

Short abstract
We propose that the phenomenon of X-chromosome inactivation in females may constitute a risk factor for loss of T-cell tolerance; specifically that skewed X-chromosome inactivation in the thymus may lead to inadequate thymic deletion. Using a DNA methylation assay, we have examined the X-chromosome inactivation patterns in peripheral blood from normal females (n = 30), female patients with a variety of autoimmune diseases (n = 167). No differences between patients and controls were observed. However, locally skewed X-chromsome inactivation may exist in the thymus, and therefore the underlying hypothesis remains to be disproved.
Keywords: autoimmunity, gender, immune tolerance, X chromosome
Abstract
Introduction:
A reduction in the sex ratio (male : female) is characteristic of most autoimmune disorders. The increased prevalence in females ranges from a modest 2:1 for multiple sclerosis [1], to approximately 10:1 for systemic lupus erythematosus [2]. This tendency toward autoimmunity in females is often ascribed to hormonal differences, because in a number of experimental disease models estrogens exacerbated disease, and androgens can inhibit disease activity [3,4]. However, human studies have failed to demonstrate a clear-cut influence of hormonal environment on disease susceptibility to lupus or other autoimmune disorders. In addition, many childhood forms of autoimmunity, such as juvenile rheumatoid arthritis, exhibit female predominance [5]. Interestingly, juvenile (type 1) diabetes is an exception to this general trend, with a sex ratio close to 1 in most studies [6]. Therefore, it is reasonable to consider alternative explanations for the increased prevalence of autoimmune diseases in human females.
A unifying feature of autoimmune disorders appears to be the loss of immunologic tolerance to self-antigens, and in many of these diseases there is evidence that T-cell tolerance has been broken. The most profound form of T-cell tolerance involves deletion of potentially self-reactive T cells during thymic selection. Thus, lack of exposure to a self-antigen in the thymus may lead to the presence of autoreactive T cells and may increase the risk of autoimmunity. An elegant example of this has recently been reported [7].
The existence of X-chromosome inactivation in females offers a potential mechanism whereby X-linked self-antigens may escape presentation in the thymus or in other peripheral sites that are involved in tolerance induction. Early in female development, one of the two X chromosomes in each cell undergoes an ordered process of inactivation, with subsequent silencing of most genes on the inactive X chromosome [8]. This phenomenon occurs at a very early embryonic stage [9], and thus all females are mosaic and may occasionally exhibit extreme skewing towards one or the other parental X chromosome. In theory, this may result in a situation in which polymorphic self-antigens on one X chromosome may fail to be expressed at sufficiently high levels in a tolerizing compartment, such as the thymus, and yet may be expressed at a considerable frequency in the peripheral soma. Thus, females may be predisposed to a situation in which they can occasionally express X-linked autoantigens in the periphery to which they have been inefficiently tolerized. Stewart [10] has recently speculated that such a mechanism may play a role in the predisposition to systemic lupus.
This hypothesis predicts that females with autoimmunity may be particularly prone to this mechanism of `inadequate tolerization' by virtue of extremely skewed X-chromosome inactivation. We therefore performed a comprehensive analysis of X-chromosome inactivation patterns in populations of females with multiple sclerosis, systemic lupus erythematosus, juvenile rheumatoid arthritis, and type 1 (insulin-dependent) diabetes mellitus, and in female control individuals. The results do not provide support for a major role for skewed X-chromosome inactivation in female predisposition to autoimmunity; however, neither is the underlying hypothesis disproved by the present data.
Materials and method:
DNA was obtained from female patients from the following sources: 45 persons with juvenile diabetes seen at the Virginia Mason Research Center in Seattle, Washington; 58 multiple sclerosis patients seen at the New York Hospital Multiple Sclerosis Center; 46 patients with systemic lupus erythematosus seen at the Hospital for Special Surgery (New York); 18 patients with juvenile rheumatoid arthritis seen at the Children's Hospital Medical Center in Cleveland. In addition, 30 healthy age-matched females were studied as normal controls.
Employing a modification of previously described methods [11], we utilized a fluorescent Hpa II/PCR assay of the androgen receptor (AR) locus to assess X-chromosome inactivation patterns. The AR gene contains a polymorphic CAG repeat, which is flanked by Hpa II sites. These Hpa II sites are methylated on the inactive X chromosome, and are unmethylated on the active X chromosome. By performing PCR amplification across this region after cutting with the methylation-sensitive enzyme Hpa II, the relative amounts of the methylated AR alleles can be quantitatively determined with a high degree of accuracy; variance on repeated assays is approximately 4% [12].
