

Short abstract
The immunodominant T-cell epitope that is involved in collagen-induced arthritis (CIA) is the glycosylated type II collagen (CII) peptide 256-270. In CII transgenic mice, which express the immunodominant CII 256-270 epitope in cartilage, the CII-specific T cells are characterized by a partially tolerant state with low proliferative activity in vitro, but with maintained effector functions, such as IFN-γ secretion and ability to provide B cell help. These mice were still susceptible to CIA. The response was mainly directed to the glycosylated form of the CII 256-270 peptide, rather than to the nonglycosylated peptide. Tolerance induction was rapid; transferred T cells encountered CII within a few days. CII immunization several weeks after thymectomy of the mice did not change their susceptibility to arthritis or the induction of partial T-cell tolerance, excluding a role for recent thymic emigrants. Thus, partially tolerant CII autoreactive T cells are maintained and are crucial for the development of CIA.
Keywords: autoimmunity, rheumatoid arthritis, T lymphocytes, tolerance, transgenic
Abstract
Introduction:
A discussion is ongoing regarding the possible role of cartilage-directed autoimmunity as a part of the pathogenesis of rheumatoid arthritis (RA). One possibility is that the association of RA with shared epitope-expressing DR molecules reflects a role for major histocompatibility complex (MHC) class II molecules as peptide receptors, and that the predilection of the inflammatory attack for the joint indicates a role for cartilage as a source of the antigenic peptides. A direct role for CII in the development of arthritis is apparent in the CIA model, in which a definite role for MHC class II molecules and a role for CII-derived peptides have been demonstrated [1,2,3]. Remarkably, it was found that the identified MHC class II molecule in the CIA model Aq has a structurally similar peptide binding pocket to that of the shared epitope, expressing DR4 molecules [4]. In fact, DR4 (DRB1*0401) and DR1 (DRB1*0101) transgenic mice are susceptible to CIA because of an immune response to a peptide that is almost identical to that which is involved in Aq-expressing mice [5,6]. They are both derived from position 260-273 of the CII molecule; the peptide binds to the Aqmolecule with isoleucine 260 in the P1 pocket, but with phenylalanine 263 in the P1 pocket of the DR4 and DR1 molecules.
Although these findings do not prove a role for CII in RA, they show that such recognition is possible and that there are structural similarities when comparing mouse with human. However, there are also strong arguments against such a possibility. First, arthritis can evolve without evidence for a cartilage-specific autoimmunity, as seen with various adjuvant-induced arthritis models [7,8] and in several observations using transgenic animals with aberrant immunity to ubiquitously expressed proteins [9,10,11]. Moreover, the MHC association in the adjuvant arthritis models correlates with severity of the disease rather than susceptibility [7,8], as has also been observed in RA [12]. Second, it has not been possible to identify the CII-reactive T cells from RA joints, or to achieve a strong and significant CII proliferative response from T cells derived from RA joints. Most recently these negative observations were corroborated using DR4+CII peptide tetramer reagents [13]. On the other hand, it has also been difficult to isolate autoreactive CII-specific T cells from CIA, and it can be anticipated that, even in the CIA model, T cells that are specific for CII will be hard to find in the joints [4].
We believe that the explanations for these observations in both experimental animals and humans are related to tolerance. The CIA model in the mouse is usually induced with heterologous CII, and is critically dependent on an immune response to the glycosylated CII peptide 256-270, which is bound to the MHC class II Aq molecule. In CII transgenic mice, expressing the heterologous (rat) form of the immunodominant CII 256-270 epitope in cartilage, we observed partial T-cell tolerance. This tolerance is characterized by a low proliferative activity, but with maintained effector functions such as production of IFN-γ and the ability to give help to B cells to produce anti-CII IgG antibodies [14]. Interestingly, these mice were susceptible to arthritis. However, a possibility was that T cells that had newly emerged from the thymus and that were not yet tolerized when the mice were immunized with CII led to the induction of arthritis. We have now addressed this possibility and found that induction of tolerance occurs within a few days, and that mice lacking recent thymic emigrants (ie thymectomized mice) display partially tolerant T cells and susceptibility to arthritis to the same extent as nonthymectomized mice. In addition we found that T cells that are reactive with the nonmodified peptides are relatively more affected by tolerance than T cells that are reactive with the more immunodominant glycosylated variants.
Objectives:
To investigate the possibility that T cells that are naturally tolerant to the cartilage protein CII are involved in the development of arthritis, and to exclude a role for nontolerized recent thymic T-cell emigrants in the development of arthritis.
Materials and methods:
A mutated mouse CII, expressing glutamic acid instead of aspartic acid at position 266, was expressed in a transgenic mouse called MMC (mutated mouse collagen) that has been described earlier [14]. The mice were thymectomized, or sham-operated, at 7 weeks of age and allowed to recover for 4 weeks before being immunized with rat CII in complete Freund's adjuvant. Arthritis development was recorded and sera analyzed for anti-CII IgG, IgG1 and IgG2a levels. To assay T-cell effector functions, other MMC and control mice were immunized in the hind footpads with rat CII in complete Freund's adjuvant, and the draining popliteal lymph nodes were taken 10 days later. The lymph node cells (LNCs) were used for proliferation assay, IFN-γ enzyme-linked immunosorbent assay (ELISA) and B-cell enzyme-linked immunospot (ELISPOT). For the proliferation assay, 106 cells were put in triplicate cultures in microtitre wells together with antigen and incubated for 72h before thymidine-labelling and harvesting 15-18h later. For IFN-γ ELISA analysis, supernatant from the proliferation plates was removed before harvesting and used in an ELISA to quantify the amount of IFN-γ produced [15]. B-cell ELISPOT was performed to enumerate the number of cells producing anti-CII IgG [16].
T-cell lines that were reactive towards rat CII were established by immunization with rat CII. An established T-cell line that was reactive with CII and specific for the CII 256-270 peptide was restimulated with freshly collected, irradiated, syngenic spleen cells and rat CII for 3 days followed by 2 weeks of IL-2 containing medium. Immediately before transfer, the cells were labelled with the cytoplasmic dye 5 (and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE) [17]. Labelled cells (107) were injected intravenously into transgenic MMC mice and nontransgenic littermates. The mice were killed 4 days after cell transfer, and the concentration of CFSE-labelled cells was determined by flow cytometry.
Results and discussion:
To investigate whether and how quickly CII-reactive T cells will encounter CII in vivo, an established T-cell line that is reactive towards rat CII was labelled with the cytoplasmic dye CFSE and transferred into MMC-QD and control mice. Four days later the mice were killed, and it was found that MMC-transgenic mice had dramatically fewer CFSE-labelled cells in the spleen than did nontransgenic littermates (0.11% compared with 0.57%). Similarly, reduced numbers of CFSE-positive cells were observed in blood. This indicates that the T cells encountered the mutated CII that was present in the cartilage of MMC mice, but not in the nontransgenic littermates. Presumably, CII from cartilage is spread by antigen-presenting cells (APCs) to peripheral lymphoid organs. This observation also suggests that newly exported T cells from the thymus will be tolerized to CII in the periphery within less than 4 days.
