

Abstract
Tumor angiogenesis has been related to the initiation as well as progression toward more aggressive behavior of human tumors. We will discuss genetic events underlying the initiation and progression of colorectal and pancreatic adenocarcinoma with a particular focus on the modulation of angiogenesis. A secreted fibroblast growth factor (FGF) binding protein (FGF-BP), which is an extracellular chaperone molecule for FGFs, has been shown to enhance FGF-mediated biochemical and biologic events and to be a crucial rate-limiting factor for tumor-dependent angiogenesis. Histochemical and in situ hybridization studies with archival samples show that FGF-BP is induced early during the initiation of colorectal and pancreatic adenocarcinoma. We will discuss the potential of this secreted protein as a serum marker to identify at-risk subjects.
Keywords: FGF-Binding Protein, Colon Cancer, Pancreatic Cancer, Dysplastic Lesions, Cancer Progression
INTRODUCTION
Colorectal cancer is the third most common type of cancer among men and women in the United States. Eighty-five percent of affected patients undergo surgical removal of the primary tumor as first-line treatment. However, disease will recur in approximately half of these patients within 5 years (1). It was estimated that in 2004, there were approximately 100,000 new cases and 57,000 deaths from colorectal cancer in the United States (2). Risk factors include age, a diet rich in fat and cholesterol, inflammatory bowel disease (especially ulcerative colitis), and genetic predisposition, including hereditary polyposis and nonpolyposis syndromes. Colorectal cancer screening tests, such as the fecal occult blood test (FOBT), flexible sigmoidoscopy, and colonoscopy, performed in asymptomatic individuals of age 50 years and older have been shown to achieve accurate detection of early stage cancer and its precursors. Reductions in morbidity and mortality can be achieved through detection and treatment of early stage colorectal cancers and the identification and removal of adenomatous polyps.
GENETIC ALTERATIONS IN COLORECTAL CANCER
Progress has been made in understanding the molecular basis of colorectal cancer predisposition and progression. The vast majority of colorectal cancers are adenocarcinomas, which develop through a series of clinical and histopathologic steps, ranging from a single crypt lesion through small benign tumors (adenomatous polyps) to malignant and invasive carcinomas (3). Over the last 15 years, Vogelstein and colleagues discovered a series of genetic alterations that contribute, through their multiplicity over many years, to the eventual initiation and progression of colorectal cancer (4) (Fig 1).
Figure 1.
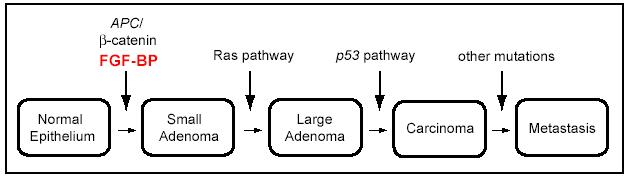
Genetic alterations during the development of colorectal cancer. Significant genetic alterations at different junctures during the transformation of colon epithelia to invasive adenocarcinoma are depicted. Induction of FGF-BP expression is an early event that is driven by mutations and activation of the WNT/beta-catenin pathway (Adapted and modified from Kinzler Vogelstein [4]).
Adenomatous Polyposis Coli (APC) Tumor Suppressor Gene
Germline mutation in the adenomatous polyposis coli (APC) tumor suppressor gene is the earliest event during the transformation process in both sporadic colorectal tumors and familial adenomatous polyposis (FAP), an autosomal dominant disorder causing extensive adenomatous polyps in the colon and early onset colorectal cancer (4-6). Carboxy-terminal truncations of the
APC protein result in impairment of the ability of APC to interact with and downregulate beta-catenin, a transcriptional activator regulated by the Wnt signaling pathway (7-13). The resulting constitutive overexpression of beta-catenin is likely responsible for the activation of growth-promoting oncogenes, such as cyclin D1 or c-myc (14-16). In 1990, Moser and colleagues identified an APC mutant mouse, namely ApcMin/+, which carries a mutation in the APC gene similar to that detected in FAP and sporadic colorectal cancers (17). ApcMin/+ mice do not survive beyond 120 days due to the occurrence of intestinal tumors that start from a rapid development of adenomatous polyps.
K-ras Proto-Oncogene
The K-ras pathway mediates the cellular response to extracellular signals, such as growth factors, cytokines, and hormones that regulate cell proliferation, differentiation, and apoptosis (18). Mutation of the ras proto-oncogene has been detected in various human cancers, including 95% of pancreatic cancers and up to 50% of large bowel adenocarcinomas (19-21). Mutation of the K-ras oncogene has been found in adenomas measuring <1 cm (10%-15%) as well as in larger adenomas and adenocarcinomas (30%-60%) (22-24). Therefore, it has been proposed that a mutation of K-ras is likely to represent a hallmark contributing to the transition from intermediate to late adenoma or adenocarcinoma (25).