Skewing of X-chromosome inactivation is expressed as percentage deviation from equal (50:50) inactivation of the upper and lower AR alleles. Therefore, the maximal possible deviation is 50%, in which case all of the X chromosomes bearing one of the AR alleles are inactivated.
Results:
We examined X-chromosome inactivation patterns in several different populations. The results are summarized in Fig. 1. A wide range of X-inactivation skewing was observed in all five groups. Approximately 5% (nine out of 197) of individuals exhibited extreme skewing (greater than 40% deviation from a 50:50 distribution). However, there was no difference between the groups, either in the overall mean skewing, or in the fraction of individuals with extreme skewing (>40%).
Although the present study was not initiated in order to examine allelic variation in the AR gene per se, the data provide an opportunity to address this question. Excessively long CAG repeats in the AR are a rare cause of spinal-bulbar muscular atrophy [13], and AR repeat length appears to have an influence on the biology of certain tumors [14,15]. In this context, it has been shown that transcription of AR correlates inversely with repeat length [16]. We therefore compared AR repeat length in control individuals and patients with autoimmunity. No differences were observed for mean repeat length, or for maximum and minimum repeat length, among the five groups.
Discussion:
The reason for the female predominance in most autoimmune diseases remains obscure. The present study was initiated in order to address the hypothesis that a nonhormonal mechanism related to X inactivation might be involved. The hypothesis rests on the idea that skewing of X inactivation might lead to a deficiency of tolerance induction in the thymus, particularly with respect to polymorphic X-linked autoantigens. The hypothesis predicts that skewed X inactivation would be more prevalent in females with autoimmune diseases than in female control individuals. This was not observed.
Nevertheless, these negative data do not rule out a role for X inactivation in female predisposition to loss of tolerance. A general model for how this mechanism might operate is shown in Fig. 2. Thymocytes undergo selection in the thymic parenchyma and, in the case of negative selection, the selecting elements appear to be derived from the bone marrow and consist mainly of thymic dendritic cells. If the thymic dendritic cell population exhibits random X inactivation, it is highly likely that differentiating thymocytes will contact dendritic cells that express self-antigens on both X chromosomes. This situation is outlined schematically on the left side of Fig. 2. However, if there is extremely skewed X inactivation in the thymic dendritic cell population, a particular thymocyte might not come into contact with dendritic cells that express one of the two X chormosomes. This would lead to a situation where T cells may undergo thymic maturation without having been negatively selected for antigens that are expressed on the predominantly inactive X chromosome. This situation is shown on the right side of Fig. 2.
In order for this mechanism to be physiologically relevant, some assumptions must be made. First, defective tolerance from skewed X inactivation should only be directed at X-linked antigens that are polymorphic, and for which the individual is heterozygous. Thus, this mechanism would not be expected to lead to lack of tolerance commonly, unless there are at least several highly polymorphic X-linked autoantigens in the population that are involved in thymic deletion events. Second, if this actually leads to autoimmunity, it also predicts that the initial break in tolerance that leads to disease should involve an X-linked autoantigen that is expressed in a peripheral nontolerizing site or circumstance.
A recent report [7] has elegantly demonstrated the importance of thymic deletion events in predisposition to autoimmune disease. The proteolipid protein (PLP) autoantigen is expressed in alternatively spliced forms, which exhibit tissue specific expression. A nonspliced variant is expressed in peripheral neural tissue. However, in the thymus a splice variant results in the lack of thymic expression of an immunodominant peptide. This results in loss of tolerace of T cells to this peptide, presumably on the basis of lack of thymic deletion of thymocytes that are reactive with this antigen. Interestingly, PLP is encoded on the X chromsome. However, there is no evidence that genetic polymorphisms control the level splicing of PLP within the thymus. Nevertheless, these data illustrate the potential importance of deficiencies in thymic deletion for autoimmune T-cell reactivity.