To further investigate whether the MMC mice harbours naïve or tolerized T cells, the mice were immunized with CII at different time points after thymectomy that were well in excess of the times required for their encounter with CII. After 10 days, the response was analyzed in vitro towards both the nonglycosylated and the glycosylated CII 256-270 peptides as well as towards purified protein derivative. The galactosylated form of the peptide (Fig. 1) was used because this is the most immunodominant modification [18]. In contrast to control mice, LNCs from transgenic mice did not proliferate significantly towards the nonglycosylated peptide, indicating that these cells have been specifically tolerized, which is in accordance with earlier observations [14]. A reduced, but still significant proliferation was also observed toward the immunodominant glycosylated CII peptide. Most important, however, was that the proliferative response in the MMC mice did not decrease after thymectomy. Similarly, a significant IFN-γ production towards the glycosylated CII peptide was observed in the MMC mice. The response was somewhat reduced compared with that observed in nontransgenic littermates, and this was especially true for the response toward the nonglycosylated peptide. Again, no decrease in the MMC response by thymectomy was observed. Taken together, the T-cell response in transgenic mice was reduced in comparison with that in the nontransgenic littermates. Furthermore, the response in transgenic animals did not decrease by thymectomy (4 or 8 weeks before immunization), showing that autoreactive T cells are still maintained (and partially tolerized) with significant effector functions at least up to 8 weeks after thymectomy, excluding a exclusive role for recent thymic emigrants in the autoimmune response towards CII. To investigate whether thymectomized mice, lacking recent CII-specific thymic emigrants, were susceptible to CIA, mice were immunized with CII 4 weeks after thymectomy and were observed for arthritis development during the following 10 weeks. Clearly, the thymectomized MMC mice were susceptible to arthritis (five out of 18 developed arthritis; Fig. 2), and no significant differences in susceptibility between thymectomized and sham-operated mice, or between males and females, were seen. In accordance with earlier results [14], MMC transgenic mice had a significantly reduced susceptibility to arthritis as compared with the nontransgenic littermates (P < 0.0001 for arthritic scores, disease onset and incidence). All mice were bled at 35 days after immunization, and the total levels of anti-CII IgG were determined. Transgenic mice developed levels of anti-CII IgG significantly above background, but the antibody titres were lower than in nontransgenic littermates (P < 0.0001). No effect on the antibody levels by thymectomy was observed, nor did thethymectomy affect the distribution of IgG1 versus IgG2a titres,indicating that the observed tolerance is not associated with a shift from a T-helper-1- to a T-helper-2-like immune response. These findings show that T cells that are specific for a tissue-specific matrix protein, CII, are partially tolerized within a few days after thymus export and that these tolerized cells are maintained after thymectomy. Most important, mice that lack newly exported CII reactive T cells are still susceptible to CIA, suggesting that the partially tolerant T cells are involved in development of arthritis.
In the light of these data it is possible to explain some of the findings in RA. T-cell reactivity to CII has been shown in RA patients, but with a very weak proliferative activity [19,20]. This is fully compatible with observations in mouse and rat CIA when autologous CII, and not heterologous CII, are used for immunization. This is particularly true if the responses are recorded during the chronic phase of disease, in which the antigen-specific T-cell responses seem to be suppressed in both humans and experimental animals. These observations were confirmed in a recent report [21] in which it was shown that CII-reactive T-cell activity could be detected in RA patients if IFN-γ production but not proliferation was measured. In the present studies in mice the strongest response is seen towards post-translational modifications of the peptide. Because the T-cell contact points are the same whether the peptide is bound to DR4 or to Aq, it is fully possible that post-translational modifications of the peptide also plays a significant role in humans [22]. The fact that IgG antibodies specific for CII are found in many RA patients could be explained by maintained B-cell helper functions of CII-reactive T cells. In fact, it has been reported [23,24] that the occurrence of IgG antibodies to CII is associated with shared epitope DR4 molecules. These observations are thus compatible with a role for CII reactivity in RA. To avoid any confusion, it needs to be stressed that RA is a heterogeneous syndrome in which not only CII, but also other cartilage proteins and other mechanisms are of importance. Such a pathogenic heterogeneity is reflected by the multitude of experimental animal models that have demonstrated how many different pathways may lead to arthritis [25].
Introduction
T cells originate from multipotent cells in the bone marrow, but mature in the thymus. They are educated in the thymus both by positive selection (ie in which only the thymocytes that are capable of interacting with self-MHC survive [26]) and by negative selection (ie in which recognition of self-antigens that are present in the thymus leads to apoptosis [27]). After approximately 2 weeks the T cells leave the thymus as mature but naïve cells [28]. Outside the thymus T cells circulate in search for a peptide-MHC complex to recognize. If a naïve T-cell fails to find such an antigenic complex its lifespan will be limited [29]. Instead, new T cells will continuously emerge from the thymus and replace the ones leaving the pool of naïve T cells.
The extent to which the lifespan is prolonged if such T cells meet their antigen is currently under debate. One possibility is that naïve T cells may only respond to antigens that are presented by professional APCs and in the proper microenvironment; potential antigens that are located in other cells and tissues are ignored [30]. Alternatively, some T cells may interact with antigens outside of the context of an immune response (eg in the physiological state by self-antigens presented for the T cells in draining lymph nodes). Recent data [31,32] suggest that a low-affinity interaction, for example by the appropriate MHC but without the relevant peptide, will lead to the prolongation of the lifespan of newly exported naïve T cells. Other findings suggest that the transgenic expression of foreign antigen in peripheral tissues will lead to maintained survival of transgenic T cells with a tolerized phenotype [33]. The importance of such T cells in autoimmune disease is of great significance, but their existence and function are still obscure.
Target antigens, which are of importance for development of autoimmune disease, may evoke either ignorance or tolerance. For example, in mice that are transgenic for a T-cell receptor (TCR) that is specific for myelin basic protein (MBP), which is a central nervous system-specific protein, the T cells ignore the existence of the antigen unless they are not activated by other means [34,35]. In other autoimmune diseases, however, the relevant self-tissue is more readily exposed to the immune system and consequently a pool of T cells that recognize the self-antigen, but which fail to be deleted, might exist. We believe that RA, as well as CIA, which is an experimental model of RA, are such diseases.
Both RA and CIA show MHC class II association, whereby CIA in mice is associated with the Aq molecule expressed by the H-2q haplotypes [36] and RA in humans with the DRB1*0401/DRA molecule in the DR4 haplotype [37,38]. Interestingly, the MHC association indicates structural relevance, because the peptide binding cleft of the Aq and the DRB1*0401/DRA molecule are very similar and bind peptides from the same CII region [39,40]. Consequently, mice that express the DRB1*0401/DRA molecule are susceptible to CIA [5,41]. The MHC class II association provides a strong indication that the model is T-cell dependent [42,43,44].
CIA is not only a useful model for RA, but is also a valuable tool for studying the interactions between the immune system and cartilage. CII is the major protein constituent of cartilage, and is highly conserved, with only minor amino acid substitution between species. When comparing the triple helical region of mouse [45] and rat [46] CII, only 13 out of 1015 amino acids were substituted. CII is further subjected to extensive post-translational modifications, including hydroxylation of prolines and lysines and glycosylation of hydroxylysines.
