

Abstract
The study of adenovirus could greatly benefit from diverse methods of virus detection. Recently, it has been demonstrated that carboxy-terminal EGFP fusions of adenovirus core proteins Mu, V, and VII properly localize to the nucleus and display novel function in the cell. Based on these observations, we hypothesized that the core proteins may serve as targets for labeling the adenovirus core with fluorescent proteins. To this end, we constructed various chimeric expression vectors with fusion core genes (Mu-EGFP, V-EGFP, preVII-EGFP, and matVII-EGFP) while maintaining expression of the native proteins. Expression of the fusion core proteins was suboptimal using E1 expression vectors with both conventional CMV and modified (with adenovirus tripartite leader sequence) CMV5 promoters, resulting in non-labeled viral particles. However, robust expression equivalent to the native protein was observed when the fusion genes were placed in the deleted E3 region. The efficient Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP expression vectors were labeled allowing visualization of purified virus and tracking of the viral core during early infection. The vectors maintained their viral function, including viral DNA replication, viral DNA encapsidation, cytopathic effect, and thermostability. Core labeling offers a means to track the adenovirus core in vector targeting studies as well as basic adenovirus virology.
Keywords: Adenovirus, Vector, Gene therapy, Core proteins, Mu, V, VII, EGFP, Fluorescent labeling
Introduction
There are 51 identified human adenovirus serotypes of which some manifest clinical relevance while others have been applied as gene delivery vectors (Russell, 2000). The study of adenoviruses with regard to virology, pathology, and gene therapy has greatly benefited from various detection methods, including quantitative and localizing techniques which can probe for viral DNA, RNA, and protein at the molecular level. In the context of adenoviral vector design, reporter gene expression has likewise played a vital role in the evaluation of vector transduction and transcriptional control both in vitro and in vivo. However, full understanding of the key steps of adenovirus host–cell interaction, cell entry, genome delivery, replication, and progeny dissemination requires detection modalities which can follow viral components and particles at the microscopic level, preferably in real time. Indeed, methods founded on this rationale have demonstrated the power of tracking individual adenoviral particles intracellularly. Studies have applied ex vivo, non-specific chemical conjugation of the adenovirus capsid with synthetic fluorophores (e.g., fluorescein isothiocyanate, Texas Red, carbocyanine 3) to evaluate viral entry (Leopold et al., 1998; Nakano and Greber, 2000), examine the effect of fiber modification on intracellular trafficking (Bernt et al., 2003; Miyazawa et al., 1999), and show the dynein-dependent nature of adenovirus interaction with microtubules (Kelkar et al., 2004).
Recently, we and others successfully developed in vivo labeling of adenovirus with the enhanced green fluorescent protein (EGFP) fused to the minor capsid protein IX (Le et al., 2004; Meulenbroek et al., 2004). These studies illustrated specific and homogeneous capsid labeling of adenovirus by genetic means to yield fluorescent adenoviruses possessing full virus function and utility in the tracking of infection as well as the detection of viral biodistribution. The ability to track the adenovirus core and genome, nevertheless, represents a potential detection strategy distinct from capsid labeling and would be operative in following adenovirus genome delivery, an imperative step in both natural infection and gene transfer. Previously in vivo genome labeling was demonstrated using a tetR-EGFP fusion protein which binds to tetO repeats incorporated into the adenovirus genome (Glotzer et al., 2001). Although the labeling was feasible, analysis by the authors showed that the tandem tetO repeat sequence was prone to deletion by recombination. Furthermore, the technique was based on an exogenous, nonviral fusion protein tetR-EGFP label expressed in a producer cell line. It would be ideal and perhaps more efficient if in vivo labeling of the adenovirus genome could be accomplished with an endogenous viral protein expressed from the vector itself without the need for a producer cell line.
Adenoviruses are nonenveloped, double-stranded DNA viruses with an icosahedral structure constructed from an array of twelve different proteins. Their approximate 36-kb genome is covalently bound to two terminal proteins and further organized through non-covalent association with the three core proteins Mu, V, and VII. All three core proteins are translated from the common L2 transcript under the control of the major late promoter (Alestrom et al., 1984). Protein VII (pVII) is the major core protein contributing over 800 copies per virion (Lehmberg et al., 1999). Expressed as a 174-amino-acid precursor from which a 24-amino-acid N-terminus is cleaved (Sung et al., 1983), its functional and sequence homology to histone H3 (Cai and Weber, 1993; Lee et al., 2003) indicates that pVII acts as a histone-like center around which viral DNA is wrapped to form nucleosome structures (Chatterjee et al., 1986; Mirza and Weber, 1982). Protein V (pV) is speculated to serve as a bridge between the genome core and the capsid through association with proteins Mu, VI, and VII as well as the viral DNA (Chatterjee et al., 1985; Matthews and Russell, 1998; Vayda and Flint, 1987). There are more than 150 copies of the minor core protein V per virion which binds non-specifically to the viral DNA (Chatterjee et al., 1985, 1986; Lehmberg et al., 1999). Mu or protein X matures into a 19-amino-acid final form (Mu) after both amino- and carboxy-terminal cleavage of the 79 amino acid precursor (preMu) by the adenovirus encoded protease. The copy number per virion of Mu is not known (Chatterjee et al., 1985). Because of its ability to precipitate negatively charged DNA in vitro via its nine positively charged arginine residues, it has been postulated to play a role in viral DNA condensation in the core (Anderson et al., 1989).
Recent intracellular localization and functional studies of Mu, pV, and pVII were performed with carboxy-terminal fusion of EGFP to the native proteins (Lee et al., 2003, 2004; Matthews, 2001). Our attention was drawn to three established observations from these reports: (i) nuclear localization of the fusion core proteins was conserved, (ii) the fusion core proteins demonstrated some function in the cell, and (iii) pVand pVII are found at ample copy numbers per virion. Based on these findings, we hypothesized that the core proteins may serve as targets for labeling of adenoviruses with fluorescent proteins. Herein, we describe the genetic labeling of the adenovirus core through the use of a chimeric expression system of both the native and fluorescent fusion core proteins. Adenovirus core labeling offers a unique way to follow the adenovirus core with potential for studying adenovirus infection and biology as well as tracking adenoviral vectors in gene therapy applications.
Results
Fluorescent fusion core protein expression and localization
We chose a chimeric system to generate recombinant adenoviruses expressing control EGFP and the fluorescent fusion core proteins: Mu-EGFP, pV-EGFP, preVII-EGFP, and matVII-EGFP. PreMu-EGFP was not chosen as a candidate for labeling because it has been shown to block late adenovirus gene expression when expressed concomitantly with adenovirus infection (Lee et al., 2004). In the chimeric expression system, both the native, unmodified core protein and the fusion counterpart were expressed from the same vector. We initially applied the AdEasy system to construct E1 expression vectors under the control of the constitutive cytomegalovirus (CMV) promoter (Fig. 1). After transient transfection of the final genomes into 911 cells, rescue of all viruses was successful. Expression and localization of the fusion core proteins from the E1 expression vectors were verified 24 h postinfection in A549 cells. Fluorescence microscopy revealed expression of all fusion core proteins (Fig. 2). Control EGFP localized diffusely throughout the cytoplasm and nucleus, while all four fusion core proteins demonstrated strong nuclear localization. MuEGFP showed diffuse cytoplasmic signal along with nucleoplasmic and what appears to be nucleolar targeting. Protein V-EGFP, like Mu-EGFP, localized to both the nucleoplasm and the nucleoli; however, little protein was found in the cytoplasm. preVII-EGFP distributed exclusively to the nucleus with signals detected in the nucleoplasm, the nucleoli, and areas where the DNA Hoechst stain associated with the cellular chromosomes. matVII-EGFP colocalized almost entirely with the host cell DNA, while some of the protein was also visualized in the nucleoplasm. The image of matVII-EGFP was nearly indistinguishable from the picture captured for Hoechst stain of the same field. In 911 cells in which the viruses actively replicated, we observed the same pattern of fusion core protein localization as that seen in A549 cells (data not shown). These results agree with published data obtained from plasmid-derived transient expression of the same fusion core proteins (Lee et al., 2003, 2004; Matthews, 2001).
Fig. 1.