The present results suggest that if skewed X inactivation is relevant to thymic tolerance induction, then the effect does not depend on global skewing of X-chromosome inactivation, at least in the hematopoietic compartment. In this study we examined X-inactivation patterns in peripheral blood mononuclear cells, and the results should reflect the state of X inactivation in all mesenchymal tissues, including dendritic cells. X inactivation occurs at a very early time point in development, and thus the results in one tissue should reflect the general situation in the rest of the body. However, there may be exceptions to this. We have occasionally observed differences in X-inactivation patterns between buccal mucosa (an ectodermally derived tissue) and peripheral blood in the same individiual (unpublished observations). This could be a chance event, or it may result from selection for certain X-linked alleles during embryonic development, as has been described in carriers of X-linked immunodeficiencies [17].
Another consideration is that certain tissue microenvironments may be derived from very small numbers of founder cells, and thus may exhibit skewed utilization of one or the other X chromosome, even if the tissue as a whole is not skewed. This situation could vary over time. Thus, there may be time points at which certain thymic microenvironments are populated by dendritic cells that, for stochastic reasons, all utilize the same X chromosome. This would create a `window of opportunity' in which a given thymocyte, in a given selecting location, could escape negative selection by antigens on the inactive X chromosome. The likelihood of this happening would obviously depend on the number of dendritic cells that are usually contacted by a thymocyte during thymic selection. There is limited information on this point, although Stewart [10] has theorized that this number may be as low as 15. If this is the case, then escape from thymic deletion may still occur in females who are heterozygous for a relevant X-linked antigen, even if the hematopoietic cells in general do not exhibit extreme skewing.
In conclusion, we suggest that X-chromosome inactivation needs to be considered as a potential factor in the predominance of females in most autoimmune diseases. Our inability to show an increase in X-chromosome skewing in females with autoimmunity does not eliminate this as an etiologic contributor to loss of immunologic tolerance. Future experiments must be directed at a detailed analysis of tissue patterns of X inactivation, as well as at a search for potential X-linked autoantigens.
Introduction
A reduction in the sex ratio (male : female) is characteristic of most autoimmune disorders. The increased prevalence in females ranges from a modest 2:1 for multiple sclerosis [1], to approximately 10:1 for systemic lupus erythematosus [2]. This tendency towards autoimmunity in females is often ascribed to hormonal differences because, in a number of experimental disease models, estrogens exacerbate disease and androgens can inhibit disease activity [3,4]. However, human studies have failed to demonstrate a clear-cut influence of hormonal environment on disease susceptibility to lupus or other autoimmune disorders. In addition, many childhood forms of autoimmunity, such as juvenile rheumatoid arthritis, exhibit female predominance [5]. Interestingly, juvenile (type 1) diabetes is an exception to this general trend, with a sex ratio close to 1 in most studies [6]. Therefore, it is reasonable to consider alternative explanations for the increased prevalence of autoimmune diseases in human females.
A unifying feature of autoimmune disorders appears to be the loss of immunologic tolerance to self-antigens, and in many of these diseases there is evidence that T-cell tolerance has been broken. The most profound form of T-cell tolerance involves deletion of potentially self-reactive T cells during thymic selection. Thus, lack of exposure to a self-antigen in the thymus may lead to the presence of autoreactive T cells and increase the risk of autoimmunity. An elegant example of this has recently been reported [7].
The existence of X-chromosome inactivation in females offers a potential mechanism whereby X-linked self-antigens may escape presentation in the thymus or in other peripheral sites that are involved in tolerance induction. Early in female development, one of the two X chromosomes in each cell undergoes an ordered process of inactivation, with subsequent silencing of most genes on the inactive X chromosome [8]. This phenomenon occurs at a very early embryonic stage [9], and thus all females are mosaic and may occasionally exhibit extreme skewing towards one or the other parental X chromosome. In theory, this may result in a situation in which polymorphic self-antigens on one X chromosome may fail to be expressed at sufficiently high levels in a tolerizing compartment, such as the thymus, and yet may be expressed at considerable frequency in the peripheral soma. Thus, females may be predisposed to a situation in which they can occasionally express X-linked autoantigens in the periphery to which they have been inefficiently tolerized. Stewart [10] has recently speculated that such a mechanism may play a role in the predisposition to systemic lupus.
This hypothesis predicts that females with autoimmunity may be particularly prone to this mechanism of `inadequate tolerization' by virtue of extremely skewed X-chromosome inactivation. We therefore performed a comprehensive analysis of X-chromosome inactivation patterns in populations of females with multiple sclerosis, systemic lupus, juvenile rheumatoid arthritis, and type 1 (insulin-dependent) diabetes mellitus, and in female control individuals. The results do not provide support for a major role for skewed X-chromosome inactivation in female predisposition to autoimmunity; however, neither is the underlying hypothesis disproved by our data.
