


Short abstract
This study describes the upregulation of neurokinin 1 and bradykinin 2 receptors in dorsal root ganglion (DRG) neurons in the course of antigen-induced arthritis (AIA) in the rat knee. In the acute phase of AIA, which was characterized by pronounced hyperalgesia, there was a substantial bilateral increase in the proportion of lumbar DRG neurons that express neurokinin 1 receptors (activated by substance P) and bradykinin 2 receptors. In the chronic phase the upregulation of bradykinin 2 receptors persisted on the side of inflammation. The increase in the receptor expression is relevant for the generation of acute and chronic inflammatory pain.
Keywords: antigen-induced arthritis, bradykinin receptor, dorsal root ganglion neurons, neurokinin 1 receptor, pain
Abstract
Introduction:
Ongoing pain and hyperalgesia (enhanced pain response to stimulation of the tissue) are major symptoms of arthritis. Arthritic pain results from the activation and sensitization of primary afferent nociceptive nerve fibres ('pain fibres') supplying the tissue (peripheral sensitization) and from the activation and sensitization of nociceptive neurons in the central nervous system (central sensitization). After sensitization, nociceptive neurons respond more strongly to mechanical and thermal stimulation of the tissue, and their activation threshold is lowered. The activation and sensitization of primary afferent fibres results from the action of inflammatory mediators such as bradykinin (BK), prostaglandins and others on membrane receptors located on these neurons. BK is a potent pain-producing substance that is contained in inflammatory exudates. Up to 50% of the primary afferent nerve fibres have receptors for BK. When primary afferent nerve fibres are activated they can release neuropeptides such as substance P (SP) and calcitonin gene-related peptide from their sensory endings in the tissue. SP contributes to the inflammatory changes in the innervated tissue (neurogenic inflammation), and it might also support the sensitization of nociceptive nerve fibres by binding to neurokinin 1 (NK1) receptors. NK1 receptors are normally expressed on a small proportion of the primary afferent nerve fibres.
Aims:
Because the expression of receptors on the primary afferent neurons is essential for the pain-producing action of inflammatory mediators and neuropeptides, we investigated in the present study whether the expression of BK and NK1 receptors on primary afferent neurons is altered during the acute and chronic phases of an antigen-induced arthritis (AIA). AIA resembles in many aspects the inflammatory process of human rheumatoid arthritis. Because peptide receptors are expressed not only in the terminals of the primary afferent units but also in the cell bodies, we removed dorsal root ganglia (DRGs) of both sides from control rats and from rats with the acute or chronic phase of AIA and determined, after short-term culture of the neurons, the proportion of DRG neurons that expressed the receptors in the different phases of AIA. We also characterized the inflammatory process and the nociceptive behaviour of the rats in the course of AIA.
Materials and methods:
In 33 female Lewis rats 10 weeks old, AIA was induced in the right knee joint. First the rats were immunized in two steps with methylated bovine serum albumin (m-BSA) emulsified with Freund's complete adjuvant, and heat-inactivated Bordetella pertussis. After immunization, m-BSA was injected into the right knee joint cavity to induce arthritis. The joint swelling was measured at regular intervals. Nociceptive (pain) responses to mechanical stimulation of the injected and the contralateral knee were monitored in the course of AIA. Groups of rats were killed at different time points after the induction of AIA, and inflammation and destruction in the knee joint were graded by histological examination. The DRGs of both sides were dissected from segments L1–L5 and C1–C7 from arthritic rats, from eight immunized rats without arthritis and from ten normal control rats. Excised DRGs were dissociated into single cells which were cultured for 18 h.
The expression of the receptors was determined by assessment of the binding of SP-gold or BK-gold to the cultured neurons. For this purpose the cells were slightly fixed. Binding of SP-gold or BK-gold was detected by using enhancement with silver and subsequent densitometric analysis of the relative grey values of the neurons. Displacement controls were performed with SP, the specific NK1 receptor agonist [Sar9, Met(O2)11]-SP, BK, the specific BK 1 (B1) receptor agonist D-Arg (Hyp3-Thi5,8-D-Phe7)-BK and the specific BK 2 (B2) receptor agonist (Des-Arg10)-Lys-BK.
Results:
The inflammatory process in the injected right knee joint started on the first day after induction of AIA and persisted throughout the observation period of 84 days (Fig. 1). The initial phase of AIA was characterized by strong joint swelling and a predominantly granulocytic infiltration of the synovial membrane and the joint cavity (acute inflammatory changes). In the later phases of AIA (10–84 days after induction of AIA) the joint showed persistent swelling, and signs of chronic arthritic alterations such as infiltration of mononuclear leucocytes, hyperplasia of synovial lining layer (pannus formation) and erosions of cartilage and bone were predominant. The contralateral knee joints appeared normal at all time points. Destruction was observed only in the injected knee but some proteoglycan loss was also noted in the non-injected, contralateral knee. In the acute and initial chronic phases of AIA (1–29 days) the rats showed mechanical hyperalgesia in the inflamed knee (limping, withdrawal response to gentle pressure onto the knee). In the acute phase (up to 9 days) a pain response was also seen when gentle pressure was applied to the contralateral knee.
Figure 2 displays the changes in the receptor expression in the DRG neurons during AIA. The expression of SP–gold-binding sites in lumbar DRG neurons (Fig. 2a) was substantially increased in the acute phase of arthritis. In untreated control rats (n = 5), 7.7 ± 3.8% of the DRG neurons from the right side and 10.0 ± 1.7% of the DRG neurons from the left side showed labelling with SP–gold. The proportion of SP–gold-labelled neurons in immunized animals without knee injection (n = 3) was similar. By contrast, at days 1 (n = 2 rats) and 3 (n = 5 rats) of AIA in the right knee, approximately 50% of the DRG neurons exhibited labelling with SP–gold, and this was seen both on the side of the injected knee and on the opposite side. At day 10 of AIA (n = 3 rats), 26.3 ± 6.1% of the ipsilateral DRG neurons but only 15.7 ± 0.6% of the contralateral neurons exhibited binding of SP–gold. At days 21 (n = 5 rats), 42 (n = 3 rats) and 84 (n = 5 rats) of AIA, the proportion of SP–gold-positive neurons had returned to the control values, although the arthritis, now with signs of chronic inflammation, was still present. Compared with the DRG neurons of the untreated control rats, the increase in the proportion of labelled neurons was significant on both sides in the acute phase (days 1 and 3) and the intermediate phase (day 10) of AIA (Mann–Whitney U-test). The size distribution of the neurons was similar in the DRG neurons of all experimental groups. Under all conditions and at all time points, SP–gold binding was found mainly in small and medium-sized (less than 700 μm2) neurons. In the cervical DRGs the expression of NK1 receptors did not change in the course of AIA. The binding of SP–gold to the neurons was suppressed by the coadministration of the specific NK1 receptor agonist [Sar9, Met(O2)11]–SP in three experiments, showing that SP–gold was bound to NK1 receptors.
The expression of BK–gold-binding sites in the lumbar DRG neurons showed also changes in the course of AIA, but the pattern was different (Fig. 2b). In untreated control rats (n = 5), 42.3 ± 3.1% of the DRG neurons of the right side and 39.6 ± 2.6% of the DRG neurons of the left side showed binding of BK–gold. At days 1 (n = 2 rats) and 3 (n = 5 rats) of AIA, approximately 80% of the DRG neurons on the side of the knee injection (ipsilateral) and approximately 70% on the opposite side were labelled. In comparison with the untreated control group, the increase in the proportion of labelled neurons was significant on both sides. The proportion of labelled neurons in the ipsilateral DRGs remained significantly increased in both the intermediate phase (day 10, n = 3 rats) and chronic phase (days 21, n = 5 rats, and 42, n = 3 rats) of inflammation. At 84 days after the induction of AIA (n = 5 rats), 51.0 ± 12.7% of the neurons showed an expression of BK–gold-binding sites and this was close to the prearthritic values. However, in the contralateral DRG of the same animals the proportion of BK–gold-labelled neurons declined in the intermediate phase (day 10) and chronic phase (days 21–84) of AIA and was not significantly different from the control value. Thus the increase in BK–gold-labelled neurons was persistent on the side where the inflammation had been induced, and transient on the opposite side. The size distribution of the DRG neurons of the different experimental groups was similar. In the cervical DRGs the expression of BK receptors did not change in the course of AIA. In another series of experiments, we determined the subtype(s) of BK receptor(s) that were expressed in DRGs L1–L5 in different experimental groups. In neither untreated control animals (n = 5) nor immunized rats without knee injection (n = 5) nor in rats at 3 days (n = 5) and 42 days (n = 5) of AIA was the binding of BK–gold decreased by the coadministration of BK–gold and the B1 agonist. By contrast, in these experimental groups the binding of BK–gold was suppressed by the coadministration of the B2 agonist. These results show that B2 receptors, but not B1 receptors, were expressed in both normal animals and in animals with AIA.
Discussion:
These results show that in AIA in the rat the expression of SP-binding and BK-binding sites in the perikarya of DRGs L1–L5 is markedly upregulated in the course of knee inflammation. Although the inflammation was induced on one side only, the initial changes in the binding sites were found in the lumbar DRGs of both sides. No upregulation of SP-binding or BK-binding sites was observed in the cervical DRGs. The expression of SP-binding sites was upregulated only in the first days of AIA, that is, in the acute phase, in which the pain responses to mechanical stimulation were most pronounced. By contrast, the upregulation of BK-binding sites on the side of AIA persisted for up to 42 days, that is, in the acute and chronic phase of AIA. Only the B2 receptor, not the B1 receptor, was upregulated. The coincidence of the enhanced expression of NK1 and BK receptors on sensory neurons and the pain behaviour suggests that the upregulation of these receptors is relevant for the generation and maintenance of arthritic pain.
In the acute phase of AIA, approximately 50% of the lumbar DRG neurons showed an expression of SP-binding sites. Because peptide receptors are transported to the periphery, the marked upregulation of SP-binding receptors probably leads to an enhanced density of receptors in the sensory endings of the primary afferent units. This will permit SP to sensitize more neurons under inflammatory conditions than under normal conditions. However, the expression of NK1 receptors was upregulated only in the acute phase of inflammation, suggesting that SP and NK1 receptors are less important for the generation of hyperalgesia in the chronic phase of AIA.
Because BK is one of the most potent algesic compounds, the functional consequence of the upregulation of BK receptors is likely to be of immediate importance for the generation and maintenance of inflammatory pain. The persistence of the upregulation of BK receptors on the side of inflammation suggests that BK receptors should be an interesting target for pain treatment in the acute and chronic phases. Only B2 receptors were identified in normal animals and in rats with AIA. This is surprising because previous pharmacological studies have provided evidence that, during inflammation, B1 receptors can be newly expressed.