Deletions of Chromosome Arm 18q
Chromosome arm 18q deletions (mainly 18q21 deletions) represent a later event associated with colon cancer progression through the stages of late adenoma to carcinoma (26). These deletions account for the reduced expression of the tumor suppressor DCC (deleted in colorectal carcinoma) (27-30) as well as SMAD4/DPC4 (deleted in pancreatic carcinoma 4) (31-33).
p53 Tumor Suppressor Gene
Loss or inactivation of p53 tumor suppressor gene has been reported in a high percentage of colorectal cancers as a late event during disease progression (34). Disruption of p53 by gene targeting in human colon cancer cells results in cell resistance to different chemotherapeutic agents, as opposed to wild type cells (35). Hence, loss of p53 in human colorectal cancers may explain the inefficacy of chemotherapy and may be correlated with decreased survival in patients with colorectal adenocarcinomas (36-39).
PANCREATIC CANCER
Pancreatic cancer accounts for more than 30,000 deaths each year in the Unites States (40). Although pancreatic adenocarcinoma represents fewer than 2% of new cancer cases in the United States, it is the fifth leading cause of cancer-related death. There is no effective early screening test, and symptoms appear during late-stage disease, when the tumor has metastasized and invaded surrounding tissues. Therefore, the majority of patients are not suitable for curative resections. Additionally, pancreatic cancer cell resistance to cytotoxic agents and radiation represents a further element that accounts for the poor prognosis (41, 42). Pancreatic adenocarcinoma develops through a step-wise progression that initiates with distinct epithelial lesions in the small interlobular ducts known as pancreatic intraepithelial neoplasias (PanINs). PanINs can be flat (PanIN-1A), papillary without atypia (PanIN-1B), papillary with atypia (PanIN-2), or with characteristics of carcinoma in situ (PanIN-3) (43) (Fig. 2). Interestingly, as observed in infiltrating colon adenocarcinomas, PanINs display some genetic changes such as activation of the K-ras pathway (44, 45), as well as inactivation of tumor suppressor genes, such as p16, p53, DPC4, and BRCA2 (46).
Figure 2.
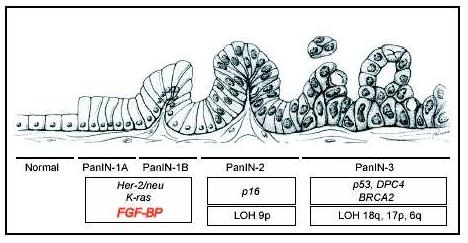
Genetic alterations during malignant transformation of pancreas epithelia. The progression from normal duct epithelium to low-grade and high-grade PanIN (Pancreatic Intraepithelial Neoplasia) and the associated accumulation of genetic alterations are shown (Adapted and modified from Hruban and colleagues [45]).
ANGIOGENESIS IN GASTROINTESTINAL CANCERS
Classically, angiogenesis is a process of endothelial sprout formation from preexisting vessels (47). In a more complex picture, angiogenesis is the mechanism mediating the growth and remodeling of a capillary network (48, 49). In the adult organism, angiogenesis is commonly restricted to conditions of tissue repair and remodeling (eg, during menstruation and mammary gland involution), and pathologic circumstances, such as wound repair, inflammation, and neoplastic growth where it plays a pivotal role (48, 50). Specific angiogenic factors can initiate a multi-step process that begins with vasodilatation, followed by the enhancement of vessel permeability and stroma degradation. Acting as chemotactic and mitogenic agents, angiogenic factors can also induce endothelial cell migration and proliferation. As a result, endothelial cells converge and tightly assemble to generate a new lumen or they intercalate with endothelial cells in a preexisting vessel to give rise to capillary elongation. The newly formed capillary-like vessels undergo final remodeling, in which they rearrange from an irregular into a structured plexus of branching vessels and capillary loops (51). In neoplastic tissues, endothelial cells present various morphologic and physiologic abnormalities and produce anomalous vasculature, characterized by tortuous, elongated, and dilated vessels, atypical enlargement of the vascular lumen, and excessive branching. The newly formed vessels are randomly fused with either arterioles or venules and create an atypical microcirculation. As a result, neoplastic regions are often hypoxic and acidic due to the chaotic and slow blood flow (48, 52-55). In addition, tumor vessels are leaky because they present endothelial fenestrae, vesicles and transcellular holes, widened inter-endothelial junctions, and a discontinuous or absent basement membrane (56-58). Tumor vessels may also lack functional perivascular cells (59) and their walls can be formed by a mosaic of alternating endothelial and cancer cells (vasculogenic mimicry) (60).
In 1971, Folkman proposed that new blood vessel formation was required for tumor growth and metastasis (61, 62) and proposed that solid tumors cannot grow beyond 1-2 mm3 without a massive blood vessel supply. These microscopic tumor masses can eventually activate an “angiogenic switch” by inducing the sprouting of new capillaries from surrounding mature blood vessels. These blood vessels will infiltrate the tumor mass, initiating the increase in size of the tumor mass and its metastatic perfusion. It is now widely accepted that the angiogenic switch is “off” when the effect of pro-angiogenic molecules is counter-weighted by that of anti-angiogenic molecules, and it is “on” when the concentration of angiogenic inducers prevail on that of the angiogenic inhibitors.