Materials and method
Subjects
DNA was obtained from female patients from the following sources: 45 persons with juvenile diabetes seen at the Virginia Mason Research Center in Seattle, Washington; 58 multiple sclerosis patients seen at the New York Hospital Multiple Sclerosis Center; 46 patients with systemic lupus erythematosus seen at the Hospital for Special Surgery (New York); 18 patients with juvenile rheumatoid arthritis seen at the Children's Hospital Medical Center in Cleveland. In addition, 30 healthy age-matched females were studied as normal controls.
Hpa II/polymerase chain reaction assay for X-chromosome inactivation
A modification to a previously described assay for AR methylation [11] was used for these studies. In the normal females the AR gene is methylated on the inactive X chromosome and is undermethylated on the active X chromosome. Furthermore, the presence of a highly polymorphic triplet repeat within the AR gene allows for the discrimination of each X chromosome in most female subjects (Fig. 3). Thus, allele-specific methylation patterns can be distinguished using this gene.
Figure 3.
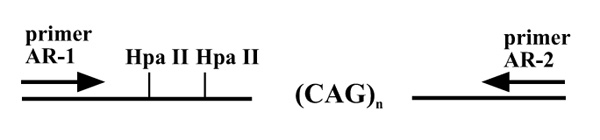
Schematic map of a portion of the first exon of the AR gene. The polymorphic CAG triplet repeat is flanked by HpaII sites, and the relative locations of primers AR1 and AR2, used for the PCR assay, are indicated.
Genomic DNA (approximately 200 ng) was digested overnight using a methylation-sensitive restriction enzyme HpaII, as per the manufacturer's instructions (Gibco BRL, Rockville, MD, USA). A mock sample as control was prepared simultaneously without the HpaII enzyme. After the digestion the samples were boiled for 10 min to inactivate the enzyme. PCR amplification was performed, with final concentration of the digest equivalent to approximately 100 ng of the DNA, 1 × PCR bufferII (Perkin-Elmer, Foster City, CA, USA), 2.5 mmol/l MgCl2, and amplitaq gold 1.25 units.
Primers used were AR1 (5'-TCCAGAATCTGTTCCA-GAGCGTGC-3') and AR2 (5'-GCTGTGAAGGTTGCT-GTTCCTCAT-3'). These primers flank the triple repeat CAG and the HpaII sites in the first exon of the AR gene (Fig. 3). The AR1 primer was fluorescence labeled with 6-Fam and TET. The 6-Fam-AR1 and the TET-AR1 were used for mock and HpaII-digested samples, respectively. This combination allowed simultaneous analyses of the PCR products of the HpaII- and mock-treated samples using the ABI310 Genetic Analyzer (Perkin-Elmer). The final concentration of the AR1 primer was 3 pmol 6-Fam-AR1+7 pmol unlabelled AR1 and 10 pmol of unlabeled AR2 in a 50 μl reaction.
The cycling conditions were as follows: denaturation at 95°C/12 min to activate the amplitaq gold, followed by 35 cycles of denaturation at 95°C/45 s, annealing at 60°C/30 s and extension at 72°C/30 s. After 35 cycles, the final extension was done at 72°C/10 min. The PCR products of each sample (mock and HpaII) were diluted 1:10 in the same vial. One microliter of the diluted product was then added to a mixture containing 12 μl deionized formamide and 0.5 μl molecular weight standard GS 350 TAMRA.The vials were denatured at 95°C/5 min, cooled on ice, and resolved using a 310 Genetic Analyzer (Perkin-Elmer).
Calculations for percentage of skewing of X inactivation
Two peaks corresponding to two alleles were obtained from the 310 Genetic Analyzer for each mock- and HpaII-treated sample. The relative intensity of the larger AR allele (higher molecular weight, peak 2) with respect to the smaller AR allele (low molecular weight, peak 1) was calculated and expressed as the ratio R (peak 2 area/peak 1 area). The ratios in the mock-digested (RM) and HpaII-digested (RH) samples were calculated separately, and were then averaged for each of the two sets of duplicate samples. For each individual, a normalized ratio (RN = RH/RM) was calculated to correct for occasional minor variation in efficiency of amplification of the two AR alleles. This normalized ratio was used to determine the percentage of inactivation of the X chromosome bearing the larger AR allele: percentage inactivation = [RN/(RN + 1)] × 100. The degree of skewing was calculated by subtracting 50 from the observed degree of inactivation.