Receptor upregulation in the acute phase of AIA was bilateral and almost symmetrical. However, hyperalgesia was much more pronounced on the inflamed side. It is most likely that receptors on the contralateral side were not readily activated because in the absence of gross inflammation the local concentration of the ligands BK and SP was probably quite low. We hypothesize that the bilateral changes in receptor expression are generated at least in part by mechanisms involving the nervous system. Symmetrical segmental changes can be produced only by the symmetrical innervation, involving either the sympathetic nervous system or the primary afferent fibres. Under inflammatory conditions, primary afferent fibres can be antidromically activated bilaterally in the entry zone of afferent fibres in the spinal cord, and it was proposed that this antidromic activation might release neuropeptides and thus contribute to neurogenic inflammation. Because both sympathetic efferent fibres and primary afferent nerve fibres can aggravate inflammatory symptoms, it is also conceivable that they are involved in the regulation of receptor expression in primary afferent neurons. A neurogenic mechanism might also have been responsible for the bilateral degradation of articular cartilage in the present study.
Introduction
Persistent pain is a major symptom of arthritis such as human rheumatoid arthritis. Inflammatory pain is caused by the activation and sensitization of primary afferent nociceptive neurons ('pain fibres') supplying the tissue (peripheral sensitization), and from the activation and sensitization of nociceptive neurons in the central nervous system (central sensitization). After sensitization, nociceptive neurons respond more strongly to mechanical and thermal stimulation of the tissue, and their activation threshold is lowered. These neuronal changes cause persistent pain as well as hyperalgesia, an enhanced pain sensitivity to mechanical and thermal stimuli [1,2,3,4,5]. The peripheral sensitization is produced by the action of inflammatory mediators such as bradykinin (BK) and prostaglandins on the primary afferent neurons that express receptors for these compounds in their sensory endings [5,6]. Subgroups of primary afferent neurons also express receptors for neuropeptides such as substance P (SP) that are released from primary afferent fibres.
Because the activation of primary afferent neurons by mediators is dependent on the receptors located on the neurons, we investigated in the present experiments the expression of BK and SP [neurokinin 1 (NK1)] receptors in primary afferent neurons in the acute and chronic phases of antigen-induced arthritis (AIA), which resembles in many aspects the inflammatory process of human rheumatoid arthritis [7,8,9,10]. BK is a potent pain-producing substance in animals and in humans. When it is applied to the tissue it causes many neurons to fire action potentials, and it sensitizes the neurons so that they respond more strongly to mechanical and thermal stimuli [3,5,11,12,13]. BK is produced under inflammatory conditions from a precursor and is released in the plasma; it is contained in inflammatory exudates, for example in the joint [14,15,16,17]. It acts on BK 2 (B2) receptors that are expressed in sensory neurons under normal conditions. Under inflammatory conditions an additional expression of a BK 1 (B1) receptor has been reported [6,18,19].
SP is a peptide that is produced in and released from the primary afferent fibres themselves. Indeed, SP is synthesized in 10–20% of the primary afferent neurons [20,21], most of which seem to be nociceptive neurons with high thresholds [22]. SP is transported from the cell bodies in the dorsal root ganglia (DRGs) to the sensory endings in the tissue, and the release from the sensory endings causes a neurogenic inflammation [23,24,25]. SP is also transported to the central terminals of the primary afferent neurons. At this site it is involved in the activation and in the generation of inflammation-evoked hyperexcitability of spinal cord neurons [26,27,28,29]. SP acts on NK1 receptors [30,31,32,33]. The application of SP to the tissue can sensitize a proportion of the nociceptive primary afferent neurons, suggesting that SP contributes to the generation of hyperalgesia [34,35]. However, other studies have failed to show effects of SP on primary afferent neurons [36,37]. Thus, under normal conditions, the role of SP is questionable.
Because it is very difficult to study the expression of peptide receptors in situ, we took advantage of the fact that peptide receptors are expressed not only in the terminals of the primary afferent units but also in the cell bodies [33,38,39,40]. We therefore removed DRGs of both sides from control rats and from rats with the acute or chronic phase of AIA and determined, after short-term culture of the neurons, the proportion of DRG neurons that expressed the receptors under the different experimental conditions. We also characterized the inflammatory process and the nociceptive behaviour of the rats in the course of AIA.
Materials and methods
Induction of joint inflammation
In 33 female Lewis rats 10 weeks old (Charles River, Sulzfeld, Germany), an inflammation was induced in the right knee joint. In the first step the rats received a subcutaneous injection of 500 μg of antigen [methylated bovine serum albumin (m-BSA); Sigma, Deisenhofen, Germany] in 500 μl of saline and emulsified with 500 μl of Freund's complete adjuvant [supplemented with 2 mg/ml Mycobacterium tuberculosis strain H37RA (Difco, Detroit, MI, USA] and an intraperitoneal injection of 2 × 109 heat-inactivated Bordetella pertussis (Pertussis-Referenzlabor, Krankenhaus Berlin-Friedrichshain, Berlin, Germany). The same immunization procedure was repeated seven days later. After a further 14 days a sterile solution of antigen (m-BSA) (500 μg in 50 μl of saline) was injected into the cavity of the right knee joint (day 0). Eight animals received the same immunization procedure excluding the injection of antigen into the knee joint. The mediolateral diameters of the knee joints were measured at regular intervals by means of a vernier caliper (see Fig. 1a).
Figure 1.
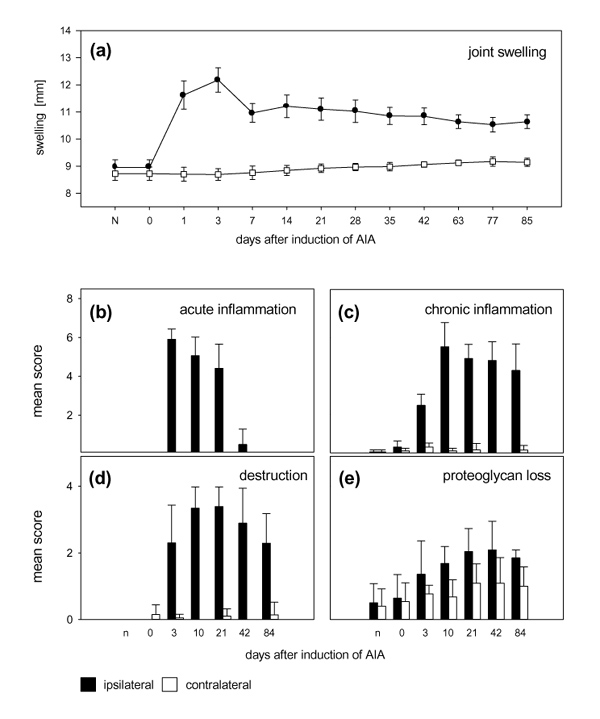
Development of antigen-induced arthritis. (a) Diameters of the injected knee joints and the contralateral knee joints at different time points of arthritis. The contralateral knees show a slight increase in their diameters owing to growth. (bStuttgart: Gustav Fischer Verlag;d) Arthritic changes of the knee joints at 3, 10, 21, 42 and 84 days after the induction of arthritis, and of knee joints from immunized non-arthritic animals (0 days) in comparison with untreated normal control animals (n). (b) Histological examination of acute inflammatory changes (fibrin exudation and granulocytic infiltration of the synovial membrane and the joint cavity) in the injected joint and the contralateral knee. (c) Histological examination of the chronic inflammatory changes (hyperplasia of synovial lining, infiltration of mononuclear leucocytes) of the injected joint and the contralateral knee. (d) Overall joint destruction (pannus formation and erosion of cartilage and bone) in the injected knee joint and in the contralateral knee joint. (e) Loss of proteoglycans (staining with safranin O) in the cartilage of the injected knee and the contralateral knee. All values are means and standard deviations.
At 1, 3, 10, 21, 42 or 84 days after the induction of inflammation in the knee joint, the rats were killed by cervical dislocation during anaesthesia with ether. A total of 10 untreated rats of the same age and sex were used as normal control animals. The immunized rats without arthritis induction were killed 14 days after the second immunization (day 0).
Two series of experiments were performed. In the first series (n = 31 rats), we determined the expression of SP-binding and BK-binding sites in the DRGs of lumbar segments L1–L5. The nerves innervating the knee joint are contained in these segments. In the second series of experiments (n = 20 rats) we again took DRGs from segments L1–L5 to determine the BK receptor subtypes, and we removed DRGs from cervical segments C1–C7 to determine the expression of NK1 and BK receptors in the primary afferent neurons at the cervical level. In addition, a detailed documentation of the behaviour was performed in the second series. All procedures complied with the regulations of the Thuringian Commission for Animal Protection.
Histology and grading of arthritis
After the rats had been killed, both knee joints were removed, skinned, fixed in 4% buffered formalin, decalcified in EDTA, embedded in paraffin, cut into 5 μm frontal sections and stained with haematoxylin–eosin for microscopic examination. Three sections per knee joint were examined in a blind fashion by two independent observers (PKP and RB) with the use of a semiquantitative score (0 = no, 1 = mild, 2 = moderate, 3 = severe alterations). The acute inflammatory reaction was assessed by evaluating the quantity of fibrin exudation and the relative number and density of granulocytes in the synovial membrane and in the joint space. The chronic inflammatory reaction was quantified on the basis of the relative number and density of infiltrating mononuclear leucocytes in the synovial membrane, the degree of synovial hyperplasia, and the extent of fibrosis in the synovial tissues. The histological score is the sum of the three parameters evaluated. A score from 0 to 4 was used to assess the degree of cartilage destruction: 0, no destruction; 1, unequivocal erosions of less than 10% of cartilage and bone cross sections; 2, erosion of 10–25%; 3, erosion of 25–50%; 4, erosion of more than 50% of cartilage and bone cross sections. Additional sections were stained with safranin O to determine the loss of proteoglycan in the cartilage matrix, with the use of the same score as for the evaluation of inflammatory reactions.
Testing of nociceptive behaviour
At several time points after the induction of AIA, the behaviour of rats (at least n = 5 rats for each time point) was assessed by using a score. First each rat was placed in a box in which it could move freely. The severity of disturbances of walking was graded: 4, no walking; 3, walking on three legs; 2, limping with the leg with inflammation; 1, limping with the leg with inflamed knee only after pressure on the knee; 0, normal walking. Other parameters (exploratory behaviour, standing on hindlimbs) were also checked and documented. Thereafter the rats were held in the hand by one experimenter and the following tests were performed by the other experimenter: flexion/extension of the left and right knee joint as well as an application of moderate non-painful and strong, lightly painful pressure onto the ankle joints and the knee joints. In all these cases we determined whether the rat showed a nociceptive reaction, namely a withdrawal of the stimulated leg. By using a mechanical device that applied pressure to a small area, we administered pressure to the lateral side of the knee joint and determined the pressure range at which the rat withdrew the leg. Responses to pressure in the range 0–100 g were scored 3 (this stimulus evokes a touch sensation in humans), responses to pressure in the range 100–200 g were scored 2 (this stimulus evokes a pressure sensation), responses to 200–250 g were scored 1 and a lack of response to 250 g was scored 0 (the application of 250 g evokes a weak pain sensation). For analysis, the scores of all animals were added and divided by the number of animals tested in the group.
