B. stearothermophilus RNase P (PDB ID #2A64) were included in the electrostatic calculation (similar results were obtained for this region when using a larger segment of RNase P). The nonlinear Poisson–Boltzmann equation was used to calculate the potential using the Delphi (Accelrys) module of Insight II (Molecular Simulations, Inc). The Debye–Huckel equation and full Coulombic boundary conditions were used to approximate the boundary potentials. Partial charges were obtained from the AMBER force field (Cornell et al. 1995). Input parameters for the calculations were as described previously (Chin et al. 1999). A monovalent salt concentration of 0.01 M was used to match experimental conditions. The three-dimensional molecular surfaces (Fig. 6C) were displayed using GRASP, provided by the Honig laboratory (http://honiglab.cpmc.columbia.edu; Nicholls et al. 1991).
ACKNOWLEDGMENTS
We thank Qi Zhang for help with the measurement and analysis of relaxation data, Dr. Meredith Lambert for help in RNA assignments, and Dr. Alex Kurochkin for his expertise and for maintenance of the NMR instruments. We also thank Michael Schmitz for providing NMR assignments of the E. coli P4 element. We thank the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for support of the purchase of the 600 MHz spectrometer. This work was funded by NIH grants awarded to H.M.A.-H. (RO1 Al066975) and C.A.F. (RO1 GM55387).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.264207.
REFERENCES
- Accelrys, Inc. FELIX user guide. Accelrys, Inc; San Diego, CA: 2002. [Google Scholar]
- Al-Hashimi, H.M. Dynamics-based amplification of RNA function and its characterization by using NMR spectroscopy. ChemBioChem. 2005;6:1506–1519. doi: 10.1002/cbic.200500002. [DOI] [PubMed] [Google Scholar]
- Al-Hashimi, H.M., Gorin, A., Majumdar, A., Gosser, Y., Patel, D.J. Towards structural genomics of RNA: Rapid NMR resonance assignment and simultaneous RNA tertiary structure determination using residual dipolar couplings. J. Mol. Biol. 2002a;318:637–649. doi: 10.1016/S0022-2836(02)00160-2. [DOI] [PubMed] [Google Scholar]
- Al-Hashimi, H.M., Gosser, Y., Gorin, A., Hu, W., Majumdar, A., Patel, D.J. Concerted motions in HIV-1 TAR RNA may allow access to bound state conformations: RNA dynamics from NMR residual dipolar couplings. J. Mol. Biol. 2002b;315:95–102. doi: 10.1006/jmbi.2001.5235. [DOI] [PubMed] [Google Scholar]
- Beebe, J.A., Kurz, J.C., Fierke, C.A. Magnesium ions are required by Bacillus subtilis ribonuclease P for both binding and cleaving precursor tRNAAsp . Biochemistry. 1996;35:10493–10505. doi: 10.1021/bi960870m. [DOI] [PubMed] [Google Scholar]
- Chen, J.L., Pace, N.R. Identification of the universally conserved core of the ribonuclease P RNA. RNA. 1997;3:557–560. [PMC free article] [PubMed] [Google Scholar]
- Chin, K., Sharp, K., Honig, B., Pyle, A. Calculating the electrostatic properties of RNA provides new insights into molecular interactions and function. Nat. Struct. Biol. 1999;6:1055–1061. doi: 10.1038/14940. [DOI] [PubMed] [Google Scholar]
- Christian, E.L., McPheeters, D.S., Harris, M.E. Identification of individual nucleotides in the bacterial ribonuclease P ribozyme adjacent to the pre-tRNA cleavage site by short-range photocrosslinking. Biochemistry. 1998;37:17618–17628. doi: 10.1021/bi982050a. [DOI] [PubMed] [Google Scholar]
- Christian, E.L., Kaye, N.M., Harris, M.E. Helix P4 is a divalent metal ion binding site in the conserved core of the ribonuclease P ribozyme. RNA. 2000;6:511–519. doi: 10.1017/s1355838200000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian, E.L., Smith, K.M.J., Perera, N., Harris, M.E. The P4 metal binding site in RNase P RNA affects active site metal affinity through substrate positioning. RNA. 2006;12:1463–1467. doi: 10.1261/rna.158606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore, G.M., Szabo, A., Bax, A., Kay, L.E., Driscoll, P.C., Gronenborn, A.M. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990;112:4989–4991. [Google Scholar]