There are only a few reports in the literature in which glycopeptides were involved in immune responses [47,48,49,50,51], and the present CII 256-270 peptide is the only one that is naturally selected [52,53]. When H-2q mice are immunized with rat CII, a T-cell response toward CII 256-270 is elicited. This T-cell epitope contains three sites for hydroxylations and two sites for glycosylations [2], and these modifications participate in the immune response. To study self-tolerance of CII 256-270 reactive T cells, we previously generated mice with transgenic, cartilage-specific expression of the rat CII epitope [14]. These MMC transgenic mice have a reduced susceptibility to rat CIA; only 45% of the mice develop a severe polyarthritis after immunization, as compared with 95% of the non-transgenic littermates. The decreased incidence of CIA can be explained by T-cell tolerance, because the immunodominant T-cell epitope from rat CII is now expressed as self in cartilage. The existence of immune tolerance towards glycopeptides has not been addressed before, and is an important issue because glycosylated CII is more arthritogenic than nonglycosylated CII [52].
The present study was designed to investigate whether tolerized T cells are capable of mediating disease or whether naïve T cells, that are not yet tolerized, make a necessary contribution to pathology. Although the thymus shrinks with age, it continues to produce fresh thymus emigrants (ie naïve T cells). If these are specific for a distant tissue-specific self-antigen we postulated that it would take time before the T cells acquired tolerance. Possibly, the arthritis in the transgenic mice could occur as a result of the encounter of such nontolerized T cells with the immunogenic CII, rather than first being tolerized by the CII derived from cartilage. To address this possibility we thymectomized adult mice and immunized them with CII after 4 weeks, thereby allowing the T cells to become tolerized. The results indicate that the tolerized T cells themselves could be involved in the development of arthritis. We also studied immune responses in lymph node cultures derived from thymectomized mice, and showed T-cell tolerance toward the glycosylated version of the CII 256-270 epitope. In spite of peripheral tolerance this peptide elicited IFN-γ production, which we believe is important for disease development.
Materials and methods
Mice
C3H.Q mice (H-2q) were originally a gift from Dr DC Shreffler, St Louis, USA; DBA/1 mice originated from the Jackson Lab (Bar Harbor, Maine, USA); and B10.Q mice (H-2q) were from Dr Jan Klein, Tübingen, Germany. All arthritis and in vitro experiments were performed on F1 animals (B10.Q×C3H.Q). The transgenic MMC-1 mice (in this work referred to as MMC mice) have previously been described [14]. Briefly, the MMC mouse, which was originally from the C3H.Q background, was back-crossed eight generations to the B10.Q mice (MMC-BQ). The MMC transgene is a mutated mouse CII gene, in which position 266 has been changed from a D into an E, thereby containing the rat CII 256-270 epitope and showing cartilage-restricted expression. Nontransgenic QD mice (B10.Q×DBA/1) F1 were used for the establishment of a CII-specific T-cell line. MMC-QD mice were used in transfer experiments as recipient mice for migration studies. All animals were bred and used in our animal facilities.
Antigens
Rat CII was prepared from the SWARM chondrosarcoma by pepsin digestion [54], and further purified as previously described [55]. The peptides were synthesized as previously described [2], the glycosylated CII 256-270 peptide with a β-D-galactopyranose residue on L-hydroxylysine at position 264 (Fig. 1) [56]. Both collagen and collagen peptides were dissolved and stored in 0.1mol/l acetic acid.
Figure 1.
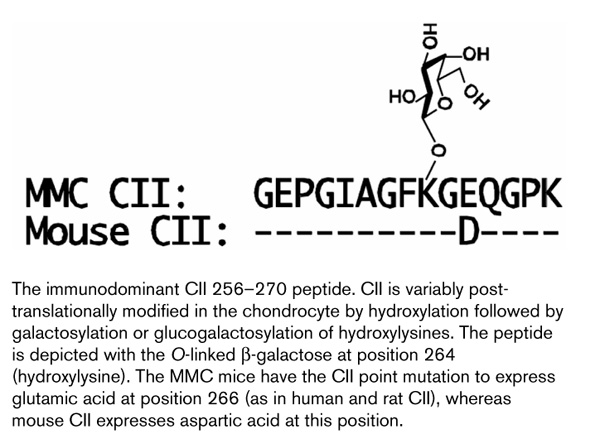
The immunodominant CII 256-270 peptide. CII is variably post-translationally modified in the chondrocyte by hydroxylation followed by galactosylation or glucogalactosylation of hydroxylysines. The peptide is depicted with the O-linked β -galactose at position 264 (hydroxylysine). The MMC mice have the CII point mutation to express glutamic acid at position 266 (as in human and rat CII), whereas mouse CII expresses aspartic acid at this position.
Thymectomy, immunization and arthritis evaluation
Adult mice (7 weeks old) were thymectomized under anaesthesia. In parallel, mice were also sham-operated under the same conditions. The mice were allowed to recover for 4 weeks before CII immunization. The mice were immunized intradermally in the base of the tail with 100 μ g rat CII emulsified 1:1 in complete Freund's adjuvant (IFA; Difco, Detroit, MI, USA). They were also boosted with 50 μ g rat CII emulsified 1:1 in IFA (Difco), 5weeks later. At that time blood samples were also taken for analysis of anti-CII antibody responses. The amounts of total anti-CII IgG as well as the IgG1 and IgG2a isotypes were determined through quantitative ELISA [57]. Using this assay no CII antibody reactivities may be found in either wild-type or MMC transgenic mice before immunization. Development of clinical arthritis was followed through visual scoring of the mice, starting 2 weeks after immunization and continuing until the end of the experiment. The arthritis was scored using a scale of 1-3 (1, one affected joint; 2, two or more arthritic joints; and 3, severe arthritis involving the entire paw), giving a maximum score of total 12 per individual.
Lymphocyteassays
To assay T-cell effector functions, mice were immunized in the hind footpads with 50 μ g rat CII emulsified 1:1 in complete Freund's adjuvant and the draining popliteal lymph nodes were taken 10 days later. The LNCs were used for proliferation assay, IFN-γ ELISA and B-cell ELISPOT. For the proliferation assay, 106 cells were put in triplicate cultures in microtitre wells together with antigen and incubated for 72h before thymidine-labelling and harvesting 15-18h later. For IFN-γ ELISA, supernatant from the proliferation plates was removed before harvesting and used in an ELISA to quantify the amount of IFN-γ produced, as previously described [15]. B-cell ELISPOT analyses were performed as previously decribed (but with minor modifications) [16] in order to enumerate the number of cells that produce anti-CII IgG. In all experiments the LNCs were assayed from individual mice and statistics were calculated from the biological variation.
T-cell lines and CFSE assay
A T-cell line that is reactive toward rat CII was established by immunization of QD mice with rat CII in the hind footpads. Eight days later draining lymph nodes were collected and restimulated in vitro with rat CII for 4 days. After 4 days of antigen stimulation, cells were collected, washed and allowed to rest in the absence of APCs and in the presence of IL-2 for 1 week. Thereafter the T cells were restimulated with freshly collected, irradiated (3000 Rad), syngenic spleen cells and rat CII for 3 days (5×105 T cells/ml, 5×106 APCs/ml, 10 μ g rat CII/ml) followed by 2 weeks of resting in IL-2-containing medium. At the time of restimulation, an aliquot of the cell line was also tested for antigen specificity. The cell line responded toward denatured CII, the nonmodified CII 256-270 peptide and the glycosylated CII 256-270 peptide with proliferation and IFN-γ production (data not shown). This scheme of 2-weekly restimulation cycle was repeated a further two times before transferring the cells to recipient mice.