Preparation of the DRGs
Because the fixation of the animals by perfusion impairs the binding of gold-labelled peptides and because the structure of DRGs in cryostat sections is not well maintained, we removed the DRGs after killing the animals and put the neurons into short-term culture. The DRGs of both sides were dissected quickly from segments L1–L5 or from segments C1–C7. The ganglia were incubated at 37°C in 0.28 U/ml collagenase type A (Boehringer, Mannheim, Germany) dissolved in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Eggenstein, Germany) for 100 min. After being washed with phosphate-buffered saline (PBS, 20 mmol, pH 7.4), the ganglia were incubated in PBS containing 25,000 U/ml trypsin (Sigma) for 11 min at 37°C. Then the ganglia were dissociated into single cells by gentle agitation and by trituration through a fire-polished Pasteur pipette. The cells were washed three times in DMEM by centrifugation (500 g, 5 min). The final cell pellet was suspended in Ham's F-12 medium (Gibco BRL) containing 10% heat-inactivated horse serum (Gibco BRL), 100 U/ml penicillin (Gibco BRL), 100 μg/ml streptomycin (Gibco BRL) and 100 ng/ml nerve growth factor (7S, recombinant human; Boehringer). Cells were plated on 13 mm glass coverslips coated with poly-(L-lysine) (200 μg/ml) and kept for 18 h at 37°C in a humidified incubator gassed with 3.5% CO2in air. Cells were fed after 14–16h with supplemented Ham's F-12 medium (see above).
Preparation of the SP–gold and BK–gold conjugates
SP (Sigma) (1 μmol) or 1 μmol of BK (Bachem, Heidelberg, Germany) was dissolved in 500 μl of HEPES (20 mmol, pH 7.5). This solution was added to 6 nmol of sulpho-N-hydroxysuccinimido Nanogold reagent (BioTrend, Köln, Germany), dissolved in 500 μl of doubly distilled water and incubated for 1 h at room temperature. To separate SP–gold or BK–gold conjugates from unbound SP or BK, membrane centrifugation (Amicon Microcon-10 system) was used. The SP–gold and BK–gold conjugates were dissolved in PBS containing 0.1% bovine serum albumin (BSA), 0.2 mol of sucrose, 4 μg/ml leupeptin and 10 mmol of sodium azide. This solution was aliquoted and stored at –20°C for a maximum of 3 months.
SP–gold and BK–gold binding to cultured neurons
The cells were pre-fixed with 2% paraformaldehyde and 0.05% glutaraldehyde in 0.1 mol of phosphate buffer (pH 7.2) for 30 min. After being washed with PBS (20 mmol, pH 7.4), the cells were pretreated with 50 mmol of glycine in PBS and thereafter with 5% BSA and 0.1% gelatine in PBS for 30min. The cells were then washed with 0.1% acetylated BSA (BSA-C) and incubated overnight with 0.3 nmol/ml SP–gold in PBS or with 0.3 nmol/ml BK–gold containing 0.1% BSA-C, bacitracin (40 μg/ml), leupeptin(4 μg/ml) and chymostatin (2 μg/ml) at 4°C in a moist chamber. After being washed with PBS plus 0.1% BSA-C and thereafter with PBS to remove unbound SP–gold or BK–gold, cells were postfixed with 2% glutaraldehyde in PBS for 10 min. After extensive washing with PBS and doubly distilled water, the gold particles were intensified with silver enhancer (R-Gent, pH 5.5; BioTrend) for 15 min at 22°C. The reaction was stopped by washing in doubly distilled water. The preparations were dehydrated and embedded in DePeX (Fluka, Neu-Ulm, Germany).
Control incubations
To determine the specificity of the SP–gold or BK–gold complex used in the binding studies, a displacement control was performed. Neurons were incubated in 1 μmol/ml unlabelled SP or 1 μmol/ml unlabelled BK together with 0.3 nmol/ml peptide–gold. The unlabelled peptides should decrease or prevent binding of the gold-labelled peptides. Furthermore, to test whether SP–gold is bound specifically to NK1 receptors, 0.3 nmol/ml SP–gold was incubated in the presence of 1 μmol/ml [Sar9, Met(O2)11]-SP (Bachem), a specific agonist at the NK1 receptor. To examine whether the binding of BK–gold is related to BK receptors, 0.3 nmol/ml BK–gold was incubated in the presence of 1 μmol/ml of D-Arg (Hyp3-Thi5,8-D-Phe7)-BK, a BK analogue specific for the B1 receptor (Bachem), and/or 1 μmol/ml of (Des-Arg10)-Lys-BK, a B2 receptor agonist (Bachem).
Data analysis
At different time points after induction of inflammation, DRG preparations of two to five rats were used to determine SP–gold-binding and BK–gold-binding sites. The DRGs of untreated rats served as the control group. DRGs were also removed from immunized but non-arthritic animals (day 0 group). To analyse the data, from every coverslip 50 or 100 structurally intact neurons were examined with a light microscope (Zeiss; Axiophot, Jena, Germany) coupled to a CCD colour video camera [AVT-BC6(0)] and an image-analysing system (Kontron, Eching, Germany). On each coverslip, neurons were randomly selected; neurons obstructed by other neurons or by tissue were not included. In total 30 500 neurons were analysed. The relative grey value (grey value of the soma/grey value of the substrate background) was determined for each soma. From each individual binding experiment one coverslip was used to perform a displacement control incubation (see above). This allowed us to determine the grey value range of neurons with no SP–gold or no BK–gold binding in each experiment (see Fig. 5, white bars). In all other coverslips of the particular experiment, neurons were considered as positive for peptide-binding sites if they had a relative grey value above that of neurons from the control incubation. For the final analysis, data from the experiments were pooled. Group results are expressed as means and standard deviations. To evaluate the cell size, the cross-sectional area was taken from each selected neuron. For statistical evaluation, four groups of DRGs were formed, namely DRGs of untreated control rats, of the acute phase (1 and 3 days), of the chronic phase (21 and 42 days) and of the intermittent phase (10 days) of AIA. For comparison of the AIA groups with the control group the Mann–Whitney U-test was used, with adjustment of significance values for multiple comparisons; significance was accepted at P <0.05 [41].
Figure 5.
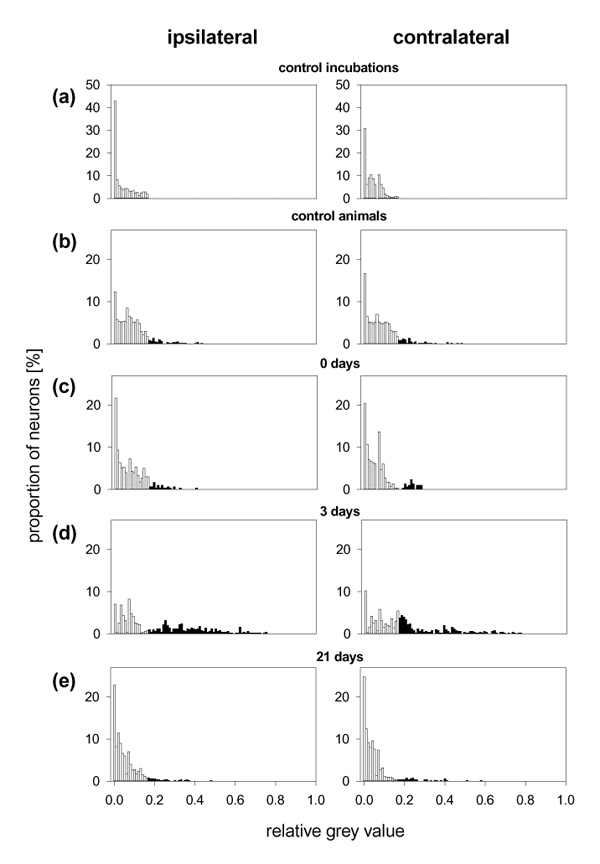
Distribution of the relative grey values of DRG neurons ipsilateral and contralateral to the injected knee with SP–gold binding. (a) Neurons (n = 800, from eight cultures) from control incubations treated with an excess of substance P that was administered together with SP–gold. (b) Neurons (n = 500, from five cultures) from untreated control animals. (c) Neurons (n = 300, from three cultures) from immunized animals without knee injection. (d) Neurons (n = 500, from five cultures) from rats 3 days after the induction of arthritis (3 days). (e) Neurons (n = 500, from five cultures) from AIA rats (21 days). White bars, neurons exhibiting grey densities that were in the range of those observed in neurons from control incubations; black bars, neurons exhibiting grey densities that were higher than those observed in the neurons from control incubations.
Results
Joint inflammation
Figure 1 shows the time course of the inflammatory process. The swelling of the injected knee is shown in Fig. 1a (dots). The diameter of the contralateral knees showed a slight increase over time owing to growth. The time course of acute inflammatory changes (a predominantly granulocytic infiltration of the synovial membrane and the joint cavity) is shown in Fig. 1b; the time course of chronic alterations (infiltration of mononuclear leucocytes, hyperplasia of synovial lining layer, extent of fibrosis) is displayed in Fig. 1c. The contralateral knee joints seemed normal at all time points. Figure 1d summarizes the destruction of the joints, taking into account the erosion of cartilage and bone and the formation of synovial pannus. Figure 1e depicts the depletion of cartilage proteoglycan (evidenced by the loss of staining with safranin O). Although destruction was observed only in the injected knee, some proteoglycan loss was also noted in the non-injected, contralateral knee.
Nociceptive behaviour (pain response)
At 1–29 days after the induction of AIA, the rats showed signs of mechanical hyperalgesia in the leg with inflammation. Most striking were the disturbances of gait (Fig. 3a). At 1–3 days most rats did not use the leg with inflammation for walking. At 9–21 days of AIA, the rats showed marked limping of the leg with the injected knee. At 29 days of AIA, limping was seen only after the knee had been pressed. Normal gait was observed after 37 days of AIA. Furthermore, at 1–9 days of AIA, rats did not stand on the hindfeet or visibly loaded only the foot of the leg with the non-injected knee. Exploring behaviour fell from day 2 to day 9 of AIA. When the inflamed knee was manually flexed and extended, the rats showed an immediate defence reaction at 1–9 days of AIA. Movements of the contralateral knee did not elicit defence behaviour. Local pressure onto the injected and non-injected knee revealed that the local threshold in the injected knee was markedly reduced at 1–9 days of AIA but a reduction was also seen on the contralateral side (Fig. 3b). Between 14 and 29 days of AIA, the rats showed a withdrawal response when the injected knee was compressed between two fingers with moderate intensity, whereas strong and painful compression was necessary to evoke a withdrawal response to pressure on the contralateral knee.
Figure 3.