Results
Fluorescent Hpa II/polymerase chain reaction assay for X-chromosome inactivation
Employing a modification of previously described methods [11], we utilized a fluorescent Hpa II/PCR assay of the AR locus to assess X-chromosome inactivation patterns. As shown in Fig. 3, the AR gene contains a polymorphic CAG repeat that is flanked by Hpa II sites. These Hpa II sites are methylated on the inactive X chromosome, and are unmethylated on the active X chromosome. By performing PCR amplification across this region after cutting with the methylation-sensitive enzyme Hpa II, the relative amounts of the methylated AR alleles can be quantitatively determined with a high degree of accuracy; variance on repeated assays is approximately 4% [12].
Two examples of this assay are shown in Fig. 4. In Fig. 4a, an individual with equivalent methylation of both AR alleles is shown. In this case, the relative peak intensity is equivalent in the mock-digested and Hpa II-digested samples. In Fig. 4b, an individual with extremely skewed X inactivation is shown, in whom the lower allele (271 bp) is relatively over-methylated compared with the upper (284 bp) allele. Thus, the X chromosome bearing the lower allele is preferentially inactivated. As described in the Materials and method section, all assays were normalized to the mock results and performed in duplicate. Skewing of X-chromosome inactivation is expressed as a percentage deviation from equal (50:50) inactivation of the upper and lower AR alleles. Therefore, the maximal possible deviation is 50%, in which case all of the X chromosome bearing one of the AR alleles are inactivated.
Figure 4.
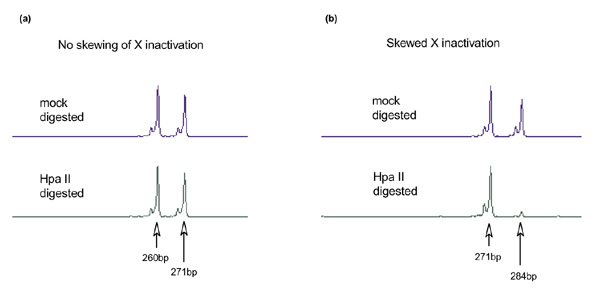
Fluorescence-based X-inactivation using PCR-based AR methylation assay. (a) An example of equivalent inactivation (51% versus 49%) of both upper (271 bp) and lower (260 bp) alleles. (b) An example of extremely skewed inactivation (10% versus 90%) of the upper (284 bp) and lower (271 bp) alleles. Mock-digested DNA (without HpaII) was used as control for peak intensities of both alleles.
Comparison of X-chromosome inactivation patterns in normal females and females with autoimmune diseases
We examined X-chromosome inactivation patterns in several different populations. The results are summarized in Fig. 1. A wide range of X-inactivation skewing was observed in all five groups. Approximately 5% (nine out of 197) of individuals exhibited extreme skewing (greater than 40% deviation from a 50:50 distribution). However, there was no difference between the groups, either in the overall mean skewing, or in the fraction of individuals with extreme skewing (>40%).
Figure 1.
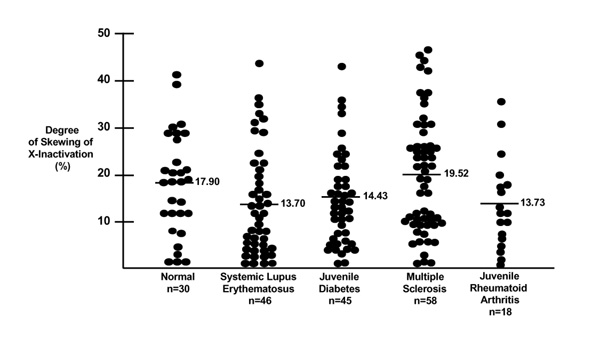
Comparison of skewing of X inactivation in normal female control individuals, and patients with various autoimmune diseases. Each dot represents one individual. The mean values for degree of skewing of X-inactivation are indicated for each population.