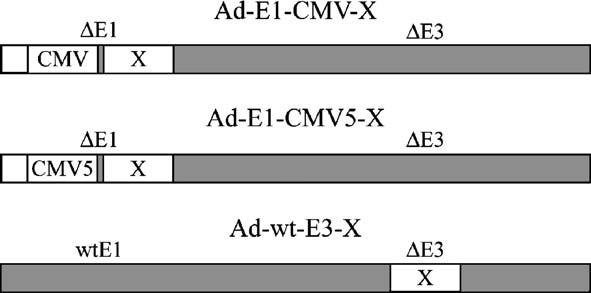
Schematic of vectors. Nomenclature and configuration of expression vectors where X represents Mu-EGFP, V-EGFP, preVII-EGFP, or matVII-EGFP. The employed expression strategies include transcription via the typical CMV promoter, a modified CMV promoter containing the adenovirus tripartite leader sequence (CMV5), and the adenovirus major late promoter (by virtue of transgene cassette insertion into the deleted E3 region).
Fig. 2.

Fluorescent core fusion protein expression and localization. A549 cells were infected with nonreplicative E1-CMV adenovirus vectors expressing the indicated genes. The cells were fixed and stained for nuclear DNA 24 h postinfection followed by visualization of the fluorescent proteins by epifluorescence microscopy.
Fluorescence of E1-CMV expression vector viral particles
After validating proper expression and intracellular trafficking of the fusion core proteins from the E1-CMV expression vectors, we analyzed the fluorescence of purified virus fractions collected from the CsCl gradient after two rounds of ultracentrifugation. During purification, both the mature and immature viral bands were retained. In our previous study involving fluorescent capsid-labeled adenovirus (Le et al., 2004), this procedure was established as a useful assay to detect fluorescence signal colocalizing with the purified viral bands. Each gradient fraction was quantitated for EGFP fluorescence and viral DNA content (optical absorbance at 260 nm, data not shown). The results were discouraging in that only minor fluorescent peaks were detected for the bottom mature virus band of Ad-E1-CMV-preVII-EGFP and the top immature virus band of Ad-E1-CMV-V-EGFP (data not shown), in contrast to our pIX-EGFP-labeled adenovirus which showed strong peaks (Le et al., 2004). We corroborated these data with fluorescence microscopy of samples from the bottom and top bands for each virus and found only occasional fluorescent particles, the number of which was disproportionately lower than the number of viral particles predicted by absorbance measurements at 260 nm (data not shown).
Western blot analysis of Ad-E1-CMV-V-EGFP purified virus and infected cells
With available anti-sera against the adenovirus pV, Western blot analysis was performed to detect pV-EGFP expression and incorporation into viral particles on a more molecular level. Purified virions collected from the top and bottom bands of control Ad-E1-CMV-GFP and Ad-E1-CMV-V-EGFP were examined. Although fluorometry analysis revealed a flat response throughout the gradient, GFP protein was unexpectedly found in the mature band of Ad-E1-CMV-GFP in contrast to our previous study (Le et al., 2004) (Fig. 3A, left panel, lane 1). Understandably, more GFP was detected in the immature band where it is expected to migrate along with unassociated cellular debris and proteins in the gradient (Fig. 3A, left panel, lane 2). A low level of pV-EGFP was detected in the bottom band, whereas significantly more was found in the top band (Fig. 3A, left panel, lanes 3 and 4, respectively), matching the results obtained in the fluorometry experiment. Probing with the pV anti-sera confirmed the findings obtained with the GFP antibody; however, it also showed that Ad-E1-CMV-V-EGFP virions contained substantially less of the fusion core protein than the native pV (Fig. 3A, right panel, lanes 3 and 4), suggesting inefficient incorporation of the fusion core pV-EGFP relative to the native version. We further analyzed the expression of pV-EGFP in 911 cells during Ad-E1-CMV-V-EGFP infection. Western blot analysis revealed that intracellular expression of the fusion core protein relative to the native protein paralleled the observations made above with the purified virus. Considerably lower expression of pV-EGFP was noted relative to natural pV (Fig. 3A, right panel, lane 6). These data indicate that the inefficient incorporation of pV-EGFP into virions may be due to the unfavorable expression of pV-EGFP in comparison to native pV, although assembly defect due to EGFP fusion could also be possible.
Fig. 3.
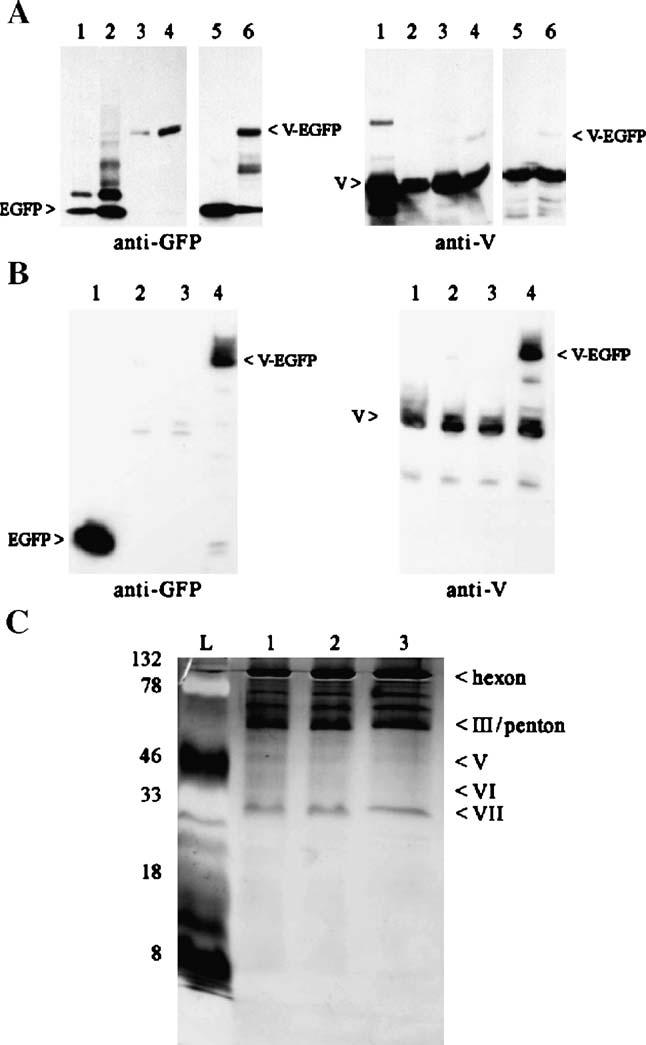
Western blot analysis of Ad-E1-CMV-V-EGFP purified virus and cells infected with various expression vectors. (A) Purified viruses (8 μg protein in each sample) and infected cell lysates (4 μg protein each sample) were subjected to SDS-PAGE and blotted with GFP and pV antibodies. Lanes (1) Ad-E1-CMV-EGFP bottom band, (2) Ad-E1-CMV-EGFP top band, (3) Ad-E1-CMV-V-EGFP bottom band, (4) Ad-E1-CMV-V-EGFP top band, (5) Ad-E1-CMV-GFP infected 911 cells, and (6) Ad-E1-CMV-V-EGFP infected 911 cells. (B) Protein lysates (10 μg) from 911 cells infected with the various V-EGFP expression vectors were subjected to SDS-PAGE and blotted with GFP and pV antibodies. Lanes (1) Ad-E1-CMV-EGFP, (2) Ad-E1-CMV-V-EGFP, (3) Ad-E1-CMV5-V-EGFP, and (4) Ad-wt-E3-V-EGFP. (C) Purified core-labeled viral particles (1.5 × 1010) were subjected to SDS-PAGE and then silver-stained. Lanes: (L) protein ladder, (1) Ad-wt-E3-EGFP, (2) Ad-wt-E3-V-EGFP, and (3) Ad-wt-E3-preVII-EGFP. The numbers on the left side indicate the molecular weight of the protein ladder (kDa). The labels on the right highlight the major Ad structural proteins visible on the SDS-PAGE.
Enhanced expression of pV-EGFP from the E3 region
As a result of the poor expression achieved with the E1-CMV vectors, we explored the possibility of increasing the fusion core protein expression relative to the native protein by using two approaches. The first strategy involved the use of the CMV5 promoter (Qbiogene, Fig. 1) to drive expression of the fusion core proteins from the E1-deleted region. This modified promoter features the adenovirus tripartite leader sequence downstream of the CMV promoter which has been previously shown to enhance transgene expression (Sheay et al., 1993). Our attempt to generate E1-CMV5 fusion core protein expression viruses unfortunately resulted in unsuccessful rescue of all candidate vectors (Ad-E1-CMV5-Mu-EGFP, Ad-E1-CMV5-preVII-EGFP, and Ad-E1-CMV5-matVII-EGFP) except the control Ad-E1-CMV5-EGFP and Ad-E1-CMV5-V-EGFP. The second strategy to generate vectors with enhanced expression exploited the deleted E3 region of the adenovirus genome to express transgenes (Fig. 1). Previous reports have demonstrated that efficient expression late in infection could be attained by using this locale for transgene insertion, likely due to transcription from the major late promoter, polyadenylation, and splicing with the tripartite leader sequence (Hawkins and Hermiston, 2001a,b; Hawkins et al., 2001; Mittal et al., 1993). Contrary to the results obtained with the E1-CMV5 expression vectors, we were successful in rescuing most of the E3 expression vectors (Ad-wt-E3-EGFP, Ad-wt-E3-Mu-EGFP, and Ad-wt-E3-V-EGFP) except Ad-wt-E3-matVII-EGFP.