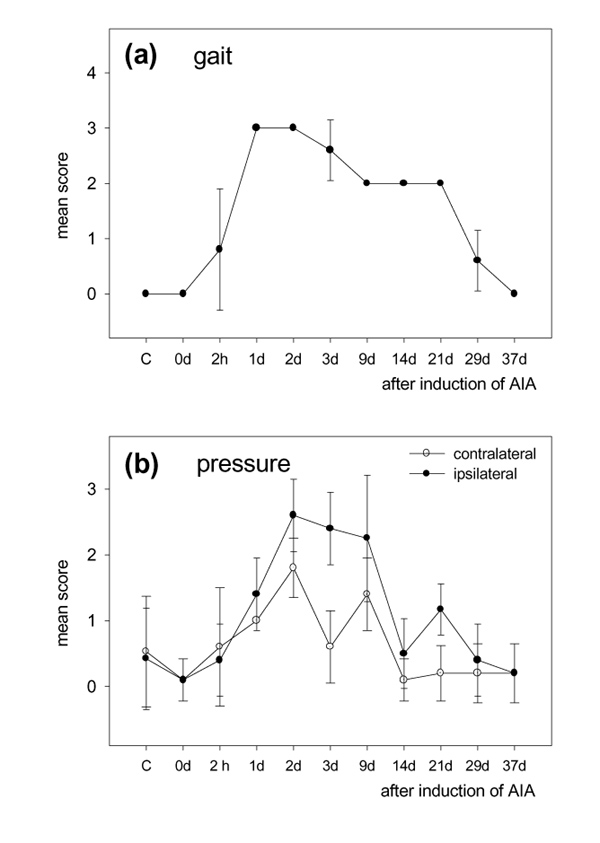
Nociceptive behaviour of the rats in different experimental groups. (a) Description of the gait, scored as follows: 4, no walking; 3, walking on three legs; 2, limping with the injected leg; 1, limping after pressure on the knee; 0, normal walking. (b) Withdrawal response of the injected and the contralateral knee on local stimulation of the knee with a mechanical device, scored as follows: 3, response to pressure in the range 75–150 g; 2, response to pressure in the range 150–200 g; 1, response to pressure in the range 200–250 g; 0, no response to pressure of more than 250 g. The symbols show mean scores and standard deviations. In (a) the standard deviation was zero at most time points.
Expression of SP–gold- and BK–gold-binding sites in DRGs from control and arthritic rats
Figure 4 shows neurons that were isolated from DRGs L1–L5 3 days after the induction of AIA and then cultured. Neurons labelled with SP–gold (Fig. 4a) or with BK–gold (Fig. 4b) appear dark as a result of the silver staining of the gold particles (arrows). The next sections will illustrate the data analysis and summarize the data on the receptor expression.
Figure 4.
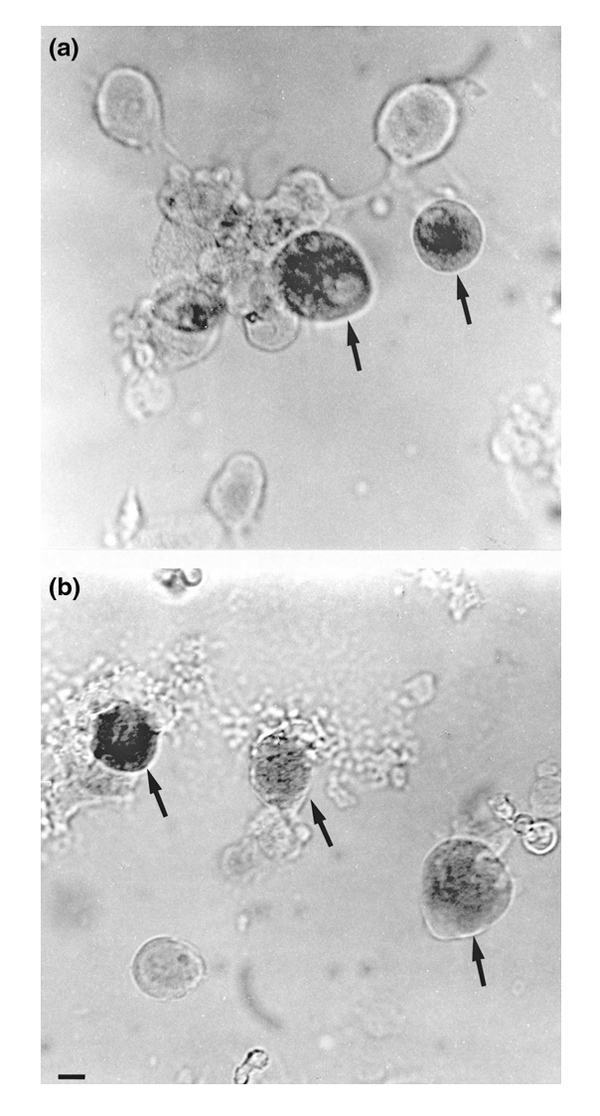
Isolated DRG neurons of the adult rat after incubation with SP–gold (a) or BK–gold (b) and subsequent enhancement with silver. The neurons were cultured for 18 h. The black staining indicates binding of SP–gold or BK–gold (arrows).
SP–gold binding sites
Lumbar DRGs
Figure 5 exemplifies the data analysis. In the DRGs of both sides of all animals, control incubations were performed in which SP was administered in excess together with SP–gold, to suppress the binding of SP–gold. Figure 5a displays the distribution of the grey values of neurons of the DRGs of both sides from these control incubations. The grey values were in the range 0.0–0.16 (white bars), and thus this range of grey values does not indicate labelling with SP–gold. In all other incubations only SP–gold was used. In the DRGs of healthy untreated control animals (n = 5) approximately 9% of the DRG neurons of both sides exhibited grey values of more than 0.16 (black bars), indicating binding of SP–gold (Fig. 5b). Similar findings were obtained in DRG neurons from immunized non-arthritic rats (n = 3; Fig. 5c). By contrast, in DRG from rats (n = 5) in which the inflammation had been induced on the ipsilateral side three days previously, a much higher proportion of DRG neurons of both sides exhibited grey values of more than 0.16, indicating binding of SP–gold (Fig. 5d). These inflammation-induced changes were reversible (Fig. 5e).
Figure 2a displays the proportions of DRG neurons that showed binding of SP–gold in the different groups of rats. In the untreated control rats (n = 5), 7.7± 3.8% of the DRG neurons from the right side (black bars) and 10.0 ± 1.7% of the DRG neurons from the left side (white bars) showed labelling with SP–gold. The proportion of SP–gold-labelled neurons in immunized animals without knee injection (n = 3) was similar. By contrast, at days 1 (n = 2 rats) and 3 (n = 5 rats) of AIA in the right knee, approximately 50% of the DRG neurons exhibited labelling with SP–gold, and this was seen both on the side of the injected knee and on the opposite side. At day 10 of AIA (n = 3 rats), 26.3 ± 6.1% of the ipsilateral DRG neurons but only 15.7 ± 0.6% of the contralateral neurons exhibited binding of SP–gold. At days 21 (n = 5 rats), 42 (n = 3 rats) and 84 (n = 5 rats) of AIA, the proportion of SP–gold-positive neurons had returned to the control values, although the inflammation was still present. In comparison with the DRG neurons of the untreated control rats, the increase in the proportion of labelled neurons was significant on both sides in the acute phase (days 1 and 3) and the intermediate phase (day 10) of AIA (Mann–Whitney U-test). The size distribution of the neurons was similar in the DRG neurons of all experimental groups. The white bars in Figure 6 show the distribution of the areas of all neurons measured, and the black insets show the subset of neurons with SP–gold-binding sites. Under all conditions and at all time points, SP–gold binding was found mainly in small and medium-sized (less than 700 μm2) neurons.
Figure 2.
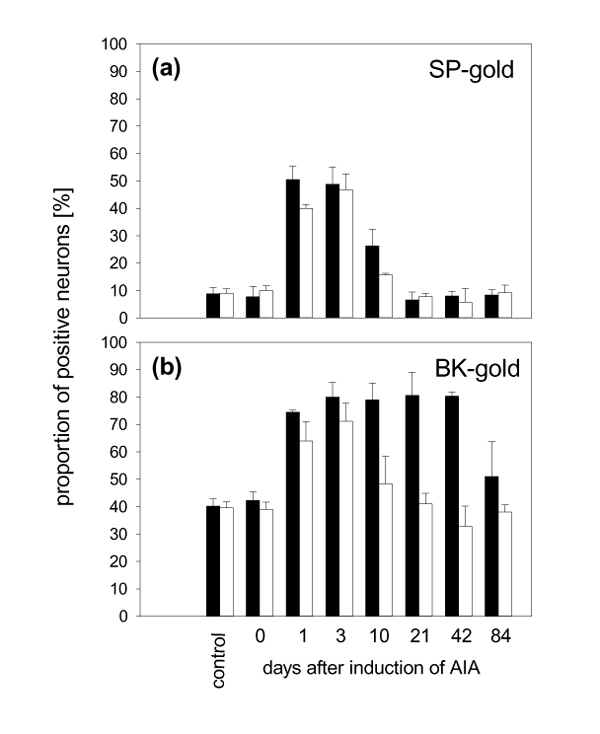
Bilateral upregulation of receptor expression during antigen-induced arthritis. (a) Proportions of DRG neurons with binding of SP–gold in control animals and in different groups of animals with AIA. (b) Proportions of DRG neurons with binding of BK–gold in control animals and in different groups of animals with AIA. Results for DRG neurons from immunized animals without knee injection are shown at 0 days. White bars, contralateral to injected knee; black bars, ipsilateral to injected knee.
Figure 6.
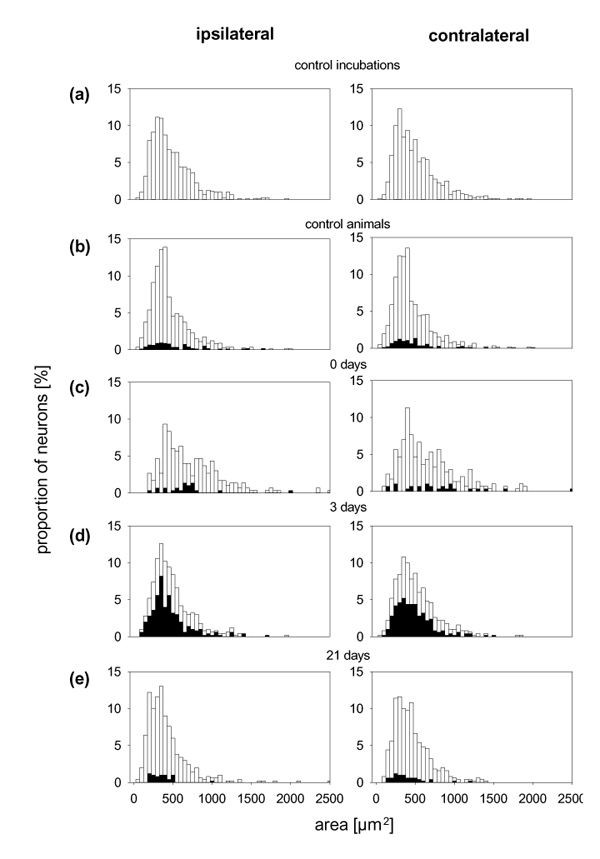
Size distribution of the cultured neurons from different experimental groups. The white bars show the proportions of neurons with different areas from experiments in which SP–gold-binding sites were determined. The neurons with SP–gold-binding sites are shown by the black bars.