- Clore, G.M., Starich, M.R., Gronenborn, A.M. Measurement of residual dipolar couplings of macromolecules aligned in the nematic phase of a colloidal suspension of rod-shaped viruses. J. Am. Chem. Soc. 1998;120:10571–10572. [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- Crary, S.M., Kurz, J.C., Fierke, C.A. Specific phosphorothioate substitutions probe the active site of Bacillus subtilis ribonuclease P. RNA. 2002;8:933–947. doi: 10.1017/s1355838202025025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dingley, A.J., Grzesiek, S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2hJNN couplings. J. Am. Chem. Soc. 1998;120:8293–8297. [Google Scholar]
- Dingley, A.J., Masse, J.E., Peterson, R.D., Barfield, M., Feigon, J., Grzesiek, S. Internucleotide scalar couplings across hydrogen bonds in Watson–Crick and Hoogsteen base pairs of a DNA triplex. J. Am. Chem. Soc. 1999;121:6019–6027. [Google Scholar]
- Duchardt, E., Schwalbe, H. Residue specific ribose and nucleobase dynamics of the cUUCGg RNA tetraloop motif by MNMR 13C relaxation. J. Biomol. NMR. 2005;32:295–308. doi: 10.1007/s10858-005-0659-x. [DOI] [PubMed] [Google Scholar]
- Ennifar, E., Nikulin, A., Tishchenko, S., Serganov, A., Nevskaya, N., Garber, M., Ehresmann, B., Ehresmann, C., Nikovov, S., Dumas, P. The crystal structure of UUCG tetraloop. J. Mol. Biol. 2000;304:35–42. doi: 10.1006/jmbi.2000.4204. [DOI] [PubMed] [Google Scholar]
- Frank, D.N., Pace, N.R. In vitro selection for altered divalent metal specificity in the RNase P RNA. Proc. Natl. Acad. Sci. 1997;94:14355–14360. doi: 10.1073/pnas.94.26.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtig, B., Richter, C., Wohnert, J., Schwalbe, H. NMR spectroscopy of RNA. ChemBioChem. 2003;4:936–962. doi: 10.1002/cbic.200300700. [DOI] [PubMed] [Google Scholar]
- Garcia de la Torre, J., Huertas, M.L., Carrasco, B. HYDRONMR: Prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- Ghosh, S., Joseph, S. Nonbridging phosphate oxygens in 16S rRNA important for 30S subunit assembly and association with the 50S ribosomal subunit. RNA. 2005;11:657–667. doi: 10.1261/rna.7224305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glemarec, C., Kufel, J., Foldesi, A., Maltseva, T., Sandstrom, A., Kirsebom, L.A., Chattopadhyaya, J. The NMR structure of 31mer RNA domain of Escherichia coli RNase P RNA using its non-uniformly deuterium labeled counterpart [the ‘NMR-window’ concept] Nucleic Acids Res. 1996;24:2022–2035. doi: 10.1093/nar/24.11.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard, T.D., Kneller, D.G. SPARKY 3. University of California; San Francisco: 2004. [Google Scholar]
- Gonzalez, R.L.J., Tinoco, I.J. Solution structure and thermodynamics of a divalent metal ion binding site in an RNA pseudoknot. J. Mol. Biol. 1999;289:1267–1282. doi: 10.1006/jmbi.1999.2841. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada, K., Gardiner, T., Marsh, N., Pace, N.R., Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada, C., Haydock, K., Allen, L., Altman, S. Metal ion requirements and other aspects of the reaction catalyzed by M1 RNA, the RNA subunit of ribonuclease P from Escherichia coli . Biochemistry. 1986;25:1509–1515. doi: 10.1021/bi00355a006. [DOI] [PubMed] [Google Scholar]