To verify enhanced expression of the fusion core proteins was indeed achieved using the E1-CMV5 and E3 chimeric expression strategies, Western blot analysis was performed with cell lysates from 911 cells infected with 1 CPEU/cell (cyptopathic effect unit) of the various vectors 24 h postinfection. Representative analysis was performed with the V-EGFP vectors. Although we observed strong expression of EGFP from the control Ad-E1-CMV-EGFP vector (Fig. 3B, left panel, lane 1), the expression of V-EGFP driven by the E1-CMV and E1-CMV5 vectors was quite low when probed with both the EGFP and pV antibodies (Fig. 3B, left and right panels, lanes 2 and 3, respectively). In our hands, the CMV5 did not provide improved transgene expression over the unmodified CMV promoter, particularly with V-EGFP as the transgene. However, pV-EGFP expression from the deleted E3 region was detected at a significant level (Fig. 3B, left and right panels, lane 4) comparable to the amount of native pV (Fig. 3B, right panel, lane 4).
SDS-PAGE was used to compare the major structural protein composition of purified Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP to that of the control vector Ad-wt-E3-EGFP. Silver staining revealed no gross differences in the protein compositions of these three viruses. Note that bands corresponding to pV-EGFP and pVII-EGFP were not apparent on SDS-PAGE (Fig. 3C).
Fluorescence of Ad-E1-CMV5-V-EGFP and E3 fusion core protein expression vectors
The fluorometry assay described above was repeated with CsCl gradient fractions of Ad-E1-CMV5-V-EGFP (the only rescued core protein E1-CMV5 vector) and the E3 expression viruses. As predicted by the Western blot analysis, no fluorescent peaks in the bottom and top viral bands were detected for Ad-E1-CMV5-V-EGFP like the included control vector Ad-E1-CMV5-EGFP (Fig. 4, middle panel). Ad-wt-E3-Mu-EGFP revealed very small peaks for both the mature virus and empty particle bands (Fig. 4, top panel). Both Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP displayed strong fluorescent peaks for the bottom bands and a couple of minor peaks above the mature particle band (Fig. 4, middle and bottom panels, respectively). All major fluorescent peaks in the bottom bands of Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP coincided with viral DNA peaks measured by absorbance at 260 nm (Fig. 4, middle and bottom panels, respectively). It is interesting to note that both Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP did not show major peaks in the top band (empty capsids with little or no viral DNA), whereas our previous capsid-labeled Ad-IXEGFP virus demonstrated a strong fluorescent peak in the top band comparable to the bottom band (Le et al., 2004). This observation may be unique to the core-labeled vectors which contain fusion core proteins with the propensity to interact with viral DNA as opposed to the capsid-labeled vector.
Fig. 4.
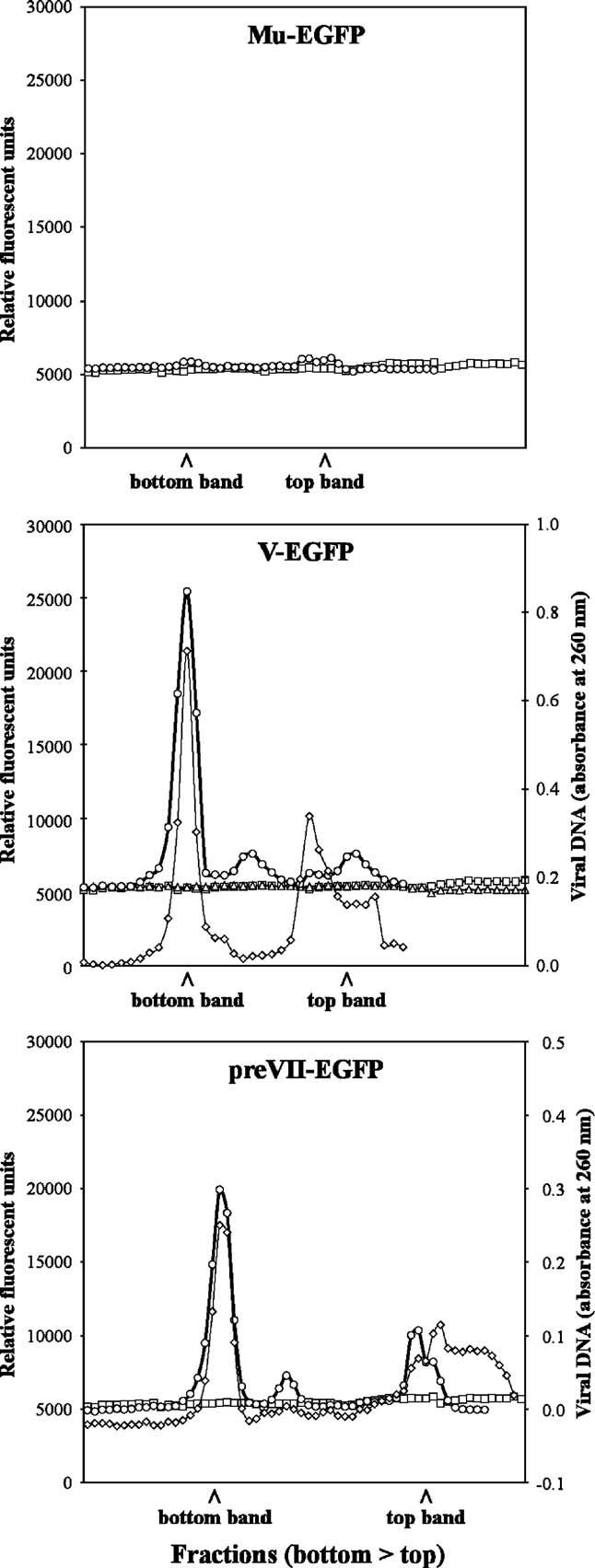
Analysis of fusion core protein viral gradients with improved expression. Fusion core protein vectors with enhanced expression were purified and fractionated to determine fluorescence of CsCl gradient fractions. For all panels, unlabeled Ad-E1-CMV5-EGFP was included as a control (–□–). Ad-E1-CMV5-X (–sh=utri–) and Ad-wt-E3-X (–○–) are shown where X is the indicated fusion protein at the top of the panel. Note that Ad-E1-CMV5-Mu-EGFP, Ad-E1-CMV5-preVII-EGFP, Ad-E1-CMV5-matVII-EGFP, and Ad-wt-E3-matVII-EGFP could not be rescued for analysis. For Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP, viral DNA content was also analyzed for the respective fractions (–◇–).
Visualization of Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP fluorescent particles
After confirming fluorescent labeling of the E3 fusion core protein vectors with the fractionation assay, the purified viral particles were examined under fluorescence microscopy. Abundant Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP fluorescent particles could be visualized (Figs. 5A and B, respectively) while Ad-wt-E3-Mu-EGFP virions could not be detected (data not shown), suggesting the poor labeling efficiency of this vector.
Fig. 5.