To show that SP-gold was bound to NK1 receptors we tried to suppress the binding of SP-gold to the neurons by the specific NK1 agonist [Sar9, Met(O2)11]-SP in three experiments. In Fig. 7 the histograms on the left side show the grey density of the neurons from two experimental groups treated only with SP-gold. A proportion of these neurons exhibited grey values of more than 0.16, indicating binding of SP-gold. The histograms on the right side show that neurons with grey values of more than 0.16 were not found when SP-gold was administered together with the specific NK1 receptor agonist. Thus SP-gold specifically labelled NK1 receptors.
Figure 7.
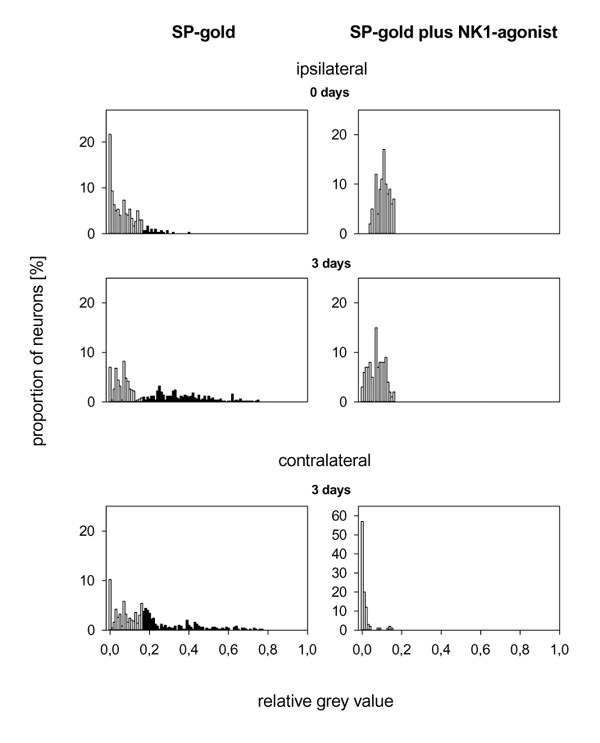
Displacement control for SP–gold with the specific NK1 receptor agonist [Sar9, Met(O2)11]-SP in DRG neurons ipsilateral and contralateral to the injected knee. The histograms on the left show the distribution of the grey values after treatment with SP–gold; the histograms on the right show the distribution of the grey values after the administration of SP–gold and [Sar9, Met(O2)11]-SP (1 μmol/ml) together.
Cervical DRGs
In the cervical DRGs the expression of NK1 receptors did not change in the course of AIA (Table 1). NK1 receptors were expressed in approximately 10% of the neurons in untreated control animals (n = 5), in immunized animals without knee injection (n = 5), in rats at 3 days of AIA in the knee (n = 5), and in 5 rats at 42 days of AIA in the knee.
Table 1.
Expression of neurokinin 1 and bradykinin receptors in cervical DRGs ipsilateral to the inflamed knee joint
| Percentage of neurons with: | ||
| Experimental group | SP–gold- | BK–gold- |
| binding sites | binding sites | |
| Untreated control animals | 10.4 ± 2.2 | 39.2 ± 3.9 |
| Immunized rats without knee injection | 11.6 ± 0.9 | 38.4 ± 4.3 |
| AIA 3 days | 10.2 ± 1.9 | 42.0 ± 3.2 |
| AIA 42 days | 8.4 ± 3.6 | 42.8 ± 4.4 |
Values shown are the means and standard deviations.
BK-gold-binding sites
Lumbar DRGs
Figure 8 shows the distribution of grey values in neurons in which labelling with BK–gold was used. Figure 8a displays the grey values from neurons of all control incubations with BK–gold and BK. The other graphs show specific BK–gold-binding sites (grey values of more than 0.16) in neurons from untreated control animals (Fig. 8b), from immunized rats without knee injection (Fig. 8c) and from rats with AIA (Fig. 8d and e).
Figure 8.
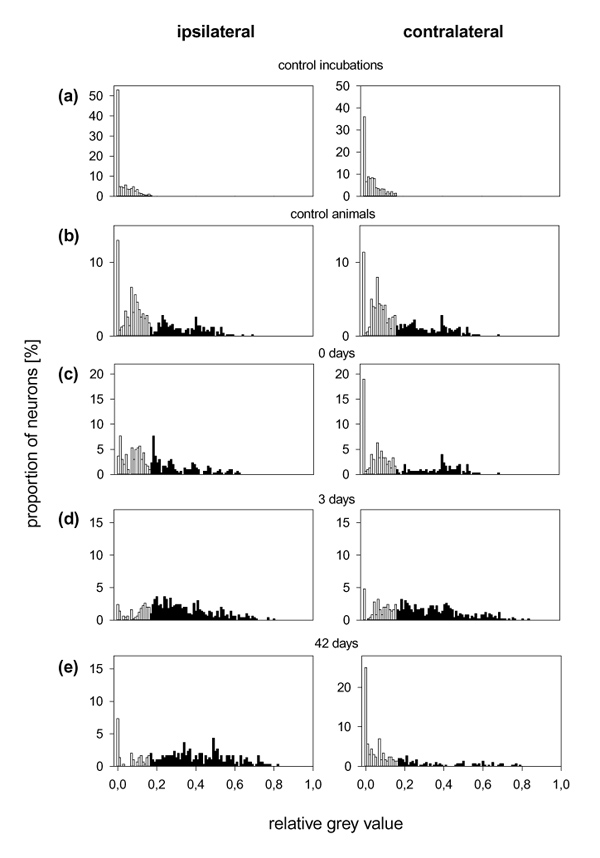
Distribution of the relative grey values of DRG neurons ipsilateral and contralateral to the injected knee with BK–gold binding. (a) Neurons (n = 800, from eight cultures) from control incubations treated with an excess of BK that was administered together with BK–gold. (b) Neurons (n = 500, from five cultures) from untreated control animals. (c) Neurons (n = 300, from three cultures) from immunized animals without knee injection. (d) Neurons (n = 500, from five cultures) from AIA rats (3 days). (e) Neurons (n = 300, from three cultures) from AIA rats (42 days). White bars, neurons exhibiting grey densities that were in the range of those observed in neurons from control incubations; black bars, neurons exhibiting grey densities that were higher than those observed in the neurons from control incubations.
Figure 2b shows the proportions of neurons with binding of BK–gold in the different groups of rats. In untreated control rats (n = 5), 42.3 ± 3.1% of the DRG neurons of the right side (black bars) and 39.6 ± 2.6% of the DRG neurons of the left side (white bars) showed binding of BK–gold. At days 1 (n = 2 rats) and 3 (n = 5 rats) of AIA, approximately 80% of the DRG neurons on the side of the knee injection (ipsilateral) and approximately 70% on the opposite side were labelled. In comparison with the untreated control group the increase in the proportion of labelled neurons was significant on both sides. The proportion of labelled neurons in the ipsilateral DRGs remained significantly increased in both the intermediate phase (day 10, n = 3 rats) and chronic phase (days 21, n = 5 rats, and 42, n = 3 rats) of inflammation. At 84 days after the induction of AIA, 51.0 ± 12.7% of the neurons showed an expression of BK–gold-binding sites and this was close to the prearthritic values. In the contralateral DRG of the same animals, however, the proportion of BK–gold-labelled neurons declined in the intermediate phase (day 10) and chronic phase (days 21–84) of AIA and was not significantly different from the control value. Thus the increase in BK–gold-labelled neurons was persistent on the side where the inflammation had been induced, and transient on the opposite side.
The size distributions of the DRG neurons of the different experimental groups were similar (Fig. 9). Under all conditions and at all time points, BK–gold binding was found mainly in small and medium-sized neurons. The binding of BK–gold in these experiments was suppressed by the administration of a mixture of both the B1 and B2 agonists (data not shown).
Figure 9.
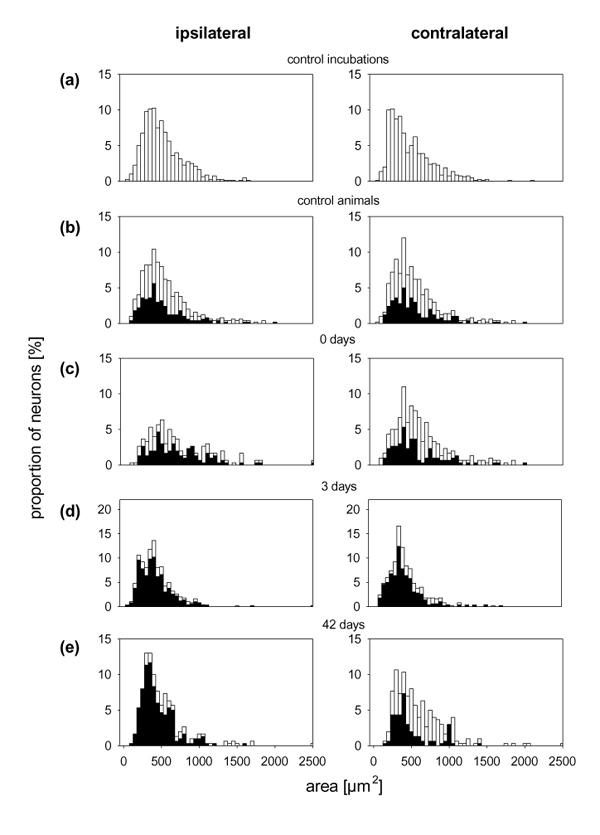
Size distribution of the cultured neurons from different experimental groups. The white bars show the proportions of neurons with different areas from experiments in which BK–gold-binding sites were determined. The neurons with BK–gold-binding sites are shown by the black bars.
In another series of experiments, we made an effort to determine the subtype(s) of BK receptor(s) that were expressed in DRGs L1–L5 in different experimental groups. The data are displayed in Table 2. In neither untreated control animals (n = 5) nor immunized rats without knee injection (n = 5) nor rats at 3 days (n = 5) and 42 days (n = 5) of AIA was the binding of BK-gold reduced by the coadministration of BK–gold and the B1 agonist. By contrast, in these experimental groups the binding of BK–gold was suppressed by the coadministration of the B2 agonist. These data show that in both normal animals and animals with AIA, B2 receptors but not B1 receptors were expressed.
Table 2.