Androgen receptor gene allele size comparisons among patients and controls
Although the present study was not initiated to examine allelic variation in the AR gene per se, the data provide an opportunity to address this question. Excessively long CAG repeats in the AR gene are a rare cause of spinal-bulbar muscular atrophy [13], and AR repeat length appears to have an influence on the biology of certain tumors [14,15]. In this context, it has been shown [16] that transcription of the AR gene correlates inversely with repeat length.We therefore compared AR repeat length in control individuals and patients with autoimmunity. No differences were observed for mean repeat length, or for maximum and minimum repeat length, among the five groups.
Discussion
The reason for the female predominance in most autoimmune diseases remains obscure. The present study was initiated in order to address the hypothesis that a nonhormonal mechanism related to X inactivation might be involved. The hypothesis rests on the idea that skewing of X inactivation might lead to a deficiency of tolerance induction in the thymus, particularly with respect to polymorphic X-linked autoantigens. The hypothesis predicts that skewed X inactivation would be more prevalent in females with autoimmune diseases than in female control individuals. This was not observed.
Nevertheless, these negative data do not rule out a role for X inactivation in female predisposition to loss of tolerance. A general model for how this mechanism might operate is shown in Fig. 1. Thymocytes undergo selection in the thymic parenchyma, and in the case of negative selection, the selecting elements appear to be derived from the bone marrow and consist mainly of thymic dendritic cells. If the thymic dendritic cell population exhibits random X inactivation, it is highly likely that differentiating thymocytes will contact dendritic cells that express self-antigens on both X chromosomes. This situation is outlined schematically on the left side of Fig. 2. However, if there is extremely skewed X inactivation in the thymic dendritic cell population, a particular thymocyte may not come into contact with dendritic cells that express one of the two X chromosomes. This would lead to a situation where T cells may undergo thymic maturation without having been negatively selected for antigens that are expressed on the predominantly inactive X chromosome. This situation is shown on the right side of Fig. 2.
Figure 2.
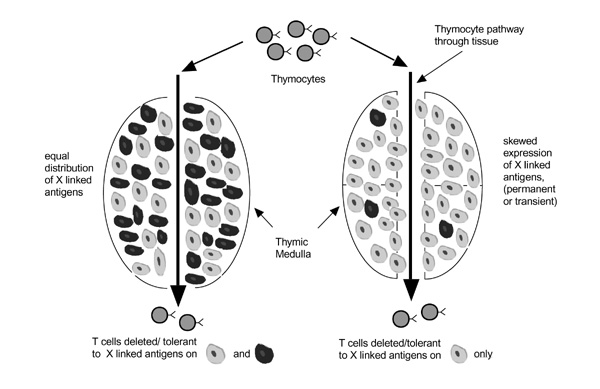
A model showing the consequence of skewed X-chromosome inactivation on tolerance induction in the thymus. Thymocytes enter the thymus and proceed on a pathway that exposes them to thymic-deleting elements, mainly dendritic cells in the thymic medulla. Dendritic cells are schematically illustrated here as grey or black, depending on which of the two parental X chromosomes are predominantly active in each cell. On the left hand side of the figure, transiting thymocytes will be exposed to dendritic cells that express both parental X chromosome, and thus will be tolerant (ie deleted) to X-linked antigens from both parents. On the right hand side of the figure, transiting thymocytes will likely be exposed only to the `grey' dendritic cells, which express only one of the two parental X chromosomes. Thus, T cells that exit the thymus will not be tolerized to X-linked antigens from the other parent (the `black' dendritic cells).
In order for this mechanism to be physiologically relevant, several assumptions must be made. First, defective tolerance from skewed X inactivation should only be directed at X-linked antigens that are polymorphic, and for which the individual is heterozygous. Thus, this mechanism would not be expected to commonly lead to lack of tolerance unless there are at least several highly polymorphic X-linked autoantigens in the population that are involved in thymic deletion events. Second, if this actually leads to autoimmunity, it also predicts that the initial break in tolerance that leads to disease should involve an X-linked autoantigen that is expressed in a peripheral nontolerizing site or circumstance.
A recent report [7] elegantly demonstrated the importance of thymic deletion events in predisposing to autoimmune disease. The PLP autoantigen is expressed in alternatively spliced forms, which exhibit tissue-specific expression. A nonspliced variant is expressed in periperal neural tissue. However, in the thymus a splice variant results in the lack of thymic expression of an immunodominant peptide. This results in loss of tolerace of T cells to this peptide, presumably on the basis of lack of thymic deletion of thymocytes that are reactive with this antigen. Interestingly, PLP is encoded on the X chromsome. However, there is no evidence that genetic polymorphisms control the level splicing of PLP within the thymus. Nevertheless, these data illustrate the potential importance of deficiencies in thymic deletion for autoimmune T-cell reactivity.