Because our purpose was to follow the migration pattern of CII-specific T cells in vivo, and not transfer of disease, the cells were allowed to rest without APCs or antigen in vitro for 7 days after restimulation in the presence of IL-2 before transfer. The cells were phenotypically characterized at this stage, and were found to be CD4-positive with high and intermediate expression of CD54 and CD69, respectively, and low expression of CD25 and CD95 ligand. Immediately before transfer, the cells were labelled with the cytoplasmic dye CFSE (Molecular Probes, Leiden, The Netherlands) [17] by a 10-min incubation of the cells with 5 μ mol/l CFSE at 37°C. Labelled cells (107) were then injected intravenously into transgenic MMC mice and nontransgenic littermates. The mice were sacrificed 4 days after cell transfer and the frequency of CFSE-labelled cells were determined by flow cytometry with a fluorescence-activated cell sorter and using CellQuest Software (Becton Dickinson, Mountain View, CA, USA).
Statistical analysis
Incidence of arthritis was analyzed using the χ2 test, and antibody levels and proliferative responses were analyzed using the Mann-Whitney U-test.
Results
Thymectomized mice develop CIA with the same features as nonthymectomized animals
Seven-week-old mice were thymectomized or sham-operated. Four weeks later the mice were immunized to induce CIA and disease was scored for the following 10 weeks. Clearly, the thymectomized MMC mice were susceptible to arthritis (five out of 18 developed arthritis). No significant differences in susceptibility between thymectomized and sham-operated mice, or between males and females, were seen (Fig. 2, Table 1). In concordance with earlier results [14], MMC transgenic mice had a significantly reduced susceptibility to arthritis as compared with the nontransgenic littermates (P < 0.0001 for arthritic scores, disease onset and incidence). Disease incidence in the transgenic mice was even lower than has previously been found, which is most likely due to the change of gene background from C3H.Q to (B10.Q×C3H.Q) F1. A similar reduction in incidence was also observed in (B10.RIII×C3H.Q) F1 MMC transgenic mice [36].
Figure 2.
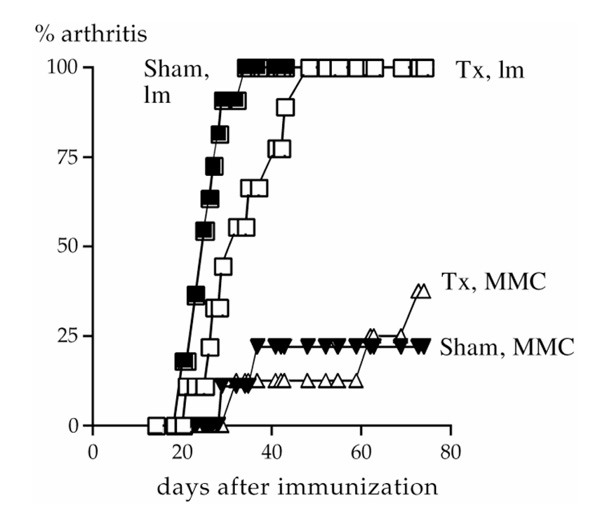
Incidence of arthritis in male mice. Triangles represent transgenic MMC mice and squares represent nontransgenic littermates. Open and filled symbols represent thymectomized (Tx) and sham-operated (Sham) mice, respectively.
Table 1.
Development of CIA and antibody responses to CII
| Arthritis | Onset (day | Anti-CII IgG (μ g/ml | ||||
| (frequency of | max score of | in serum 35 days | ||||
| Sex | Mice | CIA) | arthritic mice) | Severity | after immunization) | |
| Male | Lm | Sham | 11/11 | 25 ± 4 | 8.4 ± 1.5 | 182 ± 73 |
| Tx | 9/9 | 33 ± 9 | 9.1 ± 2.0 | 328 ± 206 | ||
| MMC | Sham | 2/8 | 34 ± 6 | 4.5 ± 5.0 | 63 ± 112 | |
| Tx | 3/8 | 56 ± 22 | 2.0 ± 1.0 | 40 ± 49 | ||
| Female | Lm | Sham | 6/7 | 35 ± 15 | 7.2 ± 2.8 | 614 ± 413 |
| Tx | 7/8 | 43 ± 16 | 7.6 ± 3.0 | 423 ± 273 | ||
| MMC | Sham | 0/8 | - | - | 30 ± 25 | |
| Tx | 2/10 | 64 ± 1.4 | 3.0 ± 2.8 | 24 ± 33 |
lm, nontransgenic littermates; Sham, sham-operated; Tx, thymectomized. Where applicable, values are expressed as mean ± standard deviation.
Antibody responses
All mice were bled at 35 days after immunization, and the total levels of anti-CII IgG determined. As shown in Table 1, transgenic mice developed significant levels of anti-CII IgG, but the antibody titres were lower than in nontransgenic littermates (P < 0.0001). An effect on the antibody levels by thymectomy could not be observed, and neither did the thymectomy affect the distribution of IgG1 titres versus those of IgG2a (Fig. 3), indicating that the observed tolerance is not associated with a shift from a T-helper-1- to a T-helper-1-like immune response. Only four animals had more IgG1 than IgG2a antibodies, two with arthritis (one transgenic and one littermate) and two without clinical disease (one transgenic and one littermate).
Figure 3.

Serum levels of antibodies specific for CII. Comparison of isotype anti-CII IgG responses in thymectomized (Tx) and sham-operated (Sham) mice as measured in sera 35 days after immunization in MMC transgenic mice and the nontransgenic littermates. Open symbols represent IgG2a and filled symbols IgG1. The transgenic mice have significantly lower isotype levels compared with their nontransgenic littermates, but thymectomy had no effect. The same pattern was seen in female (A) and male (B) mice.
Lymph node cultures
Antigen-specific T-cell activation was investigated at different time points after thymectomy to determine whether the mice harboured tolerized or naïve T cells. The mice were immunized with rat CII, and 10 days later the recall response was determined toward the nonglycosylated CII 256-270 peptide, the glycosylated CII 256-270 peptide and purified protein derivative (Fig. 4). The galactosylated form of the peptide (Fig. 1) was used because this is the most immunodominant modification [18].
Figure 4.
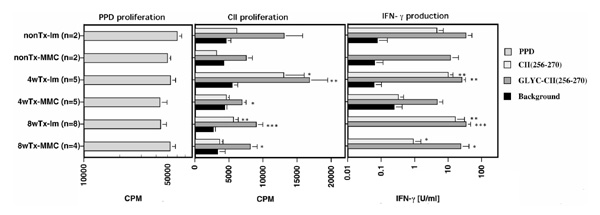
T-cell response to CII. Influence of thymectomy (Tx) on the T-cell response to CII peptides of MMC or littermate (lm) mice. Activation of lymph node cultures with different antigens; 50 μ g/ml synthetic CII 256-270 peptide, 50 μ g/ml glycosylated CII 256-270 peptide, 10 μ g/ml purified protein derivative. Backgrounds are cultures in which no antigen was added. The results after stimulation with purified protein derivative (positive control) are shown in the left, proliferative response towards CII peptides are shown in the middle and the IFN-γ production is shown in the right part of the figure, respectively. The background IFN-γ values below 0.01U/ml are not indicated in the figure. At the indicated times after thymectomy (4 weeks [4w] or 8 weeks [8w]), the indicated number of mice were immunized with rat CII and 10 days later the LNCs were isolated. Each bar represents the mean value ± standard error. Both proliferation (as measured with thymidine uptake) and IFN-γ contents in the supernatants are indicated. *P < 0.05, **P < 0.01, ***P < 0.001, versus background.