Thus, activation of the angiogenic switch during early stages of tumor development suggests that regulation of the angiogenic process is likely to be a rate-limiting step in the progression from small lesion to extensive disease (49, 63, 64). Numerous pro-angiogenic factors involved in tumor-dependent angiogenesis have been described, including, among others, members of the vascular endothelial growth factor (VEGF) family, platelet-derived growth factor (PDGF), transforming growth factor-alpha and -beta (TGF-a and TGF-b), and members of fibroblast growth factor (FGF) family.
Although pancreatic cancer is not a grossly vascular tumor, it is often characterized by enhancement of tumor-dependent angiogenesis (65). A growing line of evidence has shown that various FGFs, such as FGF-1, FGF-2, FGF-5, FGF-7 (66-69) and FGF receptors (70-73) are upregulated in pancreatic cancer tissue samples and cell lines. These findings suggest that FGF-dependent downstream biologic events are likely to play an important role in the pathobiology of pancreatic cancer.
In colorectal cancer, several studies indicate angiogenesis as a crucial event leading to colon cancer progression. As a matter of fact, colorectal cancer is one of the best-studied models of tumor angiogenesis (74). As in many other tumors, several angiogenic regulators have been recognized in colon cancer, including VEGF, PDGF, thrombospondin, and angiopoietins (74, 75). Likewise, overexpression of FGF and FGFRs in colon cancer cells and tissues, as well as increase of FGF-2 serum levels in patients with advanced colon cancer, have been extensively reported (76-81).
FIBROBLAST GROWTH FACTORS AND FIBROBLAST GROWTH FACTOR-BINDING PROTEIN
The FGF family comprises 23 distinct, structurally-related proteins described to date that exert biologic effects in different cells and organ systems, including tumor growth and angiogenesis (82-85). FGFs are heparin-binding proteins, which interact with low affinity heparan sulfate proteoglycans (HSPGs). HSPGs are ubiquitous cell-surface and extracellular matrix (ECM) proteins, which have been shown to protect FGFs from thermal denaturation and proteolysis as well as to increase FGF receptor affinity and facilitate FGF binding to cell surface receptor. In addition, ECM-associated HSPGs modulate FGF bioavailability by generating a local reservoir for the growth factor and allowing a sustained stimulation of endothelial cells (82, 84). Mobilization of FGFs from the ECM storage, and in particular of FGF-1 and FGF-2, occurs via HSPG digestion by heparanases or glycosaminoglycan-degrading enzymes (82, 84).
An alternative mechanism of FGF release from the ECM is mediated by a secreted FGF-binding protein (FGF-BP). FGF-BP was first described in 1991 by Wu et al as a low-affinity heparin binding protein, isolated from A431 human epidermoid carcinoma cells (86). Several studies conducted in our laboratory have contributed to a more extensive characterization of FGF-BP. FGF-BP binds to FGF-1 and FGF-2 in a noncovalent, reversible manner facilitating the release of the growth factor from the ECM and presenting it to FGF receptors (87-90). Moreover, FGF-BP is likely to supplement the role of heparan sulfate in mediating FGF-dependent signaling and mitogenesis (91). Indeed, we and others have shown that heparin, heparan sulfate, and other heparinoids are able to compete with FGF-BP binding to FGF-2 as well as a direct binding of FGF-BP to perlecan, thereby indicating a potential role for FGF-BP in releasing FGF proteins tightly bound to ECM molecules (90, 92). Numerous studies conducted in our laboratory have confirmed a role for FGF-BP as an extracellular chaperone for FGFs that enhances FGF-dependent biochemical and biologic functions. In murine NIH-3T3 fibroblasts as well as in bovine GM7373 endothelial cells, FGF-BP was shown to enhance FGF-2-induced MAPK signaling pathway and cell proliferation and we showed recently that the interaction domain with FGF-2 is a short domain in the C-terminus of the FGF-BP protein (90, 91, 93). Furthermore, FGF-BP was responsible for enhancing FGF-2-mediated angiogenesis in a chick chorioallantoic membrane (CAM) assay (90). Overexpression of FGF-BP in a chicken transgenic model resulted in embryonic lethality due to massive disruption of blood vessel structure and integrity and subsequent hemorrhage (94).
FGF-BP is expressed below the level of detection by Northern blotting in normal adult human tissues. However, its expression is considerably elevated in various tumors, including skin, head and neck, cervical, lung, squamous cell carcinomas, breast, colon, and pancreatic adenocarcinomas (87, 88, 95-97). Studies with FGF-BP-negative cell lines indicated FGF-BP as a rate-limiting factor for tumor growth and angiogenesis in a nude mouse model (87). Similarly, ribozyme-targeting of endogenous FGF-BP from human squamous cell carcinoma and colorectal cancer cell lines resulted in a significant reduction of tumor growth and angiogenesis (88). These findings support a potential role of FGF-BP as an angiogenic switch in human neoplasia (88).