Expression of bradykinin 1 and bradykinin 2 receptors in DRGs L1–L5
| Percentage of labelled neurons | ||||
| Experimental group | Test site | BK–gold | BK–gold + BK1 agonist | BK–gold + BK2 agonist |
| Untreated control animals | i.l. | 38.8 ± 7.0 | 38.0 ± 2.4 | 0 |
| c.l. | 40.4 ± 0.9 | 41.6 ± 4.6 | 1.2 ± 1.8 | |
| Immunized rats without knee injection | i.l. | 42.0 ± 1.4 | 38.8 ± 2.3 | 0 |
| c.l. | 41.6 ± 5.2 | 38.8 ± 6.1 | 0 | |
| Rats at 3 days of AIA | i.l. | 77.0 ± 3.4 | 83.4 ± 2.9 | 0.4 ± 0.9 |
| c.l. | 88.0 ± 4.7 | 88.0 ± 4.7 | 1.6 ± 2.1 | |
| Rats at 42 days of AIA | i.l. | 74.8 ± 4.8 | 71.6 ± 8.9 | 0.4 ± 0.9 |
| c.l. | 38.8 ± 2.3 | 38.0 ± 2.5 | 1.6 ± 3.6 | |
Values shown are the means and standard deviations. Abbreviations: i.l., ipsilateral to injected knee; c.l., contralateral to injected knee.
Cervical DRGs
In the cervical DRGs the expression of BK receptors did not change in the course of AIA (Table 1). BK receptors were expressed in approximately 40% of the neurons in untreated control animals (n = 5), in immunized animals without knee injection (n = 5), in rats at 3 days of AIA in the knee (n = 5), and in 5rats at 42 days of AIA in the knee.
Discussion
These results show that the expression of SP-binding and BK-binding sites in the perikarya of DRGs L1–L5 is markedly upregulated in the course of inflammation in the knee joint. Several aspects are noteworthy. Firstly, although the inflammation was induced on one side only, the initial changes in the binding sites were found in the lumbar DRGs of both sides. No upregulation of SP-binding and BK-binding sites was observed in the cervical DRGs. Secondly, whereas the expression of SP-binding sites was upregulated only in the first days of AIA, that is, in the acute phase, the upregulation of BK-binding sites on the side of AIA persisted for up to 42 days. Thirdly, and unexpectedly, only the B2 receptor, not the B1 receptor, was upregulated. The enhanced expression of NK1 and BK receptors on sensory neurons might be an important mechanism by which arthritis causes pain and hyperalgesia.
Inflammation and nociceptive behaviour
The limping during walking and the withdrawal responses to pressure on the joint show that the rats experienced pain during mechanical stimulation of the inflamed joint (mechanical hyperalgesia). Mechanical hyperalgesia is also a leading symptom of human arthritis, and thus AIA in the rat seems to be a suitable model for studying the mechanisms of arthritic pain. Hyperalgesia was most marked in the acute phase and less severe when the acute phase subsided. Interestingly, later stages of AIA were not obviously painful although the chronic inflammation and joint destruction persisted. Although hyperalgesia was pronounced in the knee with inflammation, signs of weak hyperalgesia were also noted on the contralateral side at 1–9 days of AIA because the mechanical thresholds were lowered. However, the pain response was quite asymmetrical; the main symptoms were on the injected side.
Methodological considerations for the use of DRG
During inflammation, pain is elicited by the activation and sensitization of the sensory endings of nociceptors in the tissue. For technical reasons it is currently impossible to study molecular nociceptive processing and quantitative changes in receptor expression in the sensory endings in situ. However, the cell body of DRG neurons is thought to serve as a valid model for the sensory endings, for three reasons:
1. The perikarya of DRG neurons express ion channels that seem also to be present in the sensory endings, for example ion channels that are activated by noxious heat and other noxious stimuli [42,43].
2. The perikarya of DRG neurons express receptors for ligands that activate the sensory endings in preparations in vivo and in vitro, for example capsaicin and BK [11,44,45,46,47].
3. Mediators can be released from the perikarya that are usually released from the sensory and spinal endings of the primary afferent fibres [48,49,50].
We therefore believe that the receptor expression in the cell bodies of DRG neurons also represents the receptor expression in the sensory terminals of the DRG neurons.
DRG neurons were cultured for 18 h after they had been removed from the rats. In the process of culturing, neurons might be lost, and cultured neurons might show changes compared with neurons in situ; this could influence the basal expression of receptors, for example. However, the neurons from different experimental groups were cultured in the same way, and the distribution of the cell sizes was similar in all preparations. Therefore changes in the expression of binding sites are most probably the outcome of pathophysiological changes in the rat during the inflammation.
Expression and upregulation of SP-binding sites during inflammation
Evidence was provided that some primary afferent neurons express receptors for SP. Low levels of NK1 receptor mRNA were identified in DRG of the mouse [30]. Immunohistochemical staining of NK1 receptors has been found in a proportion of unmyelinated axons of rat glabrous skin in situ [31]. In cultured DRG neurons, 10–20% of the perikarya show binding of SP–gold. Binding of SP–gold can be suppressed by the coadministration of a specific NK1 receptor agonist but not of specific NK2 and NK3 receptor agonists [33]. A similar proportion of cultured DRG neurons show binding of an antibody directed against the carboxy terminus of the NK1 receptor (unpublished data). The application of SP to freshly isolated DRG neurons evokes an inward current [32], and changes in intracellular Ca2+ concentration were seen after the application of SP or NK1 agonists to cultured DRG neurons [51]. However, other studies failed to identify NK1 receptors in primary afferent neurons [52,53,54]. Thus the expression of functional NK1 receptors in the perikarya of DRG neurons is not entirely settled [55]. There is still debate on whether or not the effect of SP on sensory endings is important for the generation of pain (see below).
During inflammation in the periphery, the synthesis of SP in DRGs is upregulated [56,57,58,59,60,61,62,63]. More SP is released from the sensory endings in the periphery (producing for example neurogenic inflammation) and from the synaptic endings in the spinal cord [26]. An upregulation of NK1 receptors during inflammation has been observed in spinal cord neurons [64,65,66,67]. Here we show that there is also a marked upregulation of SP-binding sites in primary afferent neurons. The low proportion of neurons with SP-binding sites in the normal animal (approximately 10–20%) could be the reason why SP was found to affect primary afferent neurons in some studies [34,35] but not in others [36,37]. In the acute phase of AIA, approximately 50% of the lumbar DRG neurons showed an expression of SP-binding sites. Because the binding of SP–gold can be suppressed by a specific NK1 receptor agonist (this study) but not by specific agonists at the NK2 and NK3 receptor [33], we believe that the SP–gold-binding sites represent NK1 receptors.
Because peptide receptors are transported to the periphery, the marked upregulation of SP-binding receptors probably leads to an enhanced density of receptors in the sensory endings of the primary afferent units. This allows SP to sensitize more neurons under inflammatory conditions than under normal conditions. It is therefore likely that the upregulation of NK1 receptors contributes to hyperalgesia during arthritis. Indeed, the upregulation of NK1 receptors coincided with the marked pain responses that were seen in the acute phase of inflammation. However, the final proof of the involvement of NK1 receptors in the arthritic pain will require additional functional experiments. In the chronic phase NK1 receptors seem to be less important.
Expression and upregulation of BK-binding sites during inflammation
As shown by several methods, primary afferent neurons express BK receptors in their perikarya in the DRGs and also in their peripheral sensory and spinal synaptic endings [11,38,39,40,67,68]. Although the functional effects of SP on nociceptive afferent neurons are still under discussion (see the last paragraph), there is full agreement that BK causes pain in animals and humans. In the present study, DRGs from AIA rats exhibited more neurons with BK-binding sites. Thus, during AIA more primary afferent neurons can be activated and sensitized by the inflammatory mediator BK, and thus the upregulation of the BK-binding sites is likely to be an important mechanism by which arthritis causes severe pain. Because the increased expression of BK-binding sites persisted for up to 42 days on the side of inflammation, BK-binding sites should be an interesting target for pain treatment in the acute and chronic phases of arthritis.
Only the specific B2 agonist, not the specific B1 agonist, suppressed the binding of BK–gold, suggesting that the BK receptors in the DRG in the present study were B2 receptors. This is surprising because previous pharmacological studies have provided evidence that, during inflammation, B1 receptors are newly expressed [6,18,19]. It is unlikely that the specific B1 agonist did not work in the present experiments for technical reasons, because this B1 agonist reduced the binding of BK–gold in another study with the same technique [68]. Indeed, when BK receptors are upregulated after nerve lesion, a bilateral increase in BK receptors involves the upregulation of B2 receptors and an expression of B1 receptors de novo [68]. We therefore conclude that the increased expression of BK receptors in AIA is entirely due to an upregulation of the B2 receptor.
Bilateral effects and possible mechanisms of upregulation of receptors
Receptor upregulation in the acute phase of AIA was bilateral and almost symmetrical. However, the hyperalgesia was much more pronounced on the inflamed side. That the pain responses were asymmetrical in spite of a symmetrical receptor upregulation is most probably due to the fact that the receptors on the contralateral side were not readily activated because in the absence of gross inflammation the local concentration of the ligands BK and SP was probably quite low.
At present it is unclear which mechanisms cause the upregulation of BK-binding and SP-binding sites. Because the expression of BK receptors in cultured DRG neurons can be regulated, for example by nerve growth factor (NGF) [39], we assume that mediators of different sources (such as immune cells and nerve cells) could have a role. NGF could be a candidate because inflamed tissue shows a rapid enhancement of NGF [69,70,71]. NGF can also regulate the synthesis of neuropeptides such as SP [72,73]. Although such mechanisms could explain the upregulation of receptors on the side of inflammation, it is intriguing to consider which mechanisms are possibly involved in the upregulation of the BK-binding and SP-binding sites on the contralateral side. One possibility is that the process of disease was spreading to the contralateral side and thereby stimulated receptor upregulation in contralateral DRGs. Indeed, during unilateral inflammation, cellular infiltration [74], SP and calcitonin gene-related peptide upregulation [55], an increase in the interleukin-6 content [75], a loss of proteoglycans [10] and altered load-bearing [76] have been observed on the contralateral side. However, during AIA the contralateral knee did not show gross inflammatory alterations; it is therefore questionable whether the enhanced receptor expression in the contralateral DRGs was due to the spreading of the inflammatory disease to the contralateral side.
Because the upregulation was symmetrical and segmental, it is also possible that the upregulation was caused by the symmetrical innervation. In general, bilateral changes in the nervous system after unilateral pathologies such as nerve lesions and inflammation are not uncommon, although they are not always observed [77,78]. Symmetrical efferent effects on the tissue and/or the DRGs can be provided by the sympathetic nervous system [79,80] or by the primary afferent fibres in which dorsal root reflexes are generated. Dorsal root reflexes are action potentials that originate in the spinal cord entry zone of the afferent fibres and propagate from there to the periphery [81,82,83]. Both of these neuronal pathways depend on activity in the spinal cord; it has been shown that the development of joint inflammation can be attenuated by the spinal application of glutamate receptor antagonists that reduce the activity of the spinal cord neurons [84, 85]. The involvement of the central nervous system in the generation of bilateral inflammation [86] and bilateral degeneration of articular cartilage [87] has been shown; it is therefore conceivable that efferent activity from the central nervous system to the periphery might also influence the expression of receptors in DRG neurons.
