

Short abstract
We investigated the role of Fcγ receptors (FcγRs) on synovial macrophages in immune-complex-mediated arthritis (ICA). ICA elicited in knee joints of C57BL/6 mice caused a short-lasting, florid inflammation and reversible loss of proteoglycans (PGs), moderate chondrocyte death, and minor erosion of the cartilage. In contrast, when ICA was induced in knee joints of Fc receptor (FcR) γ-chain-/- C57BL/6 mice, which lack functional FcγRI and RIII, inflammation and cartilage destruction were prevented. When ICA was elicited in DBA/1 mice, a very severe, chronic inflammation was observed, and significantly more chondrocyte death and cartilage erosion than in arthritic C57BL/6 mice. The synovial lining and peritoneal macrophages of naïve DBA/1 mice expressed a significantly higher level of FcγRs than was seen in C57BL/6 mice. Moreover, elevated and prolonged expression of IL-1 was found after stimulation of these cells with immune complexes. Zymosan or streptococcal cell walls caused comparable inflammation and only mild cartilage destruction in all strains. We conclude that FcγR expression on synovial macrophages may be related to the severity of synovial inflammation and cartilage destruction during ICA.
Keywords: autoimmunity, cytokines, Fc receptors, inflammation, macrophages
Abstract
Introduction:
Fcγ receptors (FcγRs) present on cells of the haematopoietic lineage communicate with IgG-containing immune complexes that are abundant in the synovial tissue of patients with rheumatoid arthritis (RA). In mice, three classes of FcγR (RI, RII, and RIII) have been described. Binding of these receptors leads to either activation (FcγRI and RIII) or deactivation (FcγRII) of intracellular transduction pathways. Together, the expression of activating and inhibitory receptors is thought to drive immune-complex-mediated diseases.
Earlier studies in our laboratory showed that macrophages of the synovial lining are of utmost importance in the onset and propagation of immune-complex-driven arthritic diseases. Selective depletion of macrophages in the joint downregulated both inflammation and cartilage destruction. As all three classes of FcγR are expressed on synovial macrophages, these cells are among the first that come in contact with immune complexes deposited in the joint. Recently, we observed that when immune complexes were injected into the knee joints of mice, strains susceptible to collagen-type-II arthritis (DBA/1, B10.RIII) developed more severe arthritis than nonsusceptible strains did, or even developed chronic arthritis. One reason why these strains are more susceptible might be their higher levels of FcγRs on macrophage membranes. To test this hypothesis, we investigated the role of FcγRs in inflammation and cartilage damage during immune-complex-mediated arthritis (ICA). First, we studied arthritis and subsequent cartilage damage in mice lacking functional FcγRI and RIII (FcR γ-chain-/- mice). Next, DBA/1 mice, which are prone to develop collagen-type-II arthritis (`collagen-induced arthritis'; CIA) and are hypersensitive to immune complexes, were compared with control C57BL/6 mice as regards cartilage damage and the expression and function of FcγRs on their macrophages.
Aims:
To examine whether FcγR expression on macrophages is related to severity of synovial inflammation and cartilage destruction during immune-complex-mediated joint inflammation.
Methods:
ICA was induced in three strains of mice (FcR γ-chain-/-, C57BL/6, and DBA/1, which have, respectively, no functional FcγRI and RIII, intermediate basal expression of FcγRs, and high basal expression of FcγRs) by passive immunisation using rabbit anti-lysozyme antibodies, followed by poly-L-lysine lysozyme injection into the right knee joint 1 day later. In other experiments, streptococcal-cell-wall (SCW)- or zymosan-induced arthritis was induced by injecting SCW (25 μg) or zymosan (180 μg) directly into the knee joint. At several time points after arthritis induction, knee joints were dissected and studied either histologically (using haematoxylin/eosin or safranin O staining) or immuno-histochemically. The arthritis severity and the cartilage damage were scored separately on an arbitrary scale of 0-3.
FcγRs were immunohistochemically detected using the monoclonal antibody 2.4G2, which detects both FcγRII and RIII. Deposition of IgG and C3c in the arthritic joint tissue was also detected immunohistochemically. Expression of FcγRs by murine peritoneal macrophages was measured using a fluorescence-activated cell sorter (FACS).
Peritoneal macrophages were stimulated using heat-aggregated gamma globulins (HAGGs), and production of IL-1 was measured using a bioassay. To assess the levels of IL-1 and its receptor antagonist (IL-1Ra) during arthritis, tissue was dissected and washed in RPMI medium. Washouts were tested for levels of IL-1 and IL-1Ra using radioimmunoassay and enzyme-linked immunosorbent assay. mRNA was isolated from the tissue, and levels of macrophage inflammatory protein (MIP)-2, monocyte chemoattractant protein (MCP)-1, IL-1, and IL-1Ra were determined using semiquantitative reverse-transcription polymerase chain reaction (RT-PCR).