FIBROBLAST GROWTH FACTOR-BINDING PROTEIN IS AN EARLY MARKER IN GASTROINTESTINAL CANCERS
Our laboratory has recently focused on the analysis of FGF-BP expression and regulation during the progression of gastrointestinal cancers. In an initial study by Ray and colleagues (96), immunohistochemical analysis of normal and pathologic human colon biopsies using a polyclonal anti-FGF-BP antibody showed a remarkable upregulation of FGF-BP in dysplastic lesions, originating at the level of single crypts. Upregulation of FGF-BP protein was detected in a small portion (16%) of 76 apparently normal colon samples. However, expression of FGF-BP was detected in most samples with moderate to severe dysplasia (62 of 85; P < 0.0001 normal versus dysplastic mucosa) (Fig 3A). Furthermore, coincident with FGF-BP overexpression in dysplastic crypts, an increase of blood vessel density was found by immunostaining of the lamina propria with a CD31 antibody (from 80 ± 7 to 154 ± 9 vessels/field). The fact that specimens from different inflammatory bowel diseases, such as ulcerative colitis or Crohn's disease, did not show a significant FGF-BP upregulation prompted the authors to link FGF-BP overexpression in the onset of colon cancer to an early genetic occurrence.
Figure 3.
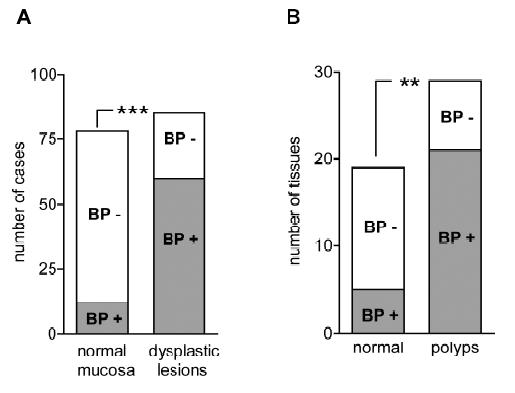
FGF-BP expression in human and mouse dysplastic lesions. (A) Frequency of FGF-BP protein expression, analyzed by immunohistochemistry (IHC) with a polyclonal anti-FGFBP antibody in 161 human specimens ***P< 0.0001. (B) Frequency of FGF-BP mRNA expression in normal and dysplastic intestinal tissues obtained from B6 APCMin/+ mice. FGF-BP expression coincides with the activation of the beta-catenin pathway in this model **P< 0.001 (96).
Analysis of FGF-BP mRNA by ISH in APCMin/+ mouse colon specimens showed a significant upregulation of FGF-BP in colon adenomas (21 of 27, >30% of the adenoma surface area). In contrast, little if any FGF-BP expression was noted in adjacent normal intestine (5 of 19; P < 0.001, normal versus adenomas) (Fig 3B). Interestingly, a prominent correlation between FGF-BP upregulation and cytoplasmic and nuclear beta-catenin staining in dysplastic colon was seen but not in a dextran sulfate-induced model of inflammatory colon disease (98). This finding further corroborated the hypothesis that FGF-BP is likely to be an early response gene during the initiation of colon cancer. Moreover, based on these findings and promoter-reporter studies, FGF-BP was shown to be a direct target of the activation of the Wnt/beta-catenin pathway (Fig 1) and therefore represents an early event for colon cancer initiation.
Tassi and colleagues (97) described the generation and characterization of a series of monoclonal antibodies against human FGF-BP. Monoclonal antibodies were applied for immunohistochemical (IHC) detection of FGF-BP in tissue microarrays containing a series of tissue samples from different stages of both colon and pancreatic cancers. In parallel, detection of FGF-BP mRNA in the same tissue slides was performed by in situ hybridization. High levels of FGF-BP protein and mRNA were found in those samples representing early stages of colon cancers, such as adenomatous polyps (Table 1 and Fig 4). However, a significant upregulation of FGF-BP was also identified in all other stages of colorectal cancer progression, such as invasive adenocarcinoma and metastases, compared to the levels obtained in normal specimens (P < 0.0001).
Table I.
Percentage of FGF-BP protein and mRNA expression analysis (%) in normal and pathologic colon and pancreatic tissues analyzed by IHC (monoclonal antibody) and ISH. n = number of cases analyzed (97).
| FGF-BP | Protein (%) | n | mRNA (%) | n | |
|---|---|---|---|---|---|
| COLON | |||||
| Normal crypts | 15 | 92 | 5 | 61 | |
| Adenomatous polyps | 90 | 29 | 100 | 9 | |
| Adenocarcinoma | 72 | 46 | 80 | 44 | |
| Metastases | 80 | 71 | 91 | 66 | |
| PANCREAS | |||||
| Normal ducts | 5 | 71 | 5 | 73 | |
| Pancreatitis | 47 | 17 | 40 | 20 | |
| PanIN LOW | 43 | 61 | 52 | 58 | |
| PanIN HIGH | 68 | 19 | 79 | 19 | |
| Adenocarcinoma | 49 | 69 | 60 | 69 | |
| Nonadenocarcinoma | 23 | 35 | 26 | 34 |
Figure 4.