Acknowledgments
Acknowledgements
R Bräuer and PK Petrow were supported by grants from the German Ministry of Education and Research (FKZ 01ZZ 9602). We thank Dr R Vollandt (Institut für Medizinische Statistik, Informatik und Dokumentation, University of Jena) for performing the statistical evaluation, and Mrs H Kümpel and Mrs H Börner for excellent technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (Scha 404/9-2).
References
- Dray A. Tasting the inflammatory soup: the role of peripheral neurones. Pain Rev. 1994;1:153–171. [Google Scholar]
- Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- Mense S. Nociception from skeletal muscle in relation to clinical muscle pain. Pain. 1993;54:241–289. doi: 10.1016/0304-3959(93)90027-M. [DOI] [PubMed] [Google Scholar]
- Millan M. The induction of pain: an integrative review. . Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- Schaible H-G, Grubb BD. Afferent and spinal mechanisms of joint pain. Pain. 1993;55:5–54. doi: 10.1016/0304-3959(93)90183-P. [DOI] [PubMed] [Google Scholar]
- Dray A, Perkins M. Bradykinin and inflammatory pain. . Trends Neurosci. 1993;16:99–104. doi: 10.1016/0166-2236(93)90133-7. [DOI] [PubMed] [Google Scholar]
- Buchner E, Bräuer R, Emmrich F, Kinne RW. Induction of flare-up reactions in rat antigen-induced arthritis. J Autoimmun. 1995;8:61–74. [PubMed] [Google Scholar]
- Dumonde DC, Glynn LE. The production of arthritis in rabbits by an immunological reaction to fibrin. Br J Exp Pathol. 1962;43:373–383. [PMC free article] [PubMed] [Google Scholar]
- Griffith RJ. Characterisation and pharmacological sensitivity of antigen arthritis induced by methylated bovine serum albumin in the rat. . Agents Actions. 1992;35:88–95. doi: 10.1007/BF01990957. [DOI] [PubMed] [Google Scholar]
- Meyer P, Burkhardt H, Palombo-Kinne E, Gründer W, Bräuer R, Stiller KJ, Kalden JR, Becker W, Kinne RW. 123I-antileukoproteinase scintigraphy reveals microscopic cartilage alterations in the contralateral knee joint of rats with 'monarticular' antigen-induced arthritis. . Arthritis Rheum. 2000;43:298–310. doi: 10.1002/1529-0131(200002)43:2<298::AID-ANR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Steranka LR, Manning DC, DeHaas CJ, Ferkany JW, Borosky SA, Connor JR, Vavrek RJ, Stewart JM, Snyder SH. Bradykinin as a pain mediator: receptors are localized to sensory neurons, and antagonists have analgesic action. Proc Natl Acad Sci USA. 1988;85:3245–3249. doi: 10.1073/pnas.85.9.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaka R, Schaible H-G, Schmidt RF. Activation of articular afferent units by bradykinin. Brain Res. 1985;327:81–90. doi: 10.1016/0006-8993(85)91501-x. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Schaible H-G, Schmidt RF. Sensitization of articular afferents to mechanical stimuli by bradykinin. Eur J Physiol. 1989;415:330–335. doi: 10.1007/BF00370884. [DOI] [PubMed] [Google Scholar]
- Di Rosa M, Giroud JP, Willoughby DA. Studies of the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J Pathol. 1971;104:15–29. doi: 10.1002/path.1711040103. [DOI] [PubMed] [Google Scholar]
- Hendersen B, Pettipher ER, Higgs GA. Mediators of rheumatoid arthritis. Br Med Bull. 1987;43:415–428. doi: 10.1093/oxfordjournals.bmb.a072191. [DOI] [PubMed] [Google Scholar]
- Melmom KL, Webster ME, Godlfinger SE, Seegmiller JE. The presence of a kinin in inflammatory synovial effusion from arhritides of varying etiologies. Arthritis Rheum. 1967;10:13–20. doi: 10.1002/art.1780100103. [DOI] [PubMed] [Google Scholar]
- Regoli D. Kinins. Br Med Bull. 1987;43:270–284. doi: 10.1093/oxfordjournals.bmb.a072182. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Perkins MN. Induction of B1 receptors in vivo in a model of persistent inflammatory mechanical hyperalgesia in the rat. Neuropharmacology. 1994;33:127–133. doi: 10.1016/0028-3908(94)90107-4. [DOI] [PubMed] [Google Scholar]
- Perkins MN, Campbell E, Dray A. Antinociceptive activity of the bradykinin B1 and B2 receptor antagonists, des-Arg9, [Leu8]-BK and HOE 140, in two models of persistent hyperalgesia in the rat. Pain. 1993;53:191–197. doi: 10.1016/0304-3959(93)90080-9. [DOI] [PubMed] [Google Scholar]
- Hökfelt T. Neuropeptides in perspective: the last ten years. . Neuron. 1991;7:867–879. doi: 10.1016/0896-6273(91)90333-u. [DOI] [PubMed] [Google Scholar]
- Willis WD, Coggeshall RE. Sensory Mechanisms of the Spinal Cord. edn 2. New York: Plenum Press; 1991.
- Lawson SN, Crepps BA, Perl ER. Relationship of substance P to afferent characteristics of dorsal root ganglion neurones in guinea-pig. . J Physiol. 1997;505:177–191. doi: 10.1111/j.1469-7793.1997.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P. Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- Lam FY, Ferrell WR. Specific neurokinin receptors mediate plasma extravasation in the rat knee joint. Br J Pharmacol. 1991;103:1263–1267. doi: 10.1111/j.1476-5381.1991.tb12334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Clark R, Devor M, Helms C, Moskowitz MA, Basbaum AI. Intraneuronal substance P contributes to the severity of experimental arthritis. Science. 1984;226:547–549. doi: 10.1126/science.6208609. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE, Simone DA. Receptor endocytosis and dendritic reshaping in spinal neurons after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Schaible H-G, Weiretter F, Freudenberger U. The involvement of substance P and neurokinin-1 receptors in the responses of rat dorsal horn neurons to noxious but not to innocuous mechanical stimuli applied to the knee joint. Brain Res. 1994;666:207–215. doi: 10.1016/0006-8993(94)90774-9. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Weiretter F, Schaible H-G. The involvement of substance P and neurokinin-1 receptors in the hyperexcitability of dorsal horn neurons during development of acute arthritis in rat's knee joint. . J Neurophysiol. 1995;73:1574–1583. doi: 10.1152/jn.1995.73.4.1574. [DOI] [PubMed] [Google Scholar]
- Thompson SWN, Dray A, Urban L. Injury-induced plasticity of spinal reflex activity: NK1 neurokinin receptor activation and enhanced A- and C-fiber mediated responses in the rat spinal cord in vitro. . J Neurosci. 1994;14:3672–3687. doi: 10.1523/JNEUROSCI.14-06-03672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh T, Nagaswa T, Kuraishi Y. Expression of tachykinin NK-1 receptor mRNA in dorsal root ganglia of the mouse. Mol Brain Res. 1996;35:329–332. doi: 10.1016/0169-328x(95)00244-m. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Coggeshall RE. Localization and activation of substance P receptors in unmyelinated axons of rat glabrous skin. Brain Res. 1996;734:103–108. [PubMed] [Google Scholar]
- Hu H-Z, Li Z-W, Si J-Q. Evidence for the existence of substance P autoreceptors in the membrane of rat dorsal root ganglion neurons. . Neuroscience. 1997;77:535–541. doi: 10.1016/s0306-4522(96)00451-4. [DOI] [PubMed] [Google Scholar]
- Segond von Banchet G, Petersen M, Schaible H-G. Expression of neurokinin 1 receptors on cultured dorsal root ganglion neurons form the adult rat. Neuroscience. 1999;90:677–684. doi: 10.1016/s0306-4522(98)00408-4. [DOI] [PubMed] [Google Scholar]
- Heppelmann B, Pawlak M. Sensitisation of articular afferents in normal and inflamed knee joints by substance P in the rat. . Neurosci Lett. 1997;223:97–100. doi: 10.1016/s0304-3940(97)13408-5. [DOI] [PubMed] [Google Scholar]
- Kessler W, Kirchhoff C, Reeh PW, Handwerker HO. Excitation of cutaneous afferent nerve endings in vitro by a combination of inflammatory mediators and conditioning effect of substance P. Exp Brain Res. 1992;91:467–476. doi: 10.1007/BF00227842. [DOI] [PubMed] [Google Scholar]
- Cohen RH, Perl ER. Contributions of arachidonic acid derivatives and substance P to the sensitization of cutaneous nociceptors. J Neurophysiol. 1990;64:457–464. doi: 10.1152/jn.1990.64.2.457. [DOI] [PubMed] [Google Scholar]
- Kumazawa T, Mizumura K. Effects of synthetic substance P on unit discharges of testicular nociceptors of dogs. Brain Res. 1979;170:553–557. doi: 10.1016/0006-8993(79)90974-0. [DOI] [PubMed] [Google Scholar]
- Petersen M, Eckert AS, Segond von Banchet G, Heppelmann B, Klusch A, Kniffki KD. Plasticity in the expression of bradykinin binding sites in sensory neurons after mechanical nerve injury. Neuroscience. 1998;83:949–959. doi: 10.1016/s0306-4522(97)00465-x. [DOI] [PubMed] [Google Scholar]
- Petersen M, Segond von Banchet G, Heppelmann B, Koltzenburg M. Nerve growth factor regulates the expression of bradykinin binding sites on adult sensory neurons via the neurotrophin receptor p75. . Neuroscience. 1998;83:161–168. doi: 10.1016/s0306-4522(97)00374-6. [DOI] [PubMed] [Google Scholar]
- Segond von Banchet G, Petersen M, Heppelmann B. Bradykinin receptors at cultured rat dorsal root ganglion cells: influence of length of time in culture. Neuroscience. 1996;75:1211–1218. doi: 10.1016/0306-4522(96)00346-6. [DOI] [PubMed] [Google Scholar]
- Horn M, Vollandt R. Multiple Tests und Auswahlverfahren. Stuttgart: Gustav Fischer Verlag; 1995.