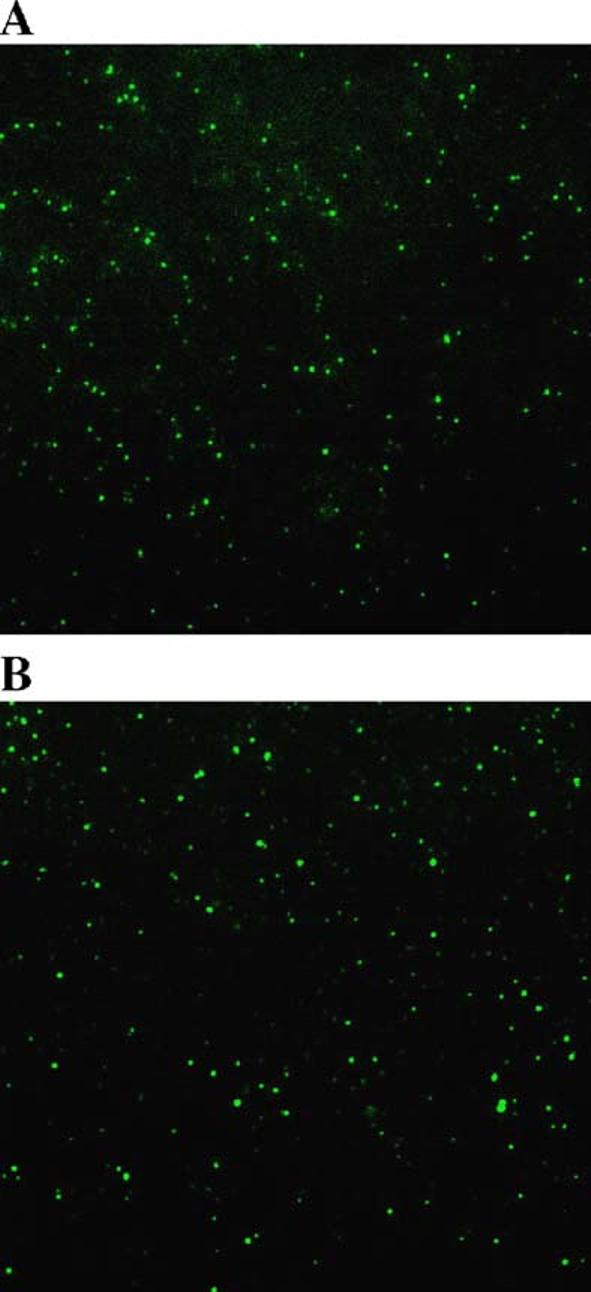
Visualization of Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP particles. Purified Ad-wt-E3-V-EGFP (A) and Ad-wt-E3-preVII-EGFP (B) were prepared on slides with coverslips and imaged with epifluorescence microscopy.
Tracking early infection with core-labeled adenovirus
To demonstrate the utility of our core labeling technique, we tracked the fate of the pV-EGFP and preVII-EGFP-labeled viruses during early infection of HeLa cells. While no fluorescent signal resembling viral particles were observed with the control Ad-wt-E3-EGFP virus, numerous fluorescent particles could be visualized in the cytoplasm (white arrows) and nuclei (orange arrows) of cells infected with Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP for 1 h at 37 °C (Fig. 6, top two rows). After 3 h of infection at 37 °C, some residual Ad-wt-E3-V-EGFP fluorescent particles were detected in the cytoplasm while very few or no fluorescent particles were seen in the nucleus. In contrast, many Ad-wt-E3-preVII-EGFP fluorescent particles remained intact in the nucleus at this time, and only a few were found in the cytoplasm. Although some cells had started to weakly express pV-EGFP or preVII-EGFP at 3 h after infection, strong EGFP expression was noted in cells infected with the control Ad-wt-E3-EGFP virus (Fig. 6, bottom two rows). By 6 h after infection, all cells strongly expressed EGFP, pV-EGFP, or preVII-EGFP with localization patterns similar to those shown in Fig. 2. The high level of newly synthesized fluorescent signal interfered with detection of the input fluorescent particles at 6 h after infection (data not shown).
Fig. 6.
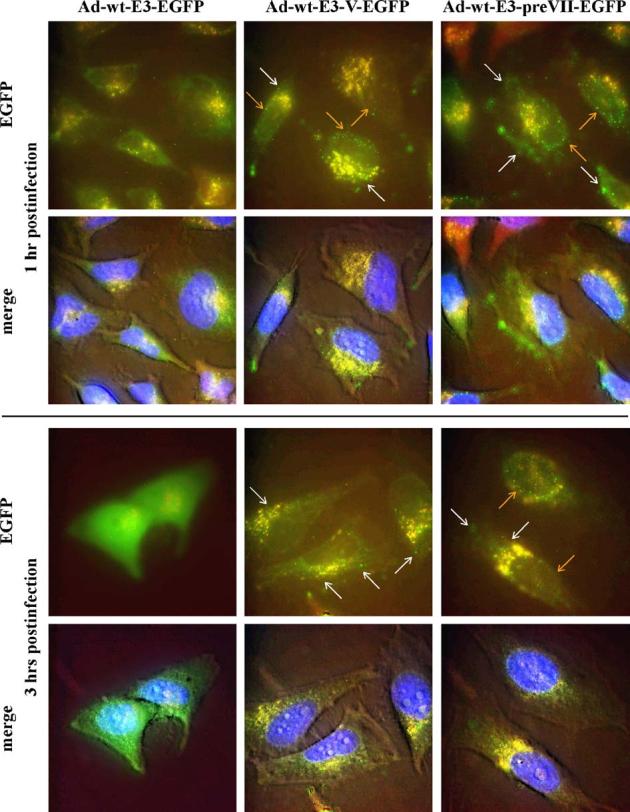
Tracking of core-labeled virus infection. HeLa cells were infected with Ad-wt-E3-EGFP (left column), Ad-wt-E3-V-EGFP (middle column), and Ad-wt-E3-preVII-EGFP (right column) at 37 °C and then imaged at 1 h (upper 2 rows) and 3 h (bottom 2 rows) postinfection using epifluorescence microscopy. Green represents EGFP fluorescence and some background autofluorescence; blue indicates nuclear DNA; and red, yellow, and brown represent background signal. White arrows = fluorescent viral particles in the cytoplasm. Orange arrows = fluorescent viral particles in the nucleus. Note that the bottom row pictures for each time period are merged color images overlaid on top of a phase contrast picture of the cells to show morphology.
DNA packaging analysis of core-labeled adenoviruses
The core proteins play essential roles in packaging of the viral genome inside the capsid. We tested whether our chimeric system of expressing the fusion core proteins in competition with the endogenous proteins would affect viral DNA packaging. For this analysis, we infected 911 cells with the labeled vectors Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP along with the control Ad-wt-E3-EGFP (1 CPEU/cell of each virus). Total intracellular viral DNA and encapsidated viral DNA inside the cell from various days postinfection were quantitated by real-time Taqman PCR using E4-specific primers. Even though infection with the three viruses was executed at the same multiplicity of infection based on infectious titer, the control Ad-wt-E3-EGFP virus showed significantly lower total viral DNA synthesis and encapsidation by about one order of magnitude compared to Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP. By day 4, however, both the total intracellular viral genome copy number and the amount of encapsidated viral DNA were nearly the same for all three viruses, indicating that the growth kinetics of the control virus may have been delayed (Fig. 6A, left and middle panels). Expressing the data as encapsidation percentage of total viral DNA further illustrates the stunted dynamics and essentially lower yield of Ad-wt-E3-EGFP while concomitantly showing equivalent behavior of Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP (Fig. 7A, right panel).
Fig. 7.
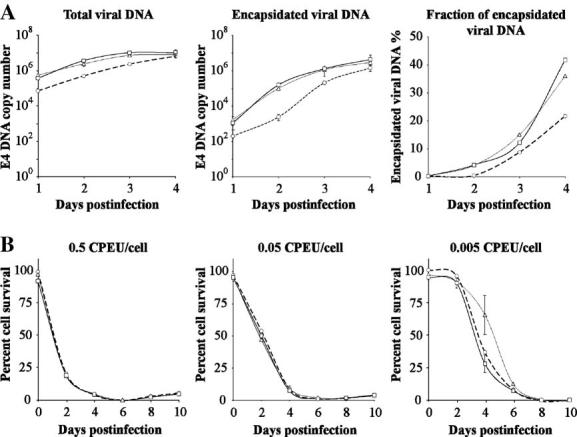
DNA packaging efficiency and cytopathic effect of core-labeled viruses. (A) 911 cells were infected over the course of 4 days with control Ad-wt-E3-EGFP (--○--), Ad-wt-E3-V-EGFP (–△–), and Ad-wt-E3-preVII-EGFP (–□–). Each day, half of the cells collected were processed for total viral DNA while the other half were processed for encapsidated viral DNA. Both viral DNA pools were quantitated by Taqman real-time quantitative PCR using E4-specific primers (n = 3). The third panel shows the percent of encapsidated viral DNA for each respective virus (encapsidated divided by total and then multiplied by 100%). (B) The cytopathic effect of the same three viruses was analyzed in 911 cells over the course of 10 days using the indicated multiplicities of infection. Every 2 days, the cell viability was quantitated by MTS assay. Cell viability is expressed as percentage relative to noninfected cells (n = 5). Ad-wt-E3-EGFP (--○--), Ad-wt-E3-V-EGFP (–△–), and Ad-wt-E3-preVII-EGFP (–□–).
Cytopathic effect of core-labeled viruses
To further evaluate the overall function of the core-labeled viruses, cytopathic effect was assessed in comparison to the control virus Ad-wt-E3-EGFP. 911 cells were infected with various amounts of each virus (0.5, 0.05, and 0.005 CPEU/cell), and their viability was assayed over the course of 10 days postinfection. The cytopathic effect was not appreciably different between the three viruses even at the lowest multiplicity of infection (Fig. 7B).