Results:
ICA induced in knee joints of C57BL/6 mice caused a florid inflammation at day 3 after induction. To investigate whether this arthritis was FcγR-mediated, ICA was induced in FcR γ-chain-/- mice, which lack functional FcγRI and RIII. At day3, virtually no inflammatory cells were found in their knee joints. Levels of mRNA of IL-1, IL-1Ra, MCP-1, and MIP-2, which are involved in the onset of this arthritis, were significantly lower in FcR γ-chain-/- mice than in control C57BL/6 mice. Levels of IL-1 protein were also measured. At 6 h after ICA induction, FcR γ-chain-/- mice and control C57BL/6 mice showed similar IL-1 production as measured by protein level. By 24 h after induction, however, IL-1 production in the FcR γ-chain-/- mice was below the detection limit, whereas the controls were still producing a significant amount. To investigate whether the difference in reaction to immune complexes between the DBA/1 and C57BL/6 mice might be due to variable expression of FcγRs in the knee joint, expression in situ of FcγRs in naïve knee joints of these mice was determined. The monoclonal antibody 2.4G2, which detects both FcγRII and RIII, stained macrophages from the synovial lining of DBA/1 mice more intensely than those from C57BL/6 mice. This finding suggests a higher constitutive expression of FcγRs by macrophages of the autoimmune-prone DBA/1 mice. To quantify the difference in FcγR expression on macrophages of the two strains, we determined the occurrence of FcγRs on peritoneal macrophages by FACS analysis. The levels of FcγR expressed by macrophages were twice as high in the DBA/1 mice as in the C57BL/6 mice (mean fluorescence, respectively, 440 ± 50 and 240 ± 30 intensity per cell). When peritoneal macrophages of both strains were stimulated with immune complexes (HAGGs), we found that the difference in basal FcγR expression was functional. The stimulated macrophages from DBA/1 mice had significantly higher IL-1α levels (120 and 135 pg/ml at 24 and 48 h, respectively) than cells from C57BL/6 mice (45 and 50 pg/ml, respectively).
When arthritis was induced using other arthritogenic triggers than immune complexes (zymosan, SCW), all the mouse strains tested (DBA/1, FcR γ-chain-/-, and C57BL/6) showed similar inflammation, indicating that the differences described above are found only when immune complexes are used to elicit arthritis.
We next compared articular cartilage damage in arthritic joints of the three mouse strains FcR γ-chain-/-, C57BL/6 (intermediate basal expression of FcγRs), and DBA/1 (high basal expression of FcγRs). Three indicators of cartilage damage were investigated: depletion of PGs, chondrocyte death, and erosion of the cartilage matrix. At day 3 after induction of ICA, there was no PG depletion in FcR γ-chain-/- mice, whereas PG depletion in the matrix of the C57BL/6 mice was marked and that in the arthritic DBA/1 mice was even greater. PG depletion was still massive at days 7 and 14 in the DBA/1 mice, whereas by day 14 the PG content was almost completely restored in knee joints of the C57BL/6 mice. Chondrocyte death and erosion of cartilage matrix, two indicators of more severe cartilage destruction, were significantly higher in the DBA/1 than in the C57BL/6 mice, while both indicators were completely absent in the FcR γ-chain-/- mice. Again, when arthritis was induced using other triggers (SCW, zymosan), all strains showed similar PG depletion and no chondrocyte death or matrix erosion. These findings underline the important role of immune complexes and FcγRs in irreversible cartilage damage.
Discussion:
Our findings indicate that inflammation and subsequent cartilage damage caused by immune complexes may be related to the occurrence of FcγRs on macrophages. The absence of functional FcγRI and RIII prevented inflammation and cartilage destruction after induction of ICA, whereas high basal expression of FcγRs on resident joint macrophages of similarly treated mice susceptible to autoimmune arthritis was correlated with markedly more synovial inflammation and cartilage destruction. The difference in joint inflammation between the three strains was not due to different susceptibilities to inflammation per se, since intra-articular injection of zymosan or SCW caused comparable inflammation. Although extensive inflammatory cell mass was found in the synovium of all strains after intra-articular injection of zymosan, no irreversible cartilage damage (chondrocyte death or matrix erosion) was found. ICA induced in C57BL/6 and DBA/1 mice did cause irreversible cartilage damage at later time points, indicating that immune complexes and FcγRs play an important role in inducing irreversible cartilage damage. Macrophages communicate with immune complexes via Fcγ receptors. Absence of functional activating receptors completely abrogates the synovial inflammation, as was shown after ICA induction in FcR γ-chain-/- mice. However, the γ-chain is essential not only in FcγRI and RIII but also for FcεRI (found on mast cells) and the T cell receptor (TcR)-CD3 (Tcells) complex of γδT cells. However, T, B, or mast cells do not play a role in this arthritis that is induced by passive immunisation. Furthermore, this effect was not caused by a difference in clearance of IgG or complement deposition in the tissue. In this study, DBA/1 mice, which are susceptible to collagen-induced autoimmune arthritis and in a recent study have been shown to react hypersensitively to immune complexes, are shown to express higher levels of FcγRs on both synovial and peritoneal macrophages. Because antibodies directed against the different subclasses of FcγR are not available, no distinction could be made between FcγRII and RIII. Genetic differences in DBA/1 mice in genes coding for or regulating FcγRs may be responsible for altered FcγR expression. If so, these mouse strains would have a heightened risk for immune-complex-mediated diseases.