The present results suggest that if skewed X inactivation is relevant to induction of thymic tolerance, then the effect does not depend on global skewing of X-chromosome inactivation, at least in the hematopoietic compartment. In this study we examined X-inactivation patterns in peripheral blood mononuclear cells, and the results should reflect the state of X inactivation in all mesenchymal tissues, including dendritic cells. X inactivation occurs at a very early time point in development, and thus the results in one tissue should reflect the general situation in the rest of the body. However, there may be exceptions to this. We have occasionally observed differences in X inactivation patterns between buccal mucosa (an ectodermally derived tissue) and peripheral blood (mesoderm) in the same individual (unpublished observations). This could be a chance event, or may result from selection for certain X-linked alleles during embryonic development, as has been described in carriers of X-linked immunodeficiencies [17]. However, in general, subsets of cells within the hematopoietic compartment do not display differences in X-inactivation patterns (unpublished observations).
Another consideration is that certain tissue microenvironments may be derived from very small numbers of founder cells, and thus may exhibit skewed utilization of one or the other X chromosome, even if the tissue as a whole is not skewed. This situation could vary over time. Thus, there may be time points at which certain thymic microenvironments are populated by dendritic cells that, for stochastic reasons, all utilize the same X chromosome. This would create a `window of opportunity' in which a given thymocyte, in a given selecting location, could escape negative selection by antigens on the inactive X chromosome. The likelihood of this happening would obviously depend on the number of dendritic cells that are usually contacted by a thymocyte during thymic selection. There is limited information on this point, although Stewart [10] has theorized that this number may be as low as 15. If this is the case, escape from thymic deletion may still occur in females who are heterozygous for a relevant X-linked antigen, even if the hematopoietic cells in general do not exhibit extreme skewing.
If the latter scenario is operative, it will be extremely difficult to document it by studying X-inactivation patterns. One might examine X-inactivation skewing specifically in thymic dendritic populations, but if the effect is at the level of microenvironment and varies over time, then it will not be possible to detect this using methods directed at large cell populations, as we have done here. Conceivably, examination of thymic tissue sections could provide support for the hypothesis. Another aspect of this is that some thymic deletion events appear to be mediated by thymic epithelial cells at the cortico-medullary junction. Interestingly, thymic epithelial cells appear to be derived from very few founder cells [18], and thus should exhibit a rather large degree of `patchiness' with respect to X inactivation. This again might lead to local epithelial cell microenvironments that fail to delete for X-linked autoantigens.
It has recently become apparent that mechanisms of peripheral tolerance also exist, and we have considered the possibility that skewed X inactivation in a peripheral tolerizing compartment might also lead to inefficient tolerance. A major mechanism of peripheral tolerance induction appears to involve the recognition of tolerizing antigens by T cells in the absence of costimulation [19]. This may specifically occur in the paracortical regions of lymph nodes, without further progression of the tolerized T cells into lymph node follicles. Because this mechanism presumably involves circulating antigen, it is difficult to invoke a role for X-inactivation skewing in altering this process, unless the skewing were virtually complete. Tolerance induction by parenchymal tissue has also been described [20], and may be a multistage process that is still not entirely understood. Conceivably, skewed expression of X-linked autoantigens could play a role here, but this would require invoking a peripheral tolerizing compartment that is limiting with respect to the dosage of tolerizing cells to which peripheral T cells can be exposed. No such compartment has yet been defined.
In conclusion, we suggest that X-chromosome inactivation needs to be considered as a potential factor in the predominance of females in most autoimmune diseases. Our inability to show an increase in X-chromosome skewing in females with autoimmunity does not eliminate this as an etiologic contributor to loss of immunologic tolerance. Future experiments must be directed at detailed analysis of tissue patterns of X inactivation, as well as at a search for potential X-linked autoantigens.
References
- Compston A. Genetic susceptibility to multiple sclerosis. McAlpine's Multiple Sclerosis. Edited by Compston A, Ebers G, Lassman H, McDonald I, Matthews B, Wekerle H. London: Churchill Livingstone, 1998. pp. 101–142.