Thermostability of core-labeled virions
We tested the thermostability of the viruses by exposing equal amounts (based on t.u., transduction unit titer) of Ad-wtE3-EGFP, Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP to 45 °C for various time periods. Following heat treatment, the samples were functionally titered in terms of transduction units. The thermostabilities of the core-labeled virions (especially Ad-wt-E3-V-EGFP) were actually better than that of the control Ad-wt-E3-EGFP (Fig. 8).
Fig. 8.
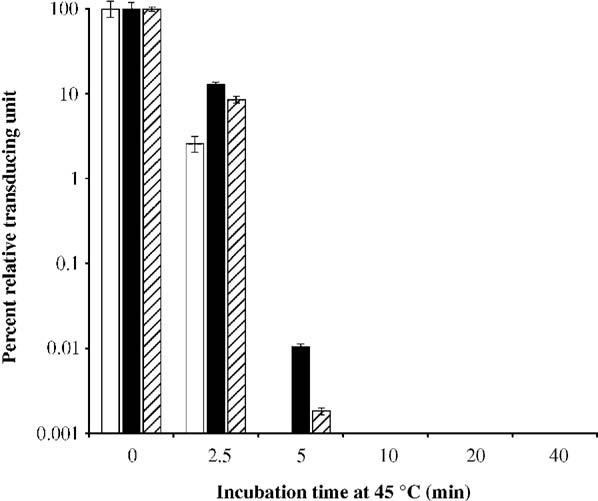
Thermostability of core-labeled viruses. Ad-wt-E3-EGFP (white), Ad-wt-V-EGFP (black), and Ad-wt-E3-preVII-EGFP (striped) were incubated at 45 °C for various times and then quantitated by transduction unit titer. Bars represent percent of remaining infectivity relative to untreated samples (n = 3).
Discussion
We have described herein successful labeling of the adenovirus core through the chimeric expression of fluorescent fusion core proteins V-EGFP and preVII-EGFP from the deleted E3 region. Because of their nonutility for gene therapy objectives and perhaps their indispensable role in formation of the virus core, modification of core proteins has not been exploited in recombinant adenoviruses. Such basis directed us to pursue a conservative approach in labeling the adenovirus core with a chimeric expression system wherein both the fluorescent fusion core proteins and their native counterparts are expressed from the same vector. This cautious strategy would safeguard against unpredictable compromise of core protein function due to the EGFP fusion.
In using a chimeric expression system, however, the relative expression levels of the exogenous and endogenous core proteins must be considered in light of the fact that each is competing with the other for assembly into virions. As well, any possibility of perturbing the structural function of the original protein through the addition of EGFP should be taken into account. Indeed, the former predicament was encountered when applying our initial method of CMV-promoter-driven expression from the deleted E1 region. The CMV promoter has been widely used as a constitutive promoter to drive strong transgene expression from nonreplicative adenoviral vectors. Nevertheless, adenoviral gene expression regulatory mechanisms during replication pose a great challenge when using the foreign CMV promoter to match the expression level of the endogenous core proteins. Late in infection, adenoviral mRNA transcripts containing the spliced 5′ tripartite leader sequence are preferentially translated over transcripts lacking this element (Berkner and Sharp, 1985; Chow et al., 1977; Logan and Shenk, 1984). Transcripts generated from the CMV promoter would not contain this tripartite leader sequence and would therefore be translated at a lower level than the endogenous adenoviral L2 transcript from which the core proteins are made.
The tripartite leader sequence has been exploited to enhance transgene expression from plasmid vectors by mating the tripartite leader sequence downstream of certain nonadenoviral promoters (Sheay et al., 1993). Moreover, a similar strategy was employed in stable packaging cell lines to achieve strong expression of a recombinant fiber protein in the context of adenovirus replication (Von Seggern et al., 2000). We pursued this approach to increase expression of the fusion core proteins from the deleted E1 region. When applying the modified CMV5 promoter which contains the tripartite leader sequence to improve transgene expression, we could only rescue Ad-E1-CMV5-EGFP and Ad-E1-CMV5-V-EGFP. Past studies have shown that pVII can interfere with adenovirus DNA synthesis (Korn and Horwitz, 1986) and mRNA transcription in vitro (Nakanishi et al., 1986). Protein Mu has the ability to precipitate negatively charged DNA in vitro and plays a role in viral DNA condensation in the core (Anderson et al., 1989). On this basis, the untimely and strong expression of Mu-EGFP, preVII-EGFP, and matVII-EGFP via the CMV5 promoter may explain the failure to rescue these respective vectors. However, all E1-CMV vectors were rescued, and yet increased gene expression of pV-EGFP with CMV5 relative to CMV was not observed under our experimental conditions. Unlike the E1-CMV5 approach, the use of the deleted E3 region to express our transgenes was more fruitful in that most viruses could be rescued (Ad-wt-E3-EGFP, Ad-wt-E3-Mu-EGFP, Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP) except Ad-wt-E3-matVII-EGFP. Timely expression in infection from the deleted E3 region (mostly via the major late promoter) may explain these results.
Strong expression of the fusions pV-EGFP and preVII-EGFP from the deleted E3 region yielded fluorescent virions under fluorescence microscopy. It should be noted that Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP particles showed qualitatively less intense and more heterogenous fluorescence than our capsid-labeled Ad-IX-EGFP vector (Le et al., 2004). The weak fluorescence seems paradoxical in the case of Ad-wt-E3-preVII-EGFP since the copy number per virion of pVII is over 800 (Lehmberg et al., 1999), whereas the copy number per virion of pIX is 240 (Lehmberg et al., 1999; van Oostrum and Burnett, 1985). The nature of core labeling may compact the fluorescent fusion core proteins within the capsid in close proximity to each other, possibly diminishing the fluorescent signal (e.g., through fluorescence energy transfer). On the other hand, the labeling of the capsid with surface-exposed pIX-EGFP would less likely result in this phenomenon. Additionally, our capsid labeling strategy was based on complete replacement of the IX gene with IX-EGFP, whereas core labeling with preVII-EGFP also involved packaging of the non-labeled, competing native pVII.
We were able to demonstrate the utility of our core-labeled vectors by using their fluorescent property to monitor early infection. Both Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP fluorescent particles could be detected in the cytoplasm and nuclei of infected cells, indicating that the core-labeled fluorescent virions were indeed infectious particles and not merely protein conglomerates. The fluorescent signal was detectable for both vectors during trafficking to the nucleus as well as after nuclear translocation. While in the cytoplasm, these fluorescent particles are likely the viral cores which remain inside a partially dismantled capsid following cell entry (Greber et al., 1993). After nuclear import, the fluorescent particles are likely the delivered genome-containing viral cores released from the capsid (Greber et al., 1997). Interestingly, the punctate fluorescence of Ad-wt-E3-preVII-EGFP was maintained in the nucleus longer than that of Ad-wt-E3-V-EGFP. This observation suggests that pV-EGFP may dissociate from the delivered viral core earlier than pVII-EGFP. A previous report has shown that protein VII remains with the input viral genome throughout the early phase of infection (Xue et al., 2005). Our data with Ad-wt-E3-preVII-EGFP corroborate this observation at least up to 3 h postinfection. One drawback of our core-labeled vectors is the fact that, at 6 h postinfection, strong de novo expression of the fusion core proteins from the delivered genomes prevented detection of the input fluorescent viral core. Although strong expression from the E3 region is attributed to the major late promoter (Mittal et al., 1993), the upstream E3 enhancer/promoter may induce expression of transgenes in this region earlier in infection. Because optimal E3 promoter activity is dependent on E1A expression (Berk, 1986), future studies employing E1-deleted core-labeled vectors may prevent early expression of the fusion core proteins to allow tracking of the input fluorescent viral cores at 6 h postinfection and perhaps even later.
Various assays were performed to evaluate the viability of the core-labeled vectors Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP. In the DNA packaging analysis, the viral DNA replication and encapsidation of the control Ad-wt-E3-EGFP were poor compared to the two core-labeled viruses (about one order lower) despite using the same multiplicity of infection for all three vectors. Interestingly, the only difference between the control virus and the other two vectors is expression of diffusely localizing EGFP for the former versus expression of nuclear localizing fusion core proteins with EGFP for the latter two. It is difficult to attribute increased DNA replication to the over-expression of the core proteins (i.e., native pV plus modified V-EGFP, Fig. 3B, right panel, lane 4) in the case of Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP since high expression of these proteins occurs late after DNA replication. On the other hand, it is plausible that the increased presence of the core proteins may lead to increased packaging efficiency late in infection resulting in a higher readout using our assay. It is also likely that the high expression of EGFP by itself may influence the kinetics of viral replication through its interactions with viral proteins or the host cell. In this respect, EGFP has been reported to cause toxicity both in living cells and in mice (Huang et al., 2000; Liu et al., 1999). We did note in the tracking experiment that EGFP was expressed much earlier and stronger (at 3 h postinfection) in most cells infected with the control Ad-wt-E3-EGFP vector relative to fusion core protein expression from Ad-wt-E3-V-EGFP and Ad-wt-E3-preVII-EGFP.