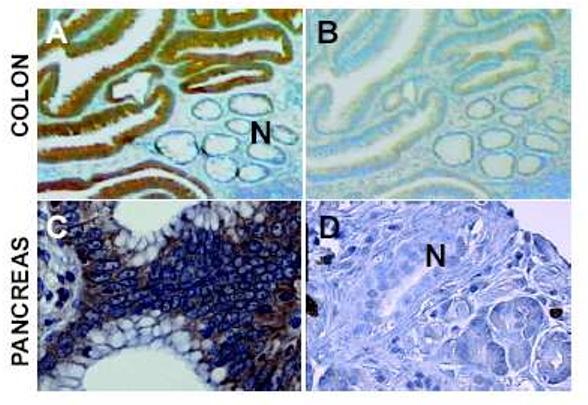
FGF-BP staining in premalignant human colon and pancreas lesions. (A) Histochemistry with a monoclonal antibody against FGF-BP shows expression (dark brown staining) in dysplastic lesions of the colon next to normal crypt epithelia (N). (B) A serial section from panel A stained with the secondary antibody only. (C) FGF-BP staining in a PanIN lesion. (D) A normal pancreatic duct shows no staining for FGF-BP. Further details in Ref. (97).
In addition, this report showed for the first time the analysis of FGF-BP expression during different progression stages of pancreatic cancer. Surprisingly, FGF-BP protein and mRNA were found dramatically upregulated during the early stages of malignant transformation, such as low-grade (PanIN1 and 2) and high-grade PanIN as well as invasive adenocarcinoma. Positive FGF-BP expression was also evidenced in some pancreatitis specimens, whereas very low or no expression was detected in normal pancreatic ducts. Nonadenocarcinoma samples showed expression of FGF-BP in 23% of samples at low levels, although this expression still represented a significant increase relative to normal specimens (P= 0.0044). A highly significant correlation resulted from comparison between protein and mRNA expression in all samples analyzed.
Based on these results, it is tempting to speculate that FGF-BP acts as an early regulatory gene, activated during the early stages of pancreatic cancer progression, most likely from the development of low-grade to high-grade PanINs (Fig 2). It is also possible that, as a consequence of FGF-BP overexpression in the early stages of colon and pancreatic cancers, an enhancement of tumor-dependent angiogenesis and invasion may occur, due to the increase of FGF-BP-mediated FGF bioavailability.
It is likely that FGF-BP, as a secreted protein, is shed into the circulation. Therefore, generation of a sensitive ELISA assay able to detect FGF-BP in patient sera may represent an important diagnostic screening method for early detection of colon and pancreatic premalignant lesions, thereby allowing early medical intervention.
CONCLUSION
Early events that initiate the stepwise progression of premalignant lesions of colon epithelia and pancreatic ducts to invasive cancer may provide circulating markers for the detection of such lesions. FGF-BP could be such a marker to identify subjects at an increased risk for developing invasive cancers.
Acknowledgements
We wish to thank Drs. Anirban Maitra (Hopkins), Ralf T. Henke and Bernadette Kim (Georgetown University) for critical discussions and contributions.
Footnotes
This work was supported in parts by grants of the NIH/NCI (R01 CA71508) and by support from the Gordon Family Foundation.
References
- 1.Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin. 2001;51(1):15–36. doi: 10.3322/canjclin.51.1.15. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Facts and Figures 2005. American Cancer Society; Atlanta: 2004. [Google Scholar]
- 3.Vogelstein B, Kinzler KW. The genetic basis for human cancer. 2nd ed. McGraw-Hill; Toronto: 2001. [Google Scholar]
- 4.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 5.Lamlum H, Ilyas M, Rowan A, et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's ‘two-hit’ hypothesis. Nat Med. 1999;5(9):1071–1075. doi: 10.1038/12511. [DOI] [PubMed] [Google Scholar]
- 6.Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54(22):5953–5958. [PubMed] [Google Scholar]
- 7.Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochem Biophys Acta. 1997;1332(3):F127–147. doi: 10.1016/s0304-419x(97)00008-5. [DOI] [PubMed] [Google Scholar]
- 8.Rubinfeld B, Souza B, Albert I, et al. Association of the APC gene product with beta-catenin. Science. 1993;262(5140):1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- 9.Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262(5140):1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 10.Bienz M. APC: the plot thickens. Curr Opin Genet Dev. 1999;9(5):595–603. doi: 10.1016/s0959-437x(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 11.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A. 1995;92(7):3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gumbiner BM. Carcinogenesis: a balance between beta-catenin and APC. Curr Biol. 1997;7(7):R443–446. doi: 10.1016/s0960-9822(06)00214-4. [DOI] [PubMed] [Google Scholar]