- Hansen, A.L., Al-Hashimi, H.M. Insight into the CSA tensors of nucleobase carbons in RNA polynucleotides from solution measurements of residual CSA: Towards new long-range orientational constraints. J. Magn. Reson. 2006;179:299–307. doi: 10.1016/j.jmr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Hansen, M.R., Mueller, L., Pardi, A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- Hardt, W.D., Warnecke, J.M., Erdmann, V.A., Hartmann, R.K. Rp-phosphorothioate modifications in RNase P RNA that interfere with tRNA binding. EMBO J. 1995;14:2935–2944. doi: 10.1002/j.1460-2075.1995.tb07293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardt, W.D., Erdmann, V.A., Hartmann, R.K. Rp-deoxy-phosphorothioate modification interference experiments identify 2′-OH groups in RNase P RNA that are crucial to tRNA binding. RNA. 1996;2:1189–1198. [PMC free article] [PubMed] [Google Scholar]
- Harris, M.E., Pace, N.R. Identification of phosphates involved in catalysis by the ribozyme RNase P RNA. RNA. 1995;1:210–218. [PMC free article] [PubMed] [Google Scholar]
- Heide, C., Pfeiffer, T., Nolan, J.M., Hartmann, R.K. Guanosine 2-NH2 groups of Escherichia coli RNase P RNA involved in intramolecular tertiary contacts and direct interactions with tRNA. RNA. 1999;5:102–116. doi: 10.1017/s1355838299981499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B.A., Blevins, R.A. NMR View—A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Kaye, N.M., Christian, E.L., Harris, M.E. NAIM and site-specific functional group modification analysis of RNase P RNA: Magnesium dependent structure within the conserved P1-P4 multihelix junction contributes to catalysis. Biochemistry. 2002a;41:4533–4545. doi: 10.1021/bi012158h. [DOI] [PubMed] [Google Scholar]
- Kaye, N.M., Zahler, N.H., Christian, E.L., Harris, M.E. Conservation of helical structure contributes to functional metal ion interactions in the catalytic domain of ribonuclease P RNA. J. Mol. Biol. 2002b;324:429–442. doi: 10.1016/s0022-2836(02)01094-x. [DOI] [PubMed] [Google Scholar]
- Kazantsev, A.V., Pace, N.R. Identification by modification-interference of purine N-7 and ribose 2′-OH groups critical for catalysis by bacterial ribonuclease P. RNA. 1998;4:937–947. doi: 10.1017/s1355838298980384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev, A.V., Krivenko, A.A., Harrington, D.J., Holbrook, S.R., Adams, P.D., Pace, N.R. Crystal structure of a bacterial ribonuclease P RNA. Proc. Natl. Acad. Sci. 2005;102:13392–13397. doi: 10.1073/pnas.0506662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz, J.C., Fierke, C.A. Ribonuclease P: A ribonucleoprotein enzyme. Curr. Opin. Chem. Biol. 2000;4:553–558. doi: 10.1016/s1367-5931(00)00131-9. [DOI] [PubMed] [Google Scholar]
- Latham, M.P., Brown, D.J., McCallum, S.A., Pardi, A. NMR methods for studying the structure and dynamics of RNA. ChemBioChem. 2005;6:1492–1505. doi: 10.1002/cbic.200500123. [DOI] [PubMed] [Google Scholar]
- Leeper, T.C., Varani, G. The structure of an enzyme-activating fragment of human telomerase RNA. RNA. 2005;11:394–403. doi: 10.1261/rna.7222505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot, N., Varani, G. Current topics in RNA–protein recognition: Control of specificity and biological function through induced fit and conformational capture. Biochemistry. 2001;40:7947–7956. doi: 10.1021/bi010680y. [DOI] [PubMed] [Google Scholar]
- Lipari, G., Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- Losonczi, J.A., Andrec, M., Fischer, M.W., Prestegard, J.H. Order matrix analysis of residual dipolar couplings using singular value decomposition. J. Magn. Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., Palmer, A.G. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- Meissner, A., Sorensen, O.W. The role of coherence transfer efficiency in design of TROSY-type multidimensional NMR experiments. J. Magn. Reson. 1999;139:439–442. doi: 10.1006/jmre.1999.1788. [DOI] [PubMed] [Google Scholar]
- Meissner, A., Duus, J.O., Sorensen, O.W. Spin-state-selective excitation. Application for E.COSY-type measurement of JHH coupling constants. J. Magn. Reson. 1997;128:92–97. [Google Scholar]