Another anomaly between control Ad-wt-E3-EGFP and the core-labeled vectors was discovered in the thermostability assay. Both modified viruses appeared more thermostable than the control. Following exposure of the viruses to 45 °C, transduction unit titer was used as an infectious index of viral function. Because the control virus was practically wild-type with respect to its capsid and core structure, the incorporation of the fusion core proteins into the modified viruses may have imparted additional stability. Whether the increased thermostability pertains to the entire adenovirus structure or just the core is uncertain. It is possible that a more thermostable core alone would produce the results we obtained since a DNA–pVII complex by itself has transfection capability (Wienhues et al., 1987). Such “nonviral” transduction capability would likely contribute to the increased t.u. titers we measured for the core-labeled vectors.
In summary, we have demonstrated the fluorescent labeling of the adenovirus core by competitive expression of fusions pV-EGFP and preVII-EGFP from the deleted E3 region. Strong expression of these fusion core proteins from the E3 region did not negatively impact DNA replication, DNA encapsidation, thermostability, and cytopathic effect of the viruses. The purified particles were labeled and fluorescent, allowing their detection under fluorescence microscopy. A future extension of the techniques described here is to generate recombinant adenoviruses with complete replacement of the native V or preVII gene with the fusion V-EGFP or preVII-EGFP. Core labeling of adenoviruses would allow the tracking of the adenovirus core and complements our capsid labeling technique in the study of adenovirus biology, pathogenesis, and vector design.
Methods
Cell culture
Human embryonic retinoblast 911 (Fallaux et al., 1996), human lung adenocarcinoma A549 (American Type Culture Collection (ATCC), Manassas, VA), human cervical carcinoma HeLa (ATCC), and Chinese hamster ovary (CHO, ATCC) cells were maintained according to the suppliers' protocols. Cells were incubated at 37 °C and 5% CO2 under humidified conditions.
Recombinant adenovirus construction
All viruses were constructed by homologous recombination in Escherichia coli (He et al., 1998). The base plasmids were acquired from the following sources: pShuttle-CMV, pAdenoVator-CMV5, and pAdEasy (Qbiogene, Irvine, CA); pcDNA3-CMV-V-EGFP (Matthews, 2001), pcDNA3-CMV-Mu-EGFP (Lee et al., 2004), pcDNA3-CMV-preVII-EGFP (Lee et al., 2003), pcDNA3-CMV-matVII-EGFP (Lee et al., 2003); shuttle plasmids for the E1-CMV/E3-deleted chimeric expression vectors were constructed as follows using restriction cloning: pShuttle-E1-CMV-V-EGFP → pShuttle-CMV/SalI/blunt/KpnI + pcDNA3-CMV-V-EGFP/ApaI/blunt/KpnI; pShuttle-E1-CMV-Mu-EGFP → pShuttle-CMV/EcoRV/KpnI + pcDNA3-CMV-Mu-EGFP/ApaI/blunt/KpnI; pShuttle-E1-CMV-preVII-EGFP → pShuttle-CMV/EcoRV/KpnI + pcDNA3-CMV-preVII-EGFP/ApaI/blunt/KpnI; pShuttle-E1-CMV-matVII-EGFP → pShuttle-CMV/EcoRV/KpnI + pcDNA3-CMV-matVII-EGFP/ApaI/blunt/KpnI. pShuttle-E1-CMV-EGFP was cloned in two steps. pcDNA3-CMV-V-EGFP was cut with BamHI and self-ligated to remove the V fusion. Then, the resulting plasmid was cut with ApaI/blunt/KpnI to liberate the EGFP gene for ligation into pShuttle-CMV cut with EcoRV/KpnI.
Shuttle plasmids for vectors expressing the fusion core proteins under the modified CMV5 promoter (containing the adenovirus tripartite leader sequence) were constructed with pAdenoVator-CMV5. For EGFP, Mu-EGFP, preVII-EGFP, and matVII-EGFP, the respective pShuttle-E1-CMV plasmids containing these genes as described above were cut with BglII/BstXI to release the transgene and a portion of the right homologous arm. These fragments were ligated into pAdenoVator-CMV5 cut with BamHI/BstXI (fragment which contains the CMV5 promoter). To generate pAdenoVator-CMV5-V-EGFP, pShuttle-E1-CMV-V-EGFP was cut with SalI/blunt/BstXI to give a fragment with the fusion gene and then ligated into pAdenoVator cut with PmeI/BstXI.
Finally, a parent E3 shuttle plasmid was made to insert the same fusion genes into the adenovirus E3 region by double selection homologous recombination. Briefly, pShuttlE3 contains the left and right homologous arms from pBHG10 (Microbix, Toronto, CA) flanking the E3-deleted region which has been replaced by a unique PacI site. The kanamycin gene along with the multiple cloning site from pABS.4 (Microbix) was cloned into the unique PacI site, eliminating it in the process, to make the final pShuttlE3. Detailed steps will be provided upon request. pShuttlE3-V-EGFP was constructed by first subcloning the KpnI/SphI V-EGFP cut from pcDNA3-CMV-V-EGFP into pABS.4 also cut with the same enzymes. An SphI/XhoI partial digest was performed on the resulting plasmid to move the V-EGFP gene into the corresponding restriction sites in pShuttlE3. All of the other pShuttlE3 plasmids were made by cutting the respective fusion core genes out of the pShuttle-E1-CMV vectors described above with BglII/HpaI and then inserting it into pShuttlE3 cut with SalI/blunt/BglII. All blunted fragments were generated with the large Klenow fragment (New England Biolabs, Beverly, MA). The plasmids were verified by restriction cutting. All E1 shuttle plasmids were recombined with the pAdEasy rescue plasmid in BJ5183 cells, whereas the E3 shuttle plasmids were recombined with the pTG3602 rescue backbone. Since the pShuttlE3 plasmids contain the kanamycin gene and the fusion core gene flanked by the homologous arms, recombination was performed using double selection for ampicillin from the rescue plasmid pTG3602 and kanamycin from pShuttlE3. Subsequently, kanamycin was cut out of the genomes with the two surrounding SwaI sites. After self-ligation, the final adenoviral genomes were linearized with PacI and then transiently transfected into 911 cells.
Virus propagation and purification
Viruses were propagated in 911 cells and purified by double cesium chloride (CsCl) ultracentrifugation followed by dialysis against phosphate-buffered saline with Mg2+, Ca2+, and 10% glycerol. Final aliquots of virus were analyzed for viral particle titer (absorbance at 260 nm) and transduction unit (t.u.) titer. Based on a previously described protocol (Alemany et al., 2000), t.u. was determined by infecting 15,000 911 cells in 96-well plates with 1:10 serial dilutions of the virus and counting the number of green cells 2 days postinfection (n = 6). In addition, all viruses were quantitated for cytopathic effect unit (CPEU) titer. 911 cells in 96-well plates (15,000 per well) were infected with 1:10 serial dilutions of the virus. Two days postinfection, cell viability was determined with an MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; Promega, Madison, WI). The TCID50 dose was determined and back-calculated to obtain the CPEU titer where theoretically 1 CPEU kills one cell in 2 days. All viruses were stored at −80 °C until use.
Fluorescence microscopy
A549 cells seeded on glass coverslips were transduced with 100 viral particles per cell of Ad-E1-CMV-EGFP, Ad-E1-CMV-Mu-EGFP, Ad-E1-CMV-V-EGFP, Ad-E1-CMV-preVII-EGFP, and Ad-E1-CMV-matVII-EGFP. Twenty-four hours postinfection, the cells were washed with PBS, fixed, stained with Hoechst 33342 (Molecular Probes, Eugene, OR), and mounted on glass slides (Fisher Scientific, Pittsburgh, PA). Epifluorescence microscopy was performed with an inverted IX-70 microscope (Olympus, Melville, NY) equipped with a Magnifire digital CCD camera (Optronics, Goleta, CA). Images were acquired with a 100× objective using oil immersion and digitally deconvoluted with Iris, version 4.15a (http://www.astrosurf.com/buil/) by applying the Richardson-Lucy algorithm with 15 iterations. An image of a single fluorescent AdIX-EGFP (Le et al., 2004) virus particle with strong signal-to-noise ratio was used to determine the point spread function as suggested by the software documentation. EGFP and Hoechst stain images were merged using Adobe Photoshop 7.0 (San Jose, CA).