To provide conclusive evidence for the roles of the various classes of FcγR during ICA, experiments are needed in which FcγRs are blocked with specific antibodies, or in which knockout mice lacking one specific class of FcγR are used. The only available specific antibody to FcγR (2.4G2) has a stimulatory effect on cells once bound to the receptor, and therefore cannot be used in blocking experiments. Experiments using specific knockout mice are now being done in our laboratory.
Macrophages are the dominant type of cell present in chronic inflammation during RA and their number has been shown to correlate well with severe cartilage destruction. Apart from that, in humans, these synovial tissue macrophages express activating FcRs, mainly FcγIIIa, which may lead to activation of these macrophages by IgG-containing immune complexes. The expression of FcRs on the surface of these cells may have important implications for joint inflammation and severe cartilage destruction and therefore FCRs may constitute a new target for therapeutic intervention.
Introduction
Rheumatoid arthritis (RA) is characterised by chronic joint inflammation eventually leading to irreversible cartilage destruction. IgG-containing immune complexes, such as rheumatoid factors, are abundant in the synovial tissue of patients with RA [1,2]. It is still debated what role these complexes play in the aetiology and pathology of the disease. Immune complexes communicate with haematopoietic cells via Fcγ receptors (FcγRs) [3,4,5]. In recent studies, the importance of these receptors in inflammation and tissue damage has been shown in various inflammatory diseases, eg autoimmune haemolytic anaemia and thrombocytopenia [6,7], autoimmune glomerulonephritis [8], and induced glomerulonephritis [9,10]. FcγRs, which belong to the immunoglobulin superfamily, bind IgG. Three classes of leucocyte FcγR have been described (FcγRI, RII, and RIII). Binding of these receptors initiates signalling cascades that can lead to either activation or deactivation of effector cells. In mice, cross-linking of FcγRI and RIII leads to activation [11,12] of intracellular signalling transduction pathways, whereas stimulation of FcγRIIb leads to their deactivation [13,14]. Coordinate expression of activating and inhibitory receptors has been suggested to drive immune-complex-mediated diseases [15].
During RA, the dominant cell type in the joint is the macrophage. Elegant studies by Bresnihan's group have shown a correlation between the number of macrophages present in the joint and severe cartilage destruction [16,17,18]. In other studies, selective elimination of synovial lining macrophages prior to induction of experimental arthritis prevented the onset of arthritis and cartilage destruction. Furthermore, selective removal of lining macrophages during chronic arthritis significantly downregulated synovial inflammation [19,20,21,22] and partly prevented exacerbation [23]. Synovial macrophages, which cover the inside of diarthrodial joints and surround blood vessels in the synovium, are among the first cells that come in contact with immune complexes. All three classes of FcγR are known to be expressed on macrophages in general and on synovial macrophages in particular [4,24,25,26] and the amount and/or ratio of the various types of FcγR expression might determine joint inflammation and cartilage destruction. Overexpression, downregulation, or functional impairment of activating FcγRI and RIII on macrophages may have consequences for inflammation and cartilage destruction. Differences in basal expression levels of FcγRs may be genetically linked or related to age [27]. Cytokines such as IFN-γ and TGF-β have been shown to upregulate, respectively, FcγRI and RIII, whereas IL-4 and IL-13 down-regulate both receptor classes [4,28,29,30,31,32,33].
Recently, we observed that when immune complexes were injected into the knee joints of mice, strains susceptible to collagen-type-II arthritis (DBA/1 and B10.RIII mice) developed more severe arthritis than nonsusceptible strains did, or even developed chronic arthritis [34]. Mouse strains that are prone to develop autoimmune arthritis may also express higher levels of FcγRs on their macrophages, thus driving joint inflammation and cartilage destruction.
To test this hypothesis, we have investigated the role of FcγRs in inflammation and cartilage damage during immune-complex-mediated arthritis (ICA). In this model, arthritis is induced by intra-articular injection of the antigen into knee joints of mice that were previously passively immunised against the antigen, by intravenous injection of specific antibodies [35]. First, synovial inflammation, cytokine production, and joint destruction were investigated in FcR γ-chain knockout (FcR γ-chain-/-) mice, lacking functional FcRI and RIII, and their controls. Then DBA/1 mice, which are prone to develop collagen-type-II autoimmune arthritis and express a higher than usual sensitivity for immune complexes, were compared with non-susceptible C57BL/6 mice, and the expression and functional relation of Fc receptors on macrophages with inflammation and cartilage destruction were investigated. The findings indicate that FcR expression on synovial-lining macrophages is related to the severity and chronicity of synovial inflammation and cartilage destruction during joint inflammation elicited by immune complexes.
Materials and methods
Animals
Male and female C57BL/6 mice were obtained from Jackson (Bar Harbor, Maine, USA). DBA/1 mice were obtained from Bomholdgard (Rye, Denmark). FcR γ-chain-/- mice were kindly given by T Saito [10]. FcR γ-chain-/- mice were backcrossed to C57BL/6 mice for 12 generations. FcR γ-chain-/- were also backcrossed to a DBA/1 background. Mice were fed