- Cesare P, Moriondo A, Vellani V, McNaughton PA. Ion channels gated by heat. Proc Natl Acad Sci USA. 1999;96:7658–7663. doi: 10.1073/pnas.96.14.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Dib-Hajj S, Cummins DR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. . Neuroscience. 1996;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress M, Reeh P, Vyklicky L. An interaction of inflammatory mediators and protons in small diameter dorsal root ganglion neurons of the rat. Neurosci Lett. 1997;224:37–40. doi: 10.1016/s0304-3940(97)13450-4. [DOI] [PubMed] [Google Scholar]
- Vyklicky L, Knotkova-Urbancova H, Vitaskova Z, Vlachova V, Kress M, Reeh PW. Inflammatory mediators at acidic pH activate capsaicin receptors in cultured sensory neurons from newborn rats. J Neurophysiol . 1998;79:670–676. doi: 10.1152/jn.1998.79.2.670. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Vasko MR. Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. . Brain Res. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3', 5'-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brechenmacher C, Larmet Y, Feltz P, Rodeau JL. Cultured rat sensory neurones express functional tachykinin receptor subtypes 1, 2 and 3. . Neurosci Lett. 1998;241:159–162. doi: 10.1016/s0304-3940(98)00045-7. [DOI] [PubMed] [Google Scholar]
- Brown JL, Liu H, Maggio JE, Vigna SR, Mantyh PW, Basbaum AI. Morphological characterization of substance P receptor-immunoreactive neurons in the rat spinal cord and trigeminal nucleus caudalis. J Comp Neurol. 1995;356:327–344. doi: 10.1002/cne.903560302. [DOI] [PubMed] [Google Scholar]
- Helke CJ, Charlton CG, Wiley RG. Studies on the cellular localization of spinal cord substance P receptors. Neuroscience. 1986;19:523–533. doi: 10.1016/0306-4522(86)90278-2. [DOI] [PubMed] [Google Scholar]
- Yashpal K, Dam TV, Quirion R. Effects of dorsal rhizotomy on neurokinin receptor subtypes in the rat spinal cord: a quantitative autoradiographic study. Brain Res. 1991;552:240–247. doi: 10.1016/0006-8993(91)90088-d. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Bowery NG. Peptide autoreceptors: does an autoreceptor for substance P exist? Trends Pharmacol Sci. 1999;20:405–407. doi: 10.1016/s0165-6147(99)01388-7. [DOI] [PubMed] [Google Scholar]
- Donaldson LF, Harmar AJ, McQueen DS, Seckl JR. Increased expression of preprotachykinin, calcitonin gene-related peptide, but not vasoactive intestinal peptide messenger RNA in dorsal root ganglia during the development of adjuvant monoarthritis in the rat. Mol Brain Res . 1992;16:143–149. doi: 10.1016/0169-328x(92)90204-o. [DOI] [PubMed] [Google Scholar]
- Hanesch U, Blecher F, Stiller RU, Emson PC, Schaible H-G, Heppelmann B. The effect of a unilateral inflammation at the rat's ankle joint on the expression of preprotachykinin-A mRNA and presomatostatin mRNA in dorsal root ganglion cells — a study using non-radioactive in situ hybridization. Brain Res. 1995;700:279–284. doi: 10.1016/0006-8993(95)01047-y. [DOI] [PubMed] [Google Scholar]
- Kar S, Rees R, Quirion R. Altered calcitonin gene-related peptide, substance P and enkephalin immunoreactivities and receptor binding sites in the dorsal spinal cord of the polyarthritic rat. Eur J Neurosci. 1994;6:345–354. doi: 10.1111/j.1460-9568.1994.tb00277.x. [DOI] [PubMed] [Google Scholar]
- Mapp PI, Terenghi G, Walsh DA, Chen ST, Cruwys SC, Garrett N, Kidd BL, Polak JM, Blake DR. Monoarthritis in the rat knee induces bilateral and time-dependent changes in substance P and calcitonin gene-related peptide immunoreactivity in the spinal cord. Neuroscience. 1993;57:1091–1096. doi: 10.1016/0306-4522(93)90051-g. [DOI] [PubMed] [Google Scholar]
- Minami M, Kuraishi Y, Kawamura M, Yamaguchi T, Masu Y, Nakanishi S, Satoh M. Enhancement of preprotachykinin A gene expression by adjuvant-induced inflammation in the rat spinal cord: possible involvement of substance P-containing spinal neurons in nociception. Neurosci Lett. 1989;98:105–110. doi: 10.1016/0304-3940(89)90382-0. [DOI] [PubMed] [Google Scholar]
- Neumann S, Doubell TP, Leslie T, Woolf CJ. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature. 1996;384:360–364. doi: 10.1038/384360a0. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Dougherty PM, Sorkin LS, Willis WD, Westlund KN. Neural changes in acute arthritis in monkeys. III. Changes in substance P, calcitonin gene-related peptide and glutamate in the dorsal horn of the spinal cord. Brain Res Rev. 1992;17:29–38. doi: 10.1016/0165-0173(92)90004-6. [DOI] [PubMed] [Google Scholar]
- Smith GD, Harmar AJ, McQueen DS, Seckl JR. Increase in substance P and CGRP, but not somatostatin content of innervating dorsal root ganglia in adjuvant monoarthritis in the rat. Neurosci Lett. 1992;137:257–260. doi: 10.1016/0304-3940(92)90417-6. [DOI] [PubMed] [Google Scholar]
- McCarson KE, Krause JE. NK-1 and NK-3 type tachykinin receptor mRNA expression in the rat spinal cord dorsal horn is increased during adjuvant or formalin-induced nociception. J Neurosci. 1994;14:712–720. doi: 10.1523/JNEUROSCI.14-02-00712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer MK-H, Nohr D, Krause JE, Weihe E. Inflammation-induced upregulation of NK-1 receptor mRNA in dorsal horn neurones. NeuroReport. 1993;4:1007–1010. doi: 10.1097/00001756-199308000-00003. [DOI] [PubMed] [Google Scholar]
- Stucky CL, Galeazza MT, Seybold VS. Time-dependent changes in Bolton-Hunter-labeled 125I-substance P binding in rat spinal cord following unilateral adjuvant-induced peripheral inflammation. Neuroscience. 1993;57:397–409. doi: 10.1016/0306-4522(93)90071-m. [DOI] [PubMed] [Google Scholar]
- Lopes P, Kar S, Chretien L, Regoli D, Quirion R, Couture R. Quantitative autoradiographic localization of [125l-Tyr8] bradykinin receptor binding sites in the rat spinal cord: effects of neonatal capsaicin, noradrenergic deafferentiation, dorsal rhizotomy and peripheral axotomy. . Neuroscience. 1995;68:867–881. doi: 10.1016/0306-4522(95)00161-b. [DOI] [PubMed] [Google Scholar]
- Eckert A, Segond von Banchet G, Sopper S, Petersen M. Spatio-temporal pattern of induction of bradykinin receptors and inflammation in rat dorsal root ganglia after unilateral nerve ligation. Pain. 1999;83:487–497. doi: 10.1016/S0304-3959(99)00152-9. [DOI] [PubMed] [Google Scholar]
- Aloe L, Tuveri MA, Levi-Montalcini R. Studies on carrageenan-induced arthritis in adult rats: presence of nerve growth factor and of sympathetic innervation. Rheumatol Int. 1992;12:213–216. doi: 10.1007/BF00302155. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Otten U. An enzyme-linked immunoassay for nerve growth factor (NGF): a tool for studying regulatory mechanisms involved in NGF production in brain and in peripheral tissues. J Neurochem. 1987;48:1779–1786. doi: 10.1111/j.1471-4159.1987.tb05736.x. [DOI] [PubMed] [Google Scholar]
- Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;33:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- Donnerer J, Schuligoi R, Stein C. Increased content and transport of substance P and calcitonin gene-related peptide in sensory nerves innervating inflamed tissue: evidence for a regulatory function of nerve growth factor in vivo. Neuroscience. 1992;49:693–698. doi: 10.1016/0306-4522(92)90237-v. [DOI] [PubMed] [Google Scholar]
- Kidd BL, Cruwys SC, Garrett NE, Mapp PI, Jolliffe VA, Blake DR. Neurogenic influences on contralateral responses during experimental rat monoarthritis. Brain Res. 1995;688:72–76. doi: 10.1016/0006-8993(95)00512-o. [DOI] [PubMed] [Google Scholar]
- Mentzel K, Bräuer R. Matrix metalloproteinases, IL-6, and nitric oxide in rat antigen-induced arthritis. Clin Exp Rheum. 1998;16:269–276. [PubMed] [Google Scholar]
- Schott E, Berge OG, Angeby-Moller K, Hammarstrom G, Dalsgaard CJ, Brodin E. Weight bearing as an objective measure of arthritic pain in the rat. J Pharmacol Toxicol Meth. 1994;31:79–83. doi: 10.1016/1056-8719(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Donaldson LF. Unilateral arthritis: contralateral effects. . Trends Neurosci. 1999;22:495–496. doi: 10.1016/s0166-2236(99)01481-2. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Wall PD, McMahon SB. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–127. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- Levine JD, Dardick SJ, Roizen MF, Helms C, Basbaum AI. Contribution of sensory afferents and sympathetic efferents to joint injury in experimental arthritis. J Neurosci. 1986;6:3423–3429. doi: 10.1523/JNEUROSCI.06-12-03423.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Goetzl EJ, Basbaum AI. Contribution of the nervous system to the pathophysiology of rheumatoid arthritis and other polyarthritides. Rheum Dis Clin N Am. 1987;13:369–383. [PubMed] [Google Scholar]
- Rees H, Sluka KA, Westlund KN, Willis WD. Dorsal root reflexes in articular nerves occur bilaterally in a chronic model of arthritis in rats. J Neurophysiol. 1996;76:4190–4193. doi: 10.1152/jn.1996.76.6.4190. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Rees H, Westlund KN, Willis WD. Fiber types contributing to dorsal root reflexes induced by joint inflammation in cats and monkeys. J Neurophysiol. 1995;74:981–989. doi: 10.1152/jn.1995.74.3.981. [DOI] [PubMed] [Google Scholar]
- Willis WD. Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res. 1999;124:395–421. doi: 10.1007/s002210050637. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Jordan HH, Westlund KN. Reduction in joint swelling and hyperalgesia following post-treatment with a non-NMDA glutamate receptor antagonist. Pain. 1994;59:95–100. doi: 10.1016/0304-3959(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Lawand NB, Westlund KN. Joint inflammation is reduced by dorsal rhizotomy and not by sympathectomy or spinal cord transection. . Ann Rheum Dis. 1994;53:309–314. doi: 10.1136/ard.53.5.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bileviciute I, Stenfors C, Theodorsson E, Lundeberg T. Unilateral injection of calcitonin gene-related peptide (CGRP) induces bilateral oedema formation and release of CGRP-like immunoreactivity in the rat hindpaw. Br J Pharmacol. 1998;125:1304–1312. doi: 10.1038/sj.bjp.0702192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaris E, Guingamp C, Chat M, Philippe L, Grillasca J-P, Abid A, Minn A, Gillet P, Netter P, Terlain B. Evidence for neurogenic transmission inducing degenerative cartilage damage distant from local inflammation. Arthritis Rheum. 1999;42:1951–1960. doi: 10.1002/1529-0131(199909)42:9<1951::AID-ANR22>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]