Western blot
Top and bottom band samples of purified control Ad-E1-CMV-EGFP and Ad-E1-CMV-V-EGFP (8 μg) were resolved with SDS-PAGE and then transferred to a polyvinylidene difluoride membrane (BioRad, Hercules, CA). Blotting was performed with a primary monoclonal GFP antibody (1:1000 dilution, BD Biosciences Clontech) followed by a secondary HRP-linked anti-mouse antibody (1:5000 dilution, Amersham Pharmacia, Piscataway, NJ) or a primary polyclonal pV antibody (1:1000 dilution, Chemicon International, Inc., Temecula, CA) followed by a secondary HRP-linked anti-rabbit antibody (1:5000 dilution, Dako, Carpinteria, CA). Bands were detected with a chemiluminescent ECL kit (Amersham Pharmacia, Piscataway, NJ). For analysis of protein expression from the V-EGFP vectors, 2 × 105 911 cells were infected with 1 CPEU/cell of Ad-E1-CMV-EGFP, Ad-E1-CMV-V-EGFP, AdE1-CMV5-V-EGFP, and Ad-wt-E3-V-EGFP in 12-well plates. Twenty-four hours postinfection, the cells were collected and lysed with RIPA buffer (100 μL). Samples (10 μg) were subjected to SDS-PAGE and blotting with GFP and pV antibodies as described above. Purified Ad-wt-E3-EGFP, Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP (1.5 × 1010 vp of each virus in RIPA buffer) were also subjected to 15% SDS-PAGE for silver staining (Silver Stain Plus, BioRad, Hercules, CA).
Characterization of virus gradient fractions
For the fractionation studies, viruses were propagated each in ten 150-mm dishes of 911 cells. Collected crude viral lysates were purified by CsCl ultracentrifugation where the top and bottom bands were retained through two centrifugation steps yielding one gradient from the 10 dishes. After the second spin, fractions (∼100 μL) were collected dropwise through a perforation at the bottom of the tube into 96-well white opaque plates. Plates with the viral fractions were measured with a microplate fluorometer (Fluostar Optima, BMG Labtechnologies, Durham, NC) using 490/10 nm excitation and 510/10 nm emission filters. To determine viral DNA content, a sample of each fraction (10 μL) was diluted in 90 μL 0.5% SDS/PBS and incubated at room temperature for 10 min to release the viral genomes. Absorbance at 260 nm was then measured for each sample (MBA 2000, Perkin Elmer, Shelton, CT).
Visualization of purified core-labeled viruses
Diluted, purified Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP viral particles (1:100 in PBS) were prepared on glass slides with coverslips and then sealed for fluorescence imaging as described above.
Tracking assay
HeLa cells (1 × 105 cells per dish) were seeded in 50/30 mm glass bottom dishes (Willco Wells B.V., Amsterdam, The Netherlands). The next day, the cells were stained for DNA with Hoechst 33342 (Molecular Probes, Eugene, OR) in normal growth medium and then washed three times with PBS. The cells were then infected with Ad-wt-E3-EGFP, Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP (100,000 viral particles per cell) in 300 μL of normal growth medium at 37 °C and under humidified conditions. Three dishes were prepared for each virus. After 1 h of infection, the dishes were washed with PBS three times to remove free virus. One of three dishes for each virus was imaged under fluorescence microscopy (described above) with PBS in the dish and noted as time point 1 h postinfection. The remaining two dishes for each virus were reincubated with growth medium (700 μL) at 37 °C. At 3 h after infection, a second dish for each virus was washed with PBS three times and then likewise imaged with the cells in PBS. The last dish was imaged as time point 6 h postinfection. Note that the timing of infection started with addition of virus to the cells. Acquired images were digitally deconvoluted as described above.
DNA packaging analysis
DNA packaging was analyzed by using a previously reported protocol (Yamamoto et al., 2003). Briefly, 911 cells (5 × 104) were infected with Ad-wt-E3-EGFP, Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP with 1 CPEU/cell in 12-well plates. On days 1, 2, 3, and 4 postinfection, the cells were collected. One half of the cell pellet was processed for total viral genome copy number, and the other half was processed for encapsidated viral genome copy number (QIAamp DNA Blood Mini Kit, QIAGEN, Valencia, CA). The viral DNA pools were quantitated with Taqman quantitative real-time PCR using E4-specific primers (LightCycler System, Roche Applied Science, Indianapolis, IN).
Cytopathic effect assay
Five thousand 911 cells were infected with Ad-wt-E3-EGFP, Ad-wt-E3-V-EGFP, and Ad-wt-E3-preVII-EGFP (0.5 CPEU/cell, 0.05 CPEU/cell, and 0.005 CPEU/cell of each vector) in 100 μL of 5% DME medium without phenol red (five replicates for each condition plus five noninfected wells as controls). Cytopathic effect was measured by an MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; Promega, Madison, WI) on days 0, 2, 4, 6, 8, and 10 postinfection. Results were calculated as viability percent of noninfected cells with blank values (medium only) subtracted beforehand.
Thermostability assay
Thermostability was analyzed using a modified version of a previously reported protocol (Dmitriev et al., 2002). Samples of Ad-CMV-EGFP and Ad-IX-EGFP (106 t.u. in 100 μL PBS) were incubated for various time periods at 45 °C (0, 5, 10, 20, and 40 min). Transduction unit infectious titers were then determined for the samples using the abovementioned protocol.
Acknowledgments
This work was supported with grants from the National Institute of Health (RO1CA083821, RO1CA94084, RO1DK063615, RO1CA111569, and P20CA101955) and the Medical Scientist Training Program of the University of Alabama at Birmingham.
References
- Alemany R, Suzuki K, Curiel DT. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000;81(Pt 11):2605–2609. doi: 10.1099/0022-1317-81-11-2605. [DOI] [PubMed] [Google Scholar]
- Alestrom P, Akusjarvi G, Lager M, Yeh-kai L, Pettersson U. Genes encoding the core proteins of adenovirus type 2. J. Biol. Chem. 1984;259(22):13980–13985. [PubMed] [Google Scholar]
- Anderson CW, Young ME, Flint SJ. Characterization of the adenovirus 2 virion protein, mu. Virology. 1989;172(2):506–512. doi: 10.1016/0042-6822(89)90193-1. [DOI] [PubMed] [Google Scholar]
- Berk AJ. Adenovirus promoters and E1A transactivation. Annu. Rev. Genet. 1986;20:45–79. doi: 10.1146/annurev.ge.20.120186.000401. [DOI] [PubMed] [Google Scholar]
- Berkner KL, Sharp PA. Effect of the tripartite leader on synthesis of a non-viral protein in an adenovirus 5 recombinant. Nucleic Acids Res. 1985;13(3):841–857. doi: 10.1093/nar/13.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Ni S, Li ZY, Shayakhmetov DM, Lieber A. The effect of sequestration by nontarget tissues on anti-tumor efficacy of systemically applied, conditionally replicating adenovirus vectors. Mol. Ther. 2003;8(5):746–755. doi: 10.1016/j.ymthe.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Cai F, Weber JM. Primary structure of the canine adenovirus PVII protein: functional implications. Virology. 1993;193(2):986–988. [PubMed] [Google Scholar]
- Chatterjee PK, Vayda ME, Flint SJ. Interactions among the three adenovirus core proteins. J. Virol. 1985;55(2):379–386. doi: 10.1128/jvi.55.2.379-386.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee PK, Vayda ME, Flint SJ. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J. Mol. Biol. 1986;188(1):23–37. doi: 10.1016/0022-2836(86)90477-8. [DOI] [PubMed] [Google Scholar]
- Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell. 1977;12(1):1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- Dmitriev IP, Kashentseva EA, Curiel DT. Engineering of adenovirus vectors containing heterologous peptide sequences in the C terminus of capsid protein IX. J. Virol. 2002;76(14):6893–6899. doi: 10.1128/JVI.76.14.6893-6899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, Van Der Eb AJ. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum. Gene Ther. 1996;7(2):215–222. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- Glotzer JB, Michou AI, Baker A, Saltik M, Cotten M. Microtubule-independent motility and nuclear targeting of adenoviruses with fluorescently labeled genomes. J. Virol. 2001;75(5):2421–2434. doi: 10.1128/JVI.75.5.2421-2434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75(3):477–486. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- Greber UF, Suomalainen M, Stidwill RP, Boucke K, Ebersold MW, Helenius A. The role of the nuclear pore complex in adenovirus DNA entry. EMBO J. 1997;16(19):5998–6007. doi: 10.1093/emboj/16.19.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins LK, Hermiston T. Gene delivery from the E3 region of replicating human adenovirus: evaluation of the E3B region. Gene Ther. 2001a;8(15):1142–1148. doi: 10.1038/sj.gt.3301509. [DOI] [PubMed] [Google Scholar]
- Hawkins LK, Hermiston TW. Gene delivery from the E3 region of replicating human adenovirus: evaluation of the ADP region. Gene Ther. 2001b;8(15):1132–1141. doi: 10.1038/sj.gt.3301508. [DOI] [PubMed] [Google Scholar]
- Hawkins LK, Johnson L, Bauzon M, Nye JA, Castro D, Kitzes GA, Young MD, Holt JK, Trown P, Hermiston TW. Gene delivery from the E3 region of replicating human adenovirus: evaluation of the 6.7 K/gp19 K region. Gene Ther. 2001;8(15):1123–1131. doi: 10.1038/sj.gt.3301507. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U.S.A. 1998;95(5):2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat. Med. 2000;6(5):482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- Kelkar SA, Pfister KK, Crystal RG, Leopold PL. Cytoplasmic dynein mediates adenovirus binding to microtubules. J. Virol. 2004;78(18):10122–10132. doi: 10.1128/JVI.78.18.10122-10132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn R, Horwitz MS. Adenovirus DNA synthesis in vitro is inhibited by the virus-coded major core protein. Virology. 1986;150(2):342–351. doi: 10.1016/0042-6822(86)90299-0. [DOI] [PubMed] [Google Scholar]
- Le LP, Everts M, Dmitriev IP, Davydova JG, Yamamoto M, Curiel DT. Fluorescently labeled adenovirus with pIX-EGFP for vector detection. Mol. Imaging. 2004;3(2):105–116. doi: 10.1162/15353500200404100. [DOI] [PubMed] [Google Scholar]
- Lee TW, Blair GE, Matthews DA. Adenovirus core protein VII contains distinct sequences that mediate targeting to the nucleus and nucleolus, and colocalization with human chromosomes. J. Gen. Virol. 2003;84(Pt 12):3423–3428. doi: 10.1099/vir.0.19546-0. [DOI] [PubMed] [Google Scholar]
- Lee TW, Lawrence FJ, Dauksaite V, Akusjarvi G, Blair GE, Matthews DA. Precursor of human adenovirus core polypeptide Mu targets the nucleolus and modulates the expression of E2 proteins. J. Gen. Virol. 2004;85(Pt 1):185–196. doi: 10.1099/vir.0.19352-0. [DOI] [PubMed] [Google Scholar]
- Lehmberg E, Traina JA, Chakel JA, Chang RJ, Parkman M, McCaman MT, Murakami PK, Lahidji V, Nelson JW, Hancock WS, Nestaas E, Pungor E., Jr. Reversed-phase high-performance liquid chromatographic assay for the adenovirus type 5 proteome. J. Chromatogr., B, Biomed. Sci. Appl. 1999;732(2):411–423. doi: 10.1016/s0378-4347(99)00316-3. [DOI] [PubMed] [Google Scholar]
- Leopold PL, Ferris B, Grinberg I, Worgall S, Hackett NR, Crystal RG. Fluorescent virions: dynamic tracking of the pathway of adenoviral gene transfer vectors in living cells. Hum. Gene Ther. 1998;9(3):367–378. doi: 10.1089/hum.1998.9.3-367. [DOI] [PubMed] [Google Scholar]
- Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 1999;260(3):712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- Logan J, Shenk T. Adenovirus tripartite leader sequence enhances translation of mRNAs late after infection. Proc. Natl. Acad. Sci. U.S.A. 1984;81(12):3655–3659. doi: 10.1073/pnas.81.12.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DA. Adenovirus protein V induces redistribution of nucleolin and B23 from nucleolus to cytoplasm. J. Virol. 2001;75(2):1031–1038. doi: 10.1128/JVI.75.2.1031-1038.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DA, Russell WC. Adenovirus core protein V is delivered by the invading virus to the nucleus of the infected cell and later in infection is associated with nucleoli. J. Gen. Virol. 1998;79(Pt 7):1671–1675. doi: 10.1099/0022-1317-79-7-1671. [DOI] [PubMed] [Google Scholar]
- Meulenbroek RA, Sargent KL, Lunde J, Jasmin BJ, Parks RJ. Use of adenovirus protein IX (pIX) to display large polypeptides on the virion-generation of fluorescent virus through the incorporation of pIX-GFP. Mol. Ther. 2004;9(4):617–624. doi: 10.1016/j.ymthe.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Mirza MA, Weber J. Structure of adenovirus chromatin. Biochim. Biophys. Acta. 1982;696(1):76–86. doi: 10.1016/0167-4781(82)90012-4. [DOI] [PubMed] [Google Scholar]
- Mittal SK, McDermott MR, Johnson DC, Prevec L, Graham FL. Monitoring foreign gene expression by a human adenovirus-based vector using the firefly luciferase gene as a reporter. Virus Res. 1993;28(1):67–90. doi: 10.1016/0168-1702(93)90090-a. [DOI] [PubMed] [Google Scholar]
- Miyazawa N, Leopold PL, Hackett NR, Ferris B, Worgall S, Falck-Pedersen E, Crystal RG. Fiber swap between adenovirus subgroups B and C alters intracellular trafficking of adenovirus gene transfer vectors. J. Virol. 1999;73(7):6056–6065. doi: 10.1128/jvi.73.7.6056-6065.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Maeda K, Ohtsuki M, Hosokawa K, Natori S. In vitro transcription of a chromatin-like complex of major core protein VII and DNA of adenovirus serotype 2. Biochem. Biophys. Res. Commun. 1986;136(1):86–93. doi: 10.1016/0006-291x(86)90880-6. [DOI] [PubMed] [Google Scholar]
- Nakano MY, Greber UF. Quantitative microscopy of fluorescent adenovirus entry. J. Struct. Biol. 2000;129(1):57–68. doi: 10.1006/jsbi.1999.4201. [DOI] [PubMed] [Google Scholar]
- Russell WC. Update on adenovirus and its vectors. J. Gen. Virol. 2000;81(Pt 11):2573–2604. doi: 10.1099/0022-1317-81-11-2573. [DOI] [PubMed] [Google Scholar]
- Sheay W, Nelson S, Martinez I, Chu TH, Bhatia S, Dornburg R. Downstream insertion of the adenovirus tripartite leader sequence enhances expression in universal eukaryotic vectors. BioTechniques. 1993;15(5):856–862. [PubMed] [Google Scholar]
- Sung MT, Cao TM, Lischwe MA, Coleman RT. Molecular processing of adenovirus proteins. J. Biol. Chem. 1983;258(13):8266–8272. [PubMed] [Google Scholar]
- van Oostrum J, Burnett RM. Molecular composition of the adenovirus type 2 virion. J. Virol. 1985;56(2):439–448. doi: 10.1128/jvi.56.2.439-448.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vayda ME, Flint SJ. Isolation and characterization of adenovirus core nucleoprotein subunits. J. Virol. 1987;61(10):3335–3339. doi: 10.1128/jvi.61.10.3335-3339.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Seggern DJ, Huang S, Fleck SK, Stevenson SC, Nemerow GR. Adenovirus vector pseudotyping in fiber-expressing cell lines: improved transduction of Epstein–Barr virus-transformed B cells. J. Virol. 2000;74(1):354–362. doi: 10.1128/jvi.74.1.354-362.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienhues U, Hosokawa K, Hoveler A, Siegmann B, Doerfler W. A novel method for transfection and expression of reconstituted DNA–protein complexes in eukaryotic cells. DNA. 1987;6(1):81–89. doi: 10.1089/dna.1987.6.81. [DOI] [PubMed] [Google Scholar]
- Xue Y, Johnson JS, Ornelles DA, Lieberman J, Engel DA. Adenovirus protein VII functions throughout early phase and interacts with cellular proteins SET and pp32. J. Virol. 2005;79(4):2474–2483. doi: 10.1128/JVI.79.4.2474-2483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Davydova J, Wang M, Siegal GP, Krasnykh V, Vickers SM, Curiel DT. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology. 2003;125(4):1203–1218. doi: 10.1016/s0016-5085(03)01196-x. [DOI] [PubMed] [Google Scholar]