LNCs from transgenic mice proliferated poorly in response to CII peptides, indicating that these cells had been specifically tolerized, which is in accordance with earlier observations (Fig. 4) [14]. Nevertheless, the proliferative response to the glycosylated peptide was still significant (P =0.002 versus background values, and P =0.003 versus the response toward the nonglycosylated peptide as analyzed in all mice), suggesting that the remaining response is directed toward this peptide. Most important, however, was that the proliferative response in the transgenic mice did not decrease after thymectomy.
The supernatants of the proliferation cultures were also analyzed for IFN-γ content (Fig. 4). The response was somewhat reduced compared with that observed in non-transgenic littermates, notably when comparing the response towards the nonglycosylated peptide. Again, no reduction in the IFN-γ response was observed after thymectomy. In fact, a significant response towards the nonglycosylated peptide was observed in mice immunized 8 weeks after thymectomy. Still, this response was very much reduced in comparison with that induced with the glycosylated peptide or with that observed in non-transgenic littermates.
In summary, the autoimmune response in transgenic animals did not decrease after thymectomy, showing that these T cells were not newly exported, but were maintained after being partially tolerized. LNCs from transgenic mice produced less IFN-γ than the nontransgenic litter-mates in response to the nonglycosylated CII 256-270 peptide, as previously reported for MMC mice with intact thymus. The time after thymectomy (4 or 8 weeks) did not attenuate the autoimmune response to the nonglycosylated or the glycosylated peptide. Thus, the responses were still significant 8 weeks after thymectomy.
In similarity with the observed T-cell responses, significant numbers of B cells producing anti-CII IgG were seen in CII-immunized MMC mice using quantitative ELISPOT assays (Fig. 5). The response did not decrease after thymectomy. Together with the data shown in Figure 3, these data show that the significant, but reduced antibody levels to CII in the MMC mouse is reflected by numbers of antibody-secreting cells. In comparison with earlier data using the MMC transgenic mice, this was a more pronounced reduction in the B-cell response. The different genetic background of the mice used in the present study may explain this, in which an F1 with the relatively low responder B10 background was used. Again the same phenomenon was observed in (B10.RIII×C3H.Q) F1 transgenic mice immunized with rat CII [46].
Figure 5.
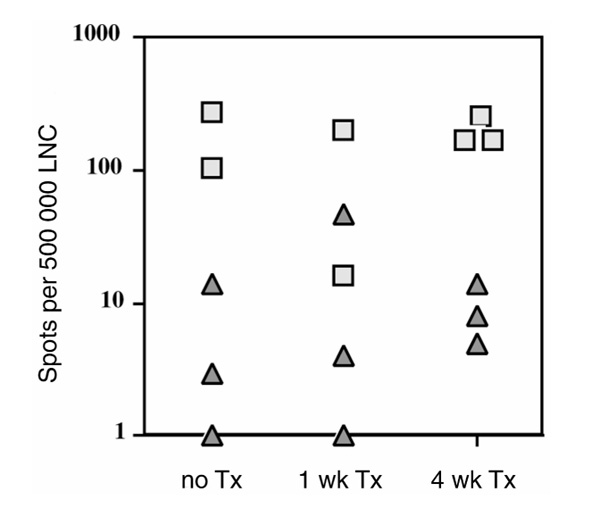
B-cell response to CII. IgG ELISPOT is an assay that enumerates the number of anti-CII antibody-producing cells present in the LNCs. Squares represent individual nontransgenic littermates and triangles represent the transgenic MMC mice. No difference before and after thymectomy was noted and the difference between MMC and wild-type mice are significant (P =0.0012 for all mice, P =0.0102 for thymectomized mice). TX, thymectomized.
In vivo transfer of rat collagen type II-reactive T cells
An activated CFSE-labelled T-cell line, which was reactive towards rat CII, was transferred into mice to follow T-cell migration and survival. Four days after cell transfer the transgenic mice had dramatically fewer CFSE-labelled cells in the spleen than did nontransgenic littermates (Fig. 6a); similar reduced numbers of CFSE-labelled cells were also seen in blood (data not shown). Only occasionally could cells be detected in the lymph nodes or in the thymus. These had only one fluorescence peak, indicating that they had not proliferated in vivo. Extracted spleen cells were analyzed for phenotypic markers, such as CD71, CD69 and CD43. CFSE-labelled cells stained positive for CD43 and negative for CD71, and approximately 50-70% were CD69 positive (Fig. 6b, and data not shown). However, there was no difference in the expression levels of these activation markers between cells transferred into MMC or nontransgenic mice.
Figure 6.
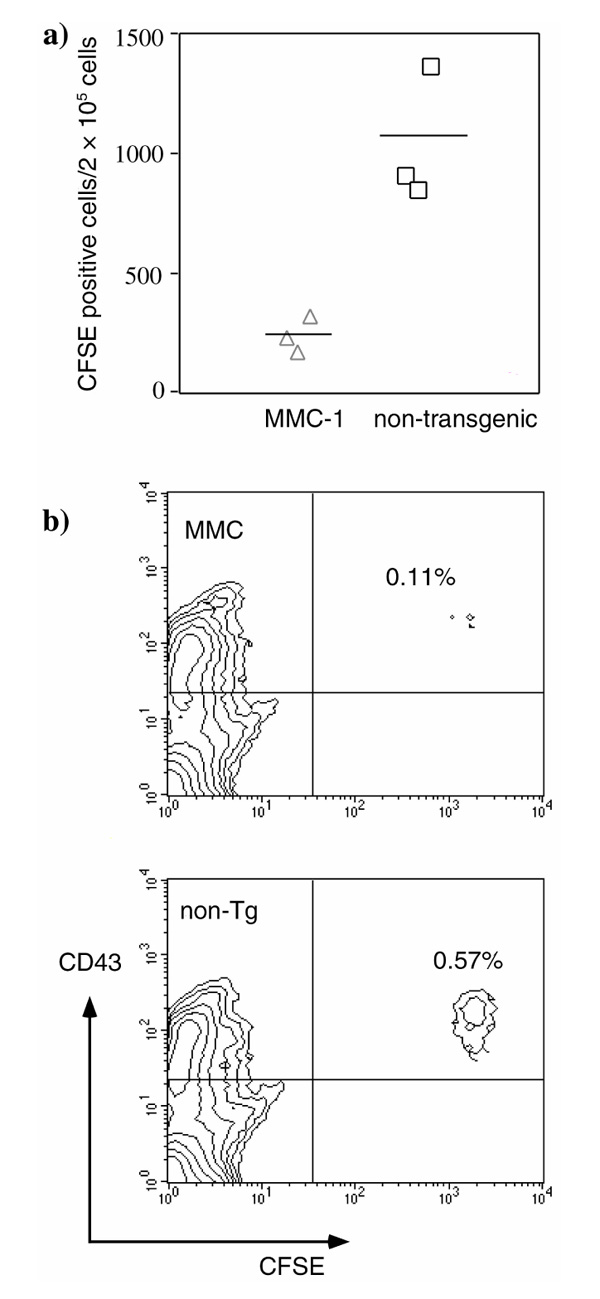
Fate of CII-reactive T cells in vivo using transferred CFSE-labelled rat CII-reactive T cells. (a) The rat CII-reactive T cells could not be seen to divide in vivo. In MMC transgenic mice, however, dramatically fewer CFSE-positive cells could be recovered, all of which were CD43-positive. (b) One representative experiment (out of three) for two-colour analysis of spleen cells from MMC transgenic mice and nontransgenic littermates for CFSE and CD43.