- Milligan, J.F., Uhlenbeck, O.C. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Mollova, E.T., Hansen, M.R., Pardi, A. Global structure of RNA determined with residual dipolar couplings. J. Am. Chem. Soc. 2000;122:11561–11562. [Google Scholar]
- Musselman, C., Pitt, S.W., Gulati, K., Foster, L.L., Andricioaei, I., Al-Hashimi, H.M. Impact of static and dynamic A-form heterogeneity on the determination of RNA global structural dynamics using NMR residual dipolar couplings. J. Biomol. NMR. 2006;36:235–249. doi: 10.1007/s10858-006-9087-9. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K., Honig, B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- Palmer, A.G. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Pan, T. Higher order folding and domain analysis of the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry. 1995;34:902–909. doi: 10.1021/bi00003a024. [DOI] [PubMed] [Google Scholar]
- Pervushin, K., Ono, A., Fernandez, C., Szyperski, T., Kainosho, M., Wüthrich, K. NMR scalar couplings across Watson–Crick base pair hydrogen bonds in DNA observed by transverse relaxation-optimized spectroscopy. Proc. Natl. Acad. Sci. 1998;95:14147–14151. doi: 10.1073/pnas.95.24.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt, S.W., Majumdar, A., Serganov, A., Patel, D.J., Al-Hashimi, H.M. Argininamide binding arrests global motions in HIV-1 TAR RNA: Comparison with Mg2+ induced conformational stabilization. J. Mol. Biol. 2004;338:7–16. doi: 10.1016/j.jmb.2004.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt, S.W., Zhang, Q., Patel, D.J., Al-Hashimi, H.M. Evidence that electrostatic interactions dictate the ligand-induced arrest of RNA global flexibility. Angew. Chem. Int. Ed. Engl. 2005;44:3412–3415. doi: 10.1002/anie.200500075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeranz Krummel, D.A., Altman, S. Multiple binding modes of substrate to the catalytic RNA subunit of the RNase P from Escherichia coli . RNA. 1999;5:1021–1033. doi: 10.1017/s1355838299990416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestegard, J.H., Al-Hashimi, H.M., Tolman, J.R. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q. Rev. Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- Richards, R.J., Wu, H., Trantirek, L., O'Connor, C.M., Collins, K., Feigon, J. Structural study of elements of Tetrahymena telomerase RNA stem–loop IV domain important for function. RNA. 2006;12:1475–1485. doi: 10.1261/rna.112306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, M., Tinoco, I.J. Solution structure and metal-ion binding of the P4 element from bacterial RNase P RNA. RNA. 2000;6:1212–1225. doi: 10.1017/s1355838200000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov, A., Polonskaia, A., Phan, A.T., Breaker, R.R., Patel, D.J. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–1171. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajani, Z., Varani, G. 13C NMR relaxation studies of RNA base and ribose nuclei reveal a complex pattern of motions in the RNA binding site for human U1A protein. J. Mol. Biol. 2005;349:699–715. doi: 10.1016/j.jmb.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Showalter, S.A., Hall, K.B. Isotropic reorientational eigenmode dynamics complements NMR relaxation measurements for RNA. Methods Enzymol. 2005;394:465–480. doi: 10.1016/S0076-6879(05)94019-6. [DOI] [PubMed] [Google Scholar]
- Sibille, N., Pardi, A., Simorre, J.P., Blackledge, M. Refinement of local and long-range structural order in theophylline-binding RNA using 13C–1H residual dipolar couplings and restrained molecular dynamics. J. Am. Chem. Soc. 2001;123:12135–12146. doi: 10.1021/ja011646+. [DOI] [PubMed] [Google Scholar]