- 13.Morin PJ. beta-catenin signaling and cancer. Bioessays. 1999;21(12):1021–30. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 14.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 15.Aoki M, Hecht A, Kruse U, Kemler R, Vogt PK. Nuclear endpoint of Wnt signaling: neoplastic transformation induced by transactivating lymphoid-enhancing factor 1. Proc Natl Acad Sci U S A. 1999;96(1):139–144. doi: 10.1073/pnas.96.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 17.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247(4940):322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 18.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17(11 Reviews):1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 19.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53(4):549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 20.Bos JL, Fearon ER, Hamilton SR, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327(6120):293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 21.Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327(6120):298–303. doi: 10.1038/327298a0. [DOI] [PubMed] [Google Scholar]
- 22.Kressner U, Bjorheim J, Westring S, et al. Ki-ras mutations and prognosis in colorectal cancer. Eur J Cancer. 1998;34(4):518–521. doi: 10.1016/s0959-8049(97)10111-3. [DOI] [PubMed] [Google Scholar]
- 23.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689. [PubMed] [Google Scholar]
- 24.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 25.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: 'it ain't over 'til it's over'. Trends Cell Biol. 2000;10(4):147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 26.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 27.Shibata D, Reale MA, Lavin P, et al. The DCC protein and prognosis in colorectal cancer. N Engl J Med. 1996;335(23):1727–1732. doi: 10.1056/NEJM199612053352303. [DOI] [PubMed] [Google Scholar]
- 28.Cho KR, Oliner JD, Simons JW, et al. The DCC gene: structural analysis and mutations in colorectal carcinomas. Genomics. 1994;19(3):525–531. doi: 10.1006/geno.1994.1102. [DOI] [PubMed] [Google Scholar]
- 29.Chen YQ, Hsieh JT, Yao F, et al. Induction of apoptosis and G2/M cell cycle arrest by DCC. Oncogene. 1999;18(17):2747–2754. doi: 10.1038/sj.onc.1202629. [DOI] [PubMed] [Google Scholar]
- 30.Tarafa G, Villanueva A, Farre L, et al. DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene. 2000;19(4):546–555. doi: 10.1038/sj.onc.1203353. [DOI] [PubMed] [Google Scholar]
- 31.Miyaki M, Iijima T, Konishi M, et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18(20):3098–3103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 32.Koyama M, Ito M, Nagai H, Emi M, Moriyama Y. Inactivation of both alleles of the DPC4/SMAD4 gene in advanced colorectal cancers: identification of seven novel somatic mutations in tumors from Japanese patients. Mutat Res. 1999;406(24):71–77. doi: 10.1016/s1383-5726(99)00003-5. [DOI] [PubMed] [Google Scholar]
- 33.Salovaara R, Roth S, Loukola A, et al. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 2002;51(1):56–59. doi: 10.1136/gut.51.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244(4901):217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 35.Bunz F, Hwang PM, Torrance C, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104(3):263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng ZS, Sarkis AS, Zhang ZF, et al. p53 nuclear overexpression: an independent predictor of survival in lymph node--positive colorectal cancer patients. J Clin Oncol. 1994;12(10):2043–2050. doi: 10.1200/JCO.1994.12.10.2043. [DOI] [PubMed] [Google Scholar]
- 37.Leahy DT, Salman R, Mulcahy H, Sheahan K, O'Donoghue DP, Parfrey NA. Prognostic significance of p53 abnormalities in colorectal carcinoma detected by PCRSSCP and immunohistochemical analysis. J Pathol. 1996;180(4):364–370. doi: 10.1002/(SICI)1096-9896(199612)180:4<364::AID-PATH683>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 38.Allegra CJ, Paik S, Colangelo LH, et al. Prognostic value of thymidylate synthase, Ki-67, and p53 in patients with Dukes' B and C colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project collaborative study. J Clin Oncol. 2003;21(2):241–250. doi: 10.1200/JCO.2003.05.044. [DOI] [PubMed] [Google Scholar]
- 39.Garrity MM, Burgart LJ, Mahoney MR, et al. Prognostic value of proliferation, apoptosis, defective DNA mismatch repair, and p53 overexpression in patients with resected Dukes' B2 or C colon cancer: a North Central Cancer Treatment Group Study. J Clin Oncol. 2004;22(9):1572–1582. doi: 10.1200/JCO.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 40.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55(1):10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 41.Bodner WR, Hilaris BS, Mastoras DA. Radiation therapy in pancreatic cancer: current practice and future trends. J Clin Gastroenterol. 2000;30(3):230–233. doi: 10.1097/00004836-200004000-00005. [DOI] [PubMed] [Google Scholar]
- 42.Berlin JD, Rothenberg M. Chemotherapy for resectable and advanced pancreatic cancer. Oncology (Williston Park) 2001;15(10):1241–9, 54. discussion 54-64. [PubMed] [Google Scholar]
- 43.Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Sessa F, Solcia E, Capella C, et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Arch. 1994;425(4):357–367. doi: 10.1007/BF00189573. [DOI] [PubMed] [Google Scholar]