The dramatic differences in detectable events in the spleen and blood between MMC and littermate recipients suggest that the T cells interacted with the antigens that were present in MMC mice, but that this did not occur in their nontransgenic littermates.
Discussion
In the present study we showed that T cells that are specific for a tissue-specific matrix protein, CII, are partially tolerized and that these cells are maintained after thymectomy. By adult thymectomy, the continuous outflow of naïve T cells in mice with an established, normal T-cell repertoire was terminated. Still, the partial tolerant state, defined by lack of proliferative capacity but with significant IFN-γ response and B-cell helping capacity, was maintained up to 8 weeks after thymectomy. In addition, thymectomy did not affect the susceptibility to CIA, suggesting that the partially tolerant T cells are involved in development of arthritis.
The loss of detectable CFSE-labelled rat CII reactive T cells in MMC mice suggests that the T cells had encountered their antigen. However, where this interaction takes place and the outcome of it was not deduced in the present study. It may be speculated that the interaction takes place in the circulation due to the inability of T cells to reach lymph nodes (the T-cell line is CD62 ligand negative; data not shown). Furthermore, the rapid decrease in cell numbers implies that this interaction occurs shortly after cells enter the periphery. However, it could also be caused by a migration of T cells into tissue, even in the absence of local inflammation, which would promote such traffic. If the T cells do migrate out into the tissue the outcome could possibly be retention and even expansion of the T-cell population, and the latter may even lead to outbreak of arthritis. Importantly, we showed that rat CII-reactive T cells interact with the antigen present in MMC mice, because fewer cells are found 4 days after cell transfer in transgenic mice compared with in control animals.
These findings suggest that the immune system normally interacts with cartilage and that the T cells that participate in such an interaction survive and have maintained function. The questions that arise are when, how and where the T cells and the CII antigen normally meet.
Presumably the interaction takes place in the lymph nodes that drain the joints, and depends on the amount of CII that is degraded and transported to these lymph nodes, the nature of the transporting APCs and the MHC-binding efficiency after processing. There is no lymphatic drainage of cartilage, but the surrounding synovial tissue contains numerous macrophages that have the capacity to engulf proteins scavenged from cartilage. It is likely, but not proven, that some of these macrophages transport CII to lymph nodes and present them to the immune system. It is not likely that synovial dendritic cells fulfil these functions, because dendritic cells do not present collagen [58,59], in contrast to the crucial role of dendritic cells to prime immune responses to other antigens [60]. Thus, the presentation of CII in the periphery is limited but must occur.
The circulation of newly exported T cells is most likely very rapid, but the possibility of T cells encountering specific antigens is of course also limited by the low numbers of CII-reactive T cells. If tolerance is an important mechanism for protection from immune-mediated inflammatory attacks on self-tissues, it should be important to allow the interactions between self-reactive T cells and the respective self-antigens to occur as quickly as possible. In this context, 4 weeks is a long time for a circulating T cell. Studies on class I restricted CD8+ T cells [31,61] have shown that naïve T cells exported from thymus will not expand unless they meet their specific antigen. However, they may survive for at least 2 weeks but will subsequently disappear. Memory cells on the other hand are more easily maintained, even in the absence of antigen. The lifespan of CD4+ T cells is more controversial, but their survival is probably more limited than for CD8+ T cells. The maintenance of memory CD4+ T cells has been suggested [62] to be dependent on the continuous presence of the specific antigen. In the case of the CII-reactive T cells, the specific antigen is in the cartilage and encounters with this antigen may lead to maintenance, which most likely is synonymous with 'partial' tolerance induction.
We describe the observed tolerance as 'partial' because we have no better word for the observed state. The description is made on the population level, and not on the cellular level. Thus, the observed partial tolerance could have different explanations. One possibility is tolerization on the cellular level, for instance, by induction of a different mode of signalling through TCR or through costimulatory molecules [63]. Alternatively, the tolerance could be achieved by deletion of T cells that express TCRs with high avidity for the CII peptide, leaving only the low-avidity T cells, which might reach the critical threshold for 'partial' activation (IFN-γ secretion and B-cell help, but no IL-2 secretion). Nevertheless, if this should be the case the triggered low avidity CII-reactive cells are maintained and play a role in arthritis.
Interestingly, the glycosylated peptide elicited a stronger recall response than the synthetic peptide, which is in accordance with the finding that glycosylated CII is more arthritogenic than nonglycosylated CII [52]. How can such a finding be explained? One possibility is that the stronger response to the glycosylated peptide reflects a greater number of various TCRs (and thus T cells) that are capable of interacting with the glycosylated peptide, possibly because of the sugar being quite flexible in its position. Alternatively, the expression level of the glycosylated epitope will also influence tolerance induction. If there is low expression of this variant, less glycopeptide-specific T cells will become tolerized in vivo. It should also be emphasized that the glycopeptide-specific T cells, which may recognize a structure much larger than any amino acid side chain, are apparently efficiently selected in the thymus, despite the absence of the relevant peptides. The structural basis of the positive selection of these T cells remains to be clarified.
The role of long-lived self-reactive T cells in mediating effector functions is of course a central issue in the understanding of chronic autoimmune disease. The location and the physical state of the relevant self-antigen will, to a large extent, determine the activity of such T cells. In experimental allergic encephalomyelitis (EAE), an animal model for multiple sclerosis, an interesting study [64] has been reported on the breakage of tolerance. Mice were administered the MBP peptide Ac1-11, and developed a severe and acute encephalomyelitis. After recovering from disease the mice were given a superantigen that was reactive to the same subtype of T cells that initially mediated disease. These T cells were now tolerant, but the superantigen was able to break this tolerance and the mice suffered a second exacerbation of disease. Thus, T cells that are specific for EAE seem normally to ignore the existence of the self-antigen unless they are activated, which leads to trafficking into the central nervous system and subsequently to induction of disease. Consequently, the recall T-cell response to MBP is as strong to self-MBP as to foreign MBP. In contrast, the recall response to CII differs dramatically between self-CII and foreign CII due to tolerance induction to the CII 256-270 peptide, which is derived from cartilage.
From the above-stated reasons it is likely that tolerance to cartilage-derived collagen peptides expressed on APCs in peripheral lymphoid organs, but not in the thymus, occurs. When tolerance is broken, for example after the introduction of adjuvant, which will activate APCs, autoimmune disease can ensue. At present we know very little regarding what can cause breakage of tolerance. In mice we often immunize with tissue-specific proteins in the presence of adjuvant or with adjuvant only [65], but what happens in humans with autoimmune diseases that are triggered by unknown causes? Peripheral tolerance towards tissue-restricted antigens in mice may include different routes of tolerance induction and maintenance [33]. Knowledge about such mechanisms would certainly help in understanding the basic mechanisms that cause autoimmune disease and will provide possibilities to control and limit the chronic development of pathologic inflammation.
Acknowledgments
Acknowledgements
The authors thank Lennart Lindström, Carlos Palestro and Yvette Sjöö for taking good care of the animals, and Andrew Cook and Alexandra Treschow for linguistic corrections. This work was supported by Crafoord Foundation, Anna Greta Crafoord Research Foundation, King Gustaf V's 80-years Foundation, Kock and Österlunds Foundations, the Swedish Rheumatism Association, the Swedish Natural Science Research Council and the Swedish Medical Research Council.