- Siew, D., Zahler, N.H., Cassano, A.G., Strobel, S.A., Harris, M.E. Identification of adenosine functional groups involved in substrate binding by the ribonuclease P ribozyme. Biochemistry. 1999;38:1873–1883. doi: 10.1021/bi982329r. [DOI] [PubMed] [Google Scholar]
- Smith, D., Pace, N.R. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Bax, A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- Tolman, J.R., Flanagan, J.M., Kennedy, M.A., Prestegard, J.H. Nuclear magnetic dipole interactions in field-oriented proteins—Information for structure determination in solution. Proc. Natl. Acad. Sci. 1995;92:9279–9283. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman, J.R., Flanagan, J.M., Kennedy, M.A., Prestegard, J.H. NMR evidence for slow collective motions in cyanometmyoglobin. Nat. Struct. Biol. 1997;4:292–297. doi: 10.1038/nsb0497-292. [DOI] [PubMed] [Google Scholar]
- Tolman, J.R., Al-Hashimi, H.M., Kay, L.E., Prestegard, J.H. Structural and dynamic analysis of residual dipolar coupling data for proteins. J. Am. Chem. Soc. 2001;123:1416–1424. doi: 10.1021/ja002500y. [DOI] [PubMed] [Google Scholar]
- Torres-Larios, A., Swinger, K.K., Krasilnikov, A.S., Pan, T., Mondragón, A. Crystal structure of the RNA component of bacterial ribonuclease P. Nature. 2005;437:584–587. doi: 10.1038/nature04074. [DOI] [PubMed] [Google Scholar]
- Torres-Larios, A., Swinger, K.K., Pan, T., Mondragón, A. Structure of ribonuclease P—A universal ribozyme. Curr. Opin. Struct. Biol. 2006;16:327–335. doi: 10.1016/j.sbi.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Tsai, H.Y., Masquida, B., Biswas, R., Westhof, E., Gopalan, V. Molecular modeling of the three-dimensional structure of the bacterial RNase P holoenzyme. J. Mol. Biol. 2003;325:661–675. doi: 10.1016/s0022-2836(02)01267-6. [DOI] [PubMed] [Google Scholar]
- Vallurupalli, P., Kay, L.E. A suite of 2H NMR spin relaxation experiments for the measurement of RNA dynamics. J. Am. Chem. Soc. 2005;127:6893–6901. doi: 10.1021/ja0427799. [DOI] [PubMed] [Google Scholar]
- Williamson, J.R. Induced fit in RNA–protein recognition. Nat. Struct. Biol. 2000;7:834–837. doi: 10.1038/79575. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. NMR of proteins and nucleic acids. John Wiley and Sons; New York: 1986. [Google Scholar]
- Yip, G.N., Zuiderweg, E.R. A phase cycle scheme that significantly suppresses offset-dependent artifacts in the R2-CPMG 15N relaxation experiment. J. Magn. Reson. 2004;171:25–36. doi: 10.1016/j.jmr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Yip, G.N., Zuiderweg, E.R. Improvement of duty-cycle heating compensation in NMR spin relaxation experiments. J. Magn. Reson. 2005;176:171–178. doi: 10.1016/j.jmr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Zacharias, M., Hagerman, P.J. Bulge-induced bends in RNA: Quantification by transient electric birefringence. J. Mol. Biol. 1995;247:486–500. doi: 10.1006/jmbi.1995.0155. [DOI] [PubMed] [Google Scholar]
- Zacharias, M., Hagerman, P.J. The influence of symmetric internal loops on the flexibility of RNA. J. Mol. Biol. 1996;257:276–289. doi: 10.1006/jmbi.1996.0162. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., Throolin, R., Pitt, S.W., Serganov, A., Al-Hashimi, H.M. Probing motions between equivalent RNA domains using magnetic field induced residual dipolar couplings: Accounting for correlations between motions and alignment. J. Am. Chem. Soc. 2003;125:10530–10531. doi: 10.1021/ja0363056. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., Sun, X., Watt, E.D., Al-Hashimi, H.M. Resolving the motional modes that code for RNA adaptation. Science. 2006;311:653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- Zweckstetter, M., Hummer, G., Bax, A. Prediction of charge-induced molecular alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys. J. 2004;86:3444–3460. doi: 10.1529/biophysj.103.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]