- 45.Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156(6):1821–1825. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hruban RH, Offerhaus GJ, Kern SE, Goggins M, Wilentz RE, Yeo CJ. Tumor-suppressor genes in pancreatic cancer. J Hepatobiliary Pancreat Surg. 1998;5(4):383–91. doi: 10.1007/s005340050062. [DOI] [PubMed] [Google Scholar]
- 47.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267(16):10931–10934. [PubMed] [Google Scholar]
- 48.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 49.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 50.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 51.Risau W. Development and differentiation of endothelium. Kidney Int Suppl. 1998;67:S3–6. doi: 10.1046/j.1523-1755.1998.06701.x. [DOI] [PubMed] [Google Scholar]
- 52.Baish JW, Jain RK. Fractals and cancer. Cancer Res. 2000;60(14):3683–8. [PubMed] [Google Scholar]
- 53.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 54.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3(2):177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 55.Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 59.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103(2):159–165. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Folberg R, Hendrix MJ, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000;156(2):361–381. doi: 10.1016/S0002-9440(10)64739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 62.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175(3):409–416. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 64.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82(1):4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 65.Korc M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer. 2003;2:8. doi: 10.1186/1476-4598-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siddiqi I, Funatomi H, Kobrin MS, Friess H, Buchler MW, Korc M. Increased expression of keratinocyte growth factor in human pancreatic cancer. Biochem Biophys Res Commun. 1995;215(1):309–315. doi: 10.1006/bbrc.1995.2467. [DOI] [PubMed] [Google Scholar]
- 67.Kuwahara K, Sasaki T, Kuwada Y, Murakami M, Yamasaki S, Chayama K. Expressions of angiogenic factors in pancreatic ductal carcinoma: a correlative study with clinicopathologic parameters and patient survival. Pancreas. 2003;26(4):344–349. doi: 10.1097/00006676-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 68.Yamanaka Y, Friess H, Buchler M, et al. Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res. 1993;53(21):5289–5296. [PubMed] [Google Scholar]
- 69.Kornmann M, Ishiwata T, Beger HG, Korc M. Fibroblast growth factor-5 stimulates mitogenic signaling and is overexpressed in human pancreatic cancer: evidence for autocrine and paracrine actions. Oncogene. 1997;15(12):1417–1424. doi: 10.1038/sj.onc.1201307. [DOI] [PubMed] [Google Scholar]
- 70.Kleeff J, Kothari NH, Friess H, Fan H, Korc M. Adenovirus-mediated transfer of a truncated fibroblast growth factor (FGF) type I receptor blocks FGF-2 signaling in multiple pancreatic cancer cell lines. Pancreas. 2004;28(1):25–30. doi: 10.1097/00006676-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 71.Kornmann M, Ishiwata T, Matsuda K, et al. IIIc isoform of fibroblast growth factor receptor 1 is overexpressed in human pancreatic cancer and enhances tumorigenicity of hamster ductal cells. Gastroenterology. 2002;123(1):301–313. doi: 10.1053/gast.2002.34174. [DOI] [PubMed] [Google Scholar]
- 72.Kornmann M, Beger HG, Korc M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas. 1998;17(2):169–175. doi: 10.1097/00006676-199808000-00010. [DOI] [PubMed] [Google Scholar]
- 73.Wagner M, Lopez ME, Cahn M, Korc M. Suppression of fibroblast growth factor receptor signaling inhibits pancreatic cancer growth in vitro and in vivo. Gastroenterology. 1998;114(4):798–807. doi: 10.1016/s0016-5085(98)70594-3. [DOI] [PubMed] [Google Scholar]
- 74.Wray CJ, Rilo HL, Ahmad SA. Colon cancer angiogenesis and antiangiogenic therapy. Expert Opin Investig Drugs. 2004;13(6):631–641. doi: 10.1517/13543784.13.6.631. [DOI] [PubMed] [Google Scholar]
- 75.Mancuso A, Sternberg CN. Colorectal cancer and antiangiogenic therapy: what can be expected in clinical practice? Crit Rev Oncol Hematol. 2005;55(1):67–81. doi: 10.1016/j.critrevonc.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 76.Dirix LY, Vermeulen PB, Hubens G, et al. Serum basic fibroblast growth factor and vascular endothelial growth factor and tumour growth kinetics in advanced colorectal cancer. Ann Oncol. 1996;7(8):843–848. doi: 10.1093/oxfordjournals.annonc.a010764. [DOI] [PubMed] [Google Scholar]