- McCarthy DJ, Manzi S, Medsger TA, Jr, Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus ertythematosus. Arthritis Rheum. 1995;38:1260–1270. doi: 10.1002/art.1780380914. [DOI] [PubMed] [Google Scholar]
- Cooper GS, Dooley MA, Treadwell EL, St Clair EW, Parks CG, Gilkeson GS. Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus. Arthritis Rheum. 1998;41:1714–1724. doi: 10.1002/1529-0131(199810)41:10<1714::AID-ART3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Cutolo N, Masi AT. Do androgens influence the pathophysiology of rheumatoid arthritis? Facts and hypotheses. J Rheumatol. 1998;25:1041–1047. [PubMed] [Google Scholar]
- Kerckhove CV, Balakrishnan K, Levinson JE, Larson MG, Glass DN. HLA and altered sex ratios in juvenile rheumatoid arthritis sibships. Hum Immunol. 1988;22:227–233. doi: 10.1016/0198-8859(88)90002-x. [DOI] [PubMed] [Google Scholar]
- Cucca F, Goy JV, Kawaguchi Y, Esposito L, Merriman ME, Wilson AJ, Cordell HJ, Bain SC, Todd JA. A male-female bias in type 1 diabetes and linkage to chromosome Xp in MHC HLA-DR3-positive patients. Nature Genet. 1998;19:301–302. doi: 10.1038/995. [DOI] [PubMed] [Google Scholar]
- Klein L, Klugmann M, Nave K-A, Tuohy V K, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nature Med. 2000;6:56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- Gartler SM, Riggs AD. Mammalian X-chromosome inactivation. Ann Rev Genet. 1983;17:155–190. doi: 10.1146/annurev.ge.17.120183.001103. [DOI] [PubMed] [Google Scholar]
- Puck JM, Stewart CC, Nussbaum RL. Maximum likelihood analysis of human T-cell X-chromosome inactivation patterns: normal women versus carriers of X-linked severe combined immunodeficiency. Am J Hum Genet. 1992;50:742–748. [PMC free article] [PubMed] [Google Scholar]
- Stewart J. The female X-inactivatrion mosaic in systemic lupus erythematosus. Immunol Today. 1998;19:352–357. doi: 10.1016/s0167-5699(98)01298-5. [DOI] [PubMed] [Google Scholar]
- Allen C, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- Monteiro J, Derom C, Vlietinck R, Kohn N, Lesser M, Gregersen PK. Commitment to X-inactivation precedes the twinning event in monochorionic monozygotic (MC-MZ) twins. Am J Hum Genet. 1998;63:339–346. doi: 10.1086/301978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Rebbeck TR, Kantoff PW, Krithivas K, Neuhausen S, Blackwood MA, Godwin AK, Daly MB, Narod SA, Garber JE, Lynch HT, Weber BL, Brown M. Modification of BRCA1-associated breast cancer risk by the polymorphic androgen-receptor CAG repeat. Am J Hum Genet. 1999;64:1371–1377. doi: 10.1086/302366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens CH, Kantoff PW. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci USA. 1997;94:3320–3323. doi: 10.1073/pnas.94.7.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain NL, Driver ED, Miesfeld RL. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994;22:3181–3186. doi: 10.1093/nar/22.15.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puck J, Krause C, Pucj S, Buckley R, Conley M. Prenatal test for X-linked severe combined immunodeficiency by analysis of maternal X-chromosome inactivation and linkage analysis. N Engl J Med. 1990;322:1063–1066. doi: 10.1056/NEJM199004123221508. [DOI] [PubMed] [Google Scholar]
- Wilcox N, Schluep M, Ritter MA. Myasthenic and nonmyasthenic thymoma. An expansion of a minor cortical epithelial cell subset? Am J Pathol. 1987;127:447–460. [PMC free article] [PubMed] [Google Scholar]
- Mondino A, Khoruts A, Jenkins MK. The anatomy of T-cell activation and tolerance. Proc Natl Acad Sci USA. 1996;93:2245–2252. doi: 10.1073/pnas.93.6.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold B, Schonrich G, Hammerling GJ. Multiple levels of peripheral tolerance. Immunol Today. 1993;14:12–14. doi: 10.1016/0167-5699(93)90317-E. [DOI] [PubMed] [Google Scholar]