References
- Brunsberg U, Gustafsson K, Jansson L, et al. Expression of a transgenic class II Ab gene confers susceptibility to collagen-induced arthritis. Eur J Immunol. 1994;24:1698–1702. doi: 10.1002/eji.1830240736. [DOI] [PubMed] [Google Scholar]
- Michaëlsson E, Andersson M, Engström A, Holmdahl R. Identification of an immunodominant type-II collagen peptide recognized by T cells in H-2q mice: self tolerance at the level of determinant selection. Eur J Immunol. 1992;22:1819–1825. doi: 10.1002/eji.1830220722. [DOI] [PubMed] [Google Scholar]
- Brand DD, Myers LK, Terato K, et al. Characterization of the T cell determinants in the induction of autoimmune arthritis by bovine alpha 1(II)-CB11 in H-2q mice. J Immunol. 1994;152:3088–3097. [PubMed] [Google Scholar]
- Holmdahl R, Andersson M, Goldschmidt TJ, et al. Type II collagen autoimmunity in animals and provocations leading to arthritis. . Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- Andersson EC, Hansen BE, Jacobsen H, et al. Definition of MHC and T cell receptor contacts in the HLA-DR4 restricted immunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. . Proc Natl Acad Sci USA. 1998;95:7574–7569. doi: 10.1073/pnas.95.13.7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosloniec EF, Brand DD, Myers LK, et al. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J Exp Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingsbo C, Sahlstrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R. Pristane-induced arthritis in rats: a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non-major histocompatibility complex genes. . Am J Pathol. 1996;149:1675–1683. [PMC free article] [PubMed] [Google Scholar]
- Kawahito Y, Cannon GW, Gulko PS, et al. Localization of quantitative trait loci regulating adjuvant-induced arthritis in rats: evidence for genetic factors common to multiple autoimmune diseases. . J Immunol. 1998;161:4411–4419. [PubMed] [Google Scholar]
- Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, Tosu M, Yoshida E, et al. Induction of inflammatory arthropathy resembling rheumatoid arthritis in mice transgenic for HTLV-I. Science. 1991;253:1026–1028. doi: 10.1126/science.1887217. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Ikeda H, Ishizu A, et al. A wide spectrum of collagen vascular and autoimmune diseases in transgenic rats carrying the env-pX gene of human T lymphocyte virus type I. Int Immunol. 1997;9:339–346. doi: 10.1093/intimm/9.2.339. [DOI] [PubMed] [Google Scholar]
- Weyand CM, McCarthy TG, Goronzy JJ. Correlation between disease phenotype and genetic heterogeneity in rheumatoid arthritis. J Clin Invest. 1995;95:2120–2126. doi: 10.1172/JCI117900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzin BL, Falta MT, Crawford F, et al. Use of soluble peptide-DR4 tetramers to detect synovial T cells specific for cartilage antigens in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:291–296. doi: 10.1073/pnas.97.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström V, Michaëlsson E, Burkhardt H, Mattsson R, Vuorio E, Holmdahl R. Systemic versus cartilage-specific expression of a type II collagen-specific T-cell epitope determines the level of tolerance and susceptibility to arthritis. Proc Natl Acad Sci USA. 1996;93:4480–4485. doi: 10.1073/pnas.93.9.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson L, Jirholt J, Holmdahl R, Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin Exp Immunol. 1998;111:521–526. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingsbo C, Larsson P, Andersson M, Holmdahl R. Association of pepsin with type II collagen (CII) breaks control of CII autoimmunity and triggers development of arthritis in rats. Scand J Immunol. 1993;37:337–342. doi: 10.1111/j.1365-3083.1993.tb02562.x. [DOI] [PubMed] [Google Scholar]
- Weston SA, Parish CR. New fluorescent dyes for lymphocyte migration studies. Analysis by flow cytometry and fluorescence microscopy. . J Immunol Methods. 1990;133:87–97. doi: 10.1016/0022-1759(90)90322-m. [DOI] [PubMed] [Google Scholar]
- Corthay A, Bäcklund J, Broddefalk J, et al. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur J Immunol. 1998;28:2580–2590. doi: 10.1002/(SICI)1521-4141(199808)28:08<2580::AID-IMMU2580>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Snowden N, Reynolds I, Morgan K, Holt L. T cell responses to human type II collagen in patients with rheumatoid arthritis and healthy controls. Arthritis Rheum. 1997;40:1210–1218. doi: 10.1002/1529-0131(199707)40:7<1210::AID-ART4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kim HY, Kim WU, Cho ML, et al. Enhanced T cell proliferative response to type II collagen and synthetic peptide CII (255-274) in patients with rheumatoid arthritis. Arthritis Rheum . 1999;42:2085–2093. doi: 10.1002/1529-0131(199910)42:10<2085::AID-ANR8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Berg L, Rönnelid J, Sanjeevi CB, Lampa J, Klareskog L. Interferon gamma production in response to in vitro stimulation with collagen type II in rheumatoid arthritis is associated with HLA-DRB1*0401 and DQ8. Arthritis Res. 2000;2:75–84. doi: 10.1186/ar71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Andersson EC, Andersen CB, Svejgaard A, Fugger L. Transgenic mouse models of rheumatoid arthritis. Immunol Rev. 1999;169:161–173. doi: 10.1111/j.1600-065x.1999.tb01314.x. [DOI] [PubMed] [Google Scholar]
- Rönnelid J, Lysholm J, Engström-Laurent A, Klareskog L, Heyman B. Local anti-type II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA-DR4-restricted IgG response. . Arthritis Rheum. 1994;37:1023–1029. doi: 10.1002/art.1780370707. [DOI] [PubMed] [Google Scholar]
- Cook AD, Stockman A, Brand CA, et al. Antibodies to type II collagen and HLA disease susceptibility markers in rheumatoid arthritis. Arthritis Rheum. 1999;42:2569–2576. doi: 10.1002/1529-0131(199912)42:12<2569::AID-ANR9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Holmdahl R. Another pathway towards arthritis. Curr Biol. 1999;9:528–530. doi: 10.1016/s0960-9822(99)80328-5. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Callahan GN, Althage A, Cooper S, Klein PA, Klein J. On the thymus in the differentiation of 'H-2 self recognition' by T-cells: evidence for dual recognition. J Exp Med. 1978;147:882–896. doi: 10.1084/jem.147.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappler JW, Staerz U, White J, Marrack PC. Self-tolerance eliminates T cells specific for Mls-modified products of the major histocompatibility complex. Nature. 1988;332:35–40. doi: 10.1038/332035a0. [DOI] [PubMed] [Google Scholar]
- Rooke R, Waltzinger C, Benoist C, Mathis D. Targeted complementation of MHC class II deficiency by intrathymic delivery of recombinant adenoviruses. Immunity. 1997;7:123–134. doi: 10.1016/s1074-7613(00)80515-4. [DOI] [PubMed] [Google Scholar]
- Ignatowicz L, Rees W, Pacholczyk R, et al. T cells can be activated by peptides that are unrelated in sequence to their selecting peptide. Immunity. 1997;7:179–186. doi: 10.1016/s1074-7613(00)80521-x. [DOI] [PubMed] [Google Scholar]
- Ohashi PS, Oehen S, Buerki K, et al. Ablation of 'tolerance' and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- Tanchot C, Rocha B. Peripheral selction of T cell repertoires; the role of continuous thymus output. J Exp Med. 1997;186:1099–1106. doi: 10.1084/jem.186.7.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämmerling GJ, Schönrich G, Ferber I, Arnold B. Peripheral tolerance as a multi-step mechanism. Immunol Rev. 1993;133:93–104. doi: 10.1111/j.1600-065x.1993.tb01511.x. [DOI] [PubMed] [Google Scholar]
- Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- Wooley PH, Luthra HS, Stuart JM, David CS. Type II collagen induced arthritis in mice. I. Major histocompatibility complex (I-region) linkage and antibody correlates. J Exp Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med. 1978;298:869–871. doi: 10.1056/NEJM197804202981602. [DOI] [PubMed] [Google Scholar]
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- Fugger L, Rothbard JB, Sonderstrup-McDevitt G. Specificity of an HLA-DRB1*0401-restricted T cell response to type II collagen. Eur J Immunol. 1996;26:928–933. doi: 10.1002/eji.1830260431. [DOI] [PubMed] [Google Scholar]
- Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 1997;7:473–481. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- Rosloniec EF, Brand DD, Myers LK, et al. Induction of autoimmune arthritis in HLA-DR4 (DRB1*0401) transgenic mice by immunization with human and bovine type II collagen. J Immunol. 1998;160:2573–2578. [PubMed] [Google Scholar]
- Ranges GE, Sriram S, Cooper SM. Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J Exp Med. 1985;162:1105–1110. doi: 10.1084/jem.162.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Klareskog L, Rubin K, Larsson E, Wigzell H. T lymphocytes in collagen II-induced arthritis in mice. Characterization of arthritogenic collagen II-specific T cell lines and clones. Scand J Immunol. 1985;22:295–306. doi: 10.1111/j.1365-3083.1985.tb01884.x. [DOI] [PubMed] [Google Scholar]
- Mauri C, Chu CQ, Woodrow D, Mori L, Londei M. Treatment of a newly established transgenic model of chronic arthritis with nondepleting anti-CD4 monoclonal antibody. J Immunol. 1997;159:5032–5041. [PubMed] [Google Scholar]
- Metsäranta M, Toman D, de Crombrugghe B, Vuorio E. Mouse type II collagen gene. Complete nucleotide sequence, exon structure, and alternative splicing. J Biol Chem. 1991;266:16862–16869. [PubMed] [Google Scholar]
- Malmström V, Kjéllen P, Holmdahl R. Type II collagen in cartilage evokes peptide-specific tolerance and skews the immune response. . J Autoimmun. 1998;11:213–221. doi: 10.1006/jaut.1998.0198. [DOI] [PubMed] [Google Scholar]
- Ishioka GY, Lamont AG, Thomson D, et al. MHC interaction and T cell recognition of carbohydrates and glycopeptides. . J Immunol. 1992;148:2446–2451. [PubMed] [Google Scholar]
- Deck B, Elofsson M, Kihlberg J, Unanue ER. Specificity of glycopeptide-specific T cells. J Immunol. 1995;155:1074–1078. [PubMed] [Google Scholar]
- Haurum JS, Arsequell G, Lellouch AC, et al. Recognition of carbohydrate by major histocompatibility complex class I-restricted, glycopeptide-specific cytotoxic T lymphocytes. J Exp Med. 1994;180:739–744. doi: 10.1084/jem.180.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Motal UM, Berg L, Rosen A, et al. Immunization with glycosylated Kb-binding peptides generates carbohydrate-specific, unrestricted cytotoxic T cells. Eur J Immunol. 1996;26:544–551. doi: 10.1002/eji.1830260307. [DOI] [PubMed] [Google Scholar]
- Jensen T, Hansen P, Galli-Stampino L, et al. Carbohydrate and peptide specificity of MHC class II-restricted T cell hybridomas raised against an O-glycosylated self peptide. J Immunol. 1997;158:3769–3778. [PubMed] [Google Scholar]
- Michaëlsson E, Malmström V, Reis S, Burkhardt H, Engström Å, Holmdahl R. T cell recognition of carbohydrates on type II collagen. . J Exp Med. 1994;30:745–749. doi: 10.1084/jem.180.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaëlsson E, Broddefalk J, Kihlberg J, Engström Å, Holmdahl R. Antigen processing and presentation of a naturally glycosylated protein elicits major histocompatibility complex class II-restricted, carbohydrate-specific T cells. Eur J Immunol. 1996;26:1906–1910. doi: 10.1002/eji.1830260835. [DOI] [PubMed] [Google Scholar]
- Smith BD, Martin GR, Dorfman A, Swarm R. Nature of the collagen synthesized by a transplanted chondrosarcoma. Arch Biochem Biophys . 1975;166:181–186. doi: 10.1016/0003-9861(75)90378-1. [DOI] [PubMed] [Google Scholar]
- Andersson M, Holmdahl R. Analysis of type II collagen reactive T cells in the mouse. I. Different regulation of autoreactive versus non-autoreactive anti-type II collagen T cells in the DBA/1 mouse. . Eur J Immunol. 1990;20:1061–1066. doi: 10.1002/eji.1830200517. [DOI] [PubMed] [Google Scholar]
- Broddefalk J, Bäcklund J, Almqvist F, Johansson M, Holmdahl R, Kihlberg J. T cells recognize a glycopeptide derived from type II collagen in a model for rheumatoid arthritis. J Am Chem Soc. 1998;120:7676–7683. [Google Scholar]
- Holmdahl R, Klareskog L, Andersson M, Hansen C. High antibody response to autologous type II collagen is restricted to H-2q. . Immunogenetics. 1986;24:84–89. doi: 10.1007/BF00373114. [DOI] [PubMed] [Google Scholar]
- Michaëlsson E, Holmdahl M, Engström Å, Burkhardt H, Scheynius A, Holmdahl R. Macrophages but not dendritic cells present collagen to T-cells. Eur J Immunol. 1995;25:2234–2241. doi: 10.1002/eji.1830250818. [DOI] [PubMed] [Google Scholar]
- Manoury Schwartz B, Chiocchia G, Fournier C. Processing and presentation of type II collagen, a fibrillar autoantigen, by H-2q antigen-presenting cells. Eur J Immunol. 1995;25:3235–3242. doi: 10.1002/eji.1830251206. [DOI] [PubMed] [Google Scholar]
- Steinman R. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Tanchot C, Rocha B. The peripheral T cell repertoire: independent homeostatic regulation of virgin and activated CD8+ T cell pools. Eur J Immunol. 1995;25:2127–2136. doi: 10.1002/eji.1830250802. [DOI] [PubMed] [Google Scholar]
- Gray D, Matzinger P. T cell memory is short-lived in the absence of antigen. J Exp Med. 1991;174:969–974. doi: 10.1084/jem.174.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Ann Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- Brocke S, Gaur A, Piercy C, et al. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial super-antigen. Nature. 1993;365:642–644. doi: 10.1038/365642a0. [DOI] [PubMed] [Google Scholar]
- Potter M, Wax JS. Genetics of susceptibility to pristane-induced plasmacytomas in BALB/cAn: reduced susceptibility in BALB/cJ with a brief description of pristane-induced arthritis. J Immunol. 1981;127:1591–1595. [PubMed] [Google Scholar]