- 77.Landriscina M, Cassano A, Ratto C, et al. Quantitative analysis of basic fibroblast growth factor and vascular endothelial growth factor in human colorectal cancer. Br J Cancer. 1998;78(6):765–770. doi: 10.1038/bjc.1998.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galzie Z, Fernig DG, Smith JA, Poston GJ, Kinsella AR. Invasion of human colorectal carcinoma cells is promoted by endogenous basic fibroblast growth factor. Int J Cancer. 1997;71(3):390–395. doi: 10.1002/(sici)1097-0215(19970502)71:3<390::aid-ijc15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 79.Stoeltzing O, Liu W, Reinmuth N, et al. Angiogenesis and antiangiogenic therapy of colon cancer liver metastasis. Ann Surg Oncol. 2003;10(7):722–733. doi: 10.1245/aso.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 80.Tahara E. Growth Factors and Oncogenes in Gastrointestinal Cancers. In: Meyers RA, editor. Encyclopedia of Molecular Cell Biology and Molecular Medicine. ed 2 Wiley-VCH Verlag GmbH & Co KGaA; Weinheim: 2005. pp. 1–31. [Google Scholar]
- 81.Akbulut H, Altuntas F, Akbulut KG, et al. Prognostic role of serum vascular endothelial growth factor, basic fibroblast growth factor and nitric oxide in patients with colorectal carcinoma. Cytokine. 2002;20(4):184–190. doi: 10.1006/cyto.2002.1993. [DOI] [PubMed] [Google Scholar]
- 82.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7(3):165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 83.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20(11):563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 84.Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16(2):159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 85.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16(2):107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 86.Wu DQ, Kan MK, Sato GH, Okamoto T, Sato JD. Characterization and molecular cloning of a putative binding protein for heparin-binding growth factors. J Biol Chem. 1991;266(25):16778–16785. [PubMed] [Google Scholar]
- 87.Czubayko F, Smith RV, Chung HC, Wellstein A. Tumor growth and angiogenesis induced by a secreted binding protein for fibroblast growth factors. J Biol Chem. 1994;269(45):28243–28248. [PubMed] [Google Scholar]
- 88.Czubayko F, Liaudet-Coopman ED, Aigner A, Tuveson AT, Berchem GJ, Wellstein A. A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat Med. 1997;3(10):1137–1140. doi: 10.1038/nm1097-1137. [DOI] [PubMed] [Google Scholar]
- 89.Kurtz A, Wang HL, Darwiche N, Harris V, Wellstein A. Expression of a binding protein for FGF is associated with epithelial development and skin carcinogenesis. Oncogene. 1997;14(22):2671–2681. doi: 10.1038/sj.onc.1201117. [DOI] [PubMed] [Google Scholar]
- 90.Tassi E, Al-Attar A, Aigner A, et al. Enhancement of fibroblast growth factor (FGF) activity by an FGF-binding protein. J Biol Chem. 2001;276(43):40247–40253. doi: 10.1074/jbc.M104933200. [DOI] [PubMed] [Google Scholar]
- 91.Ray P, Tassi E, Liu X-H, Wellstein A. Role of fibroblast growth factor-binding protein in the pathogenesis of HIV-associated hemolytic uremic syndrome. Am J Physiol. 2006;290:R105–113. doi: 10.1152/ajpregu.00492.2005. [DOI] [PubMed] [Google Scholar]
- 92.Mongiat M, Otto J, Oldershaw R, Ferrer F, Sato JD, Iozzo RV. Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J Biol Chem. 2001;276(13):10263–10271. doi: 10.1074/jbc.M011493200. [DOI] [PubMed] [Google Scholar]
- 93.Xie B, Tassi E, Swift MR, et al. Identification of the fibroblast growth factor (FGF)-interacting domain in a secreted FGF-binding protein by phage display. J Biol Chem. 2006;281(2):1137–1144. doi: 10.1074/jbc.M510754200. [DOI] [PubMed] [Google Scholar]
- 94.McDonnell K, Bowden ET, Cabal-Manzano R, Hoxter B, Riegel AT, Wellstein A. Vascular leakage in chick embryos after expression of a secreted binding protein for fibroblast growth factors. Lab Invest. 2005;85(6):747–755. doi: 10.1038/labinvest.3700269. [DOI] [PubMed] [Google Scholar]
- 95.Kagan BL, Henke RT, Cabal-Manzano R, et al. Complex regulation of the fibroblast growth factor-binding protein in MDA- MB-468 breast cancer cells by CCAAT/enhancer-binding protein beta. Cancer Res. 2003;63(7):1696–1705. [PubMed] [Google Scholar]
- 96.Ray R, Cabal-Manzano R, Moser AR, et al. Up-regulation of fibroblast growth factor-binding protein, by beta-catenin during colon carcinogenesis. Cancer Res. 2003;63(23):8085–8089. [PubMed] [Google Scholar]
- 97.Tassi E, Henke RT, Bowden ET, et al. Expression of a fibroblast growth factor-binding protein during the progression of pancreas and colorectal adenocarcinoma. Cancer Res. 2006;66:1191–1198. doi: 10.1158/0008-5472.CAN-05-2926. [DOI] [PubMed] [Google Scholar]
- 98.Herfarth H, Brand K, Rath HC, Rogler G, Scholmerich J, Falk W. Nuclear factor-kappa B activity and intestinal inflammation in dextran sulphate sodium (DSS)-induced colitis in mice is suppressed by gliotoxin. Clin Exp Immunol. 2000;120(1):59–65. doi: 10.1046/j.1365-2249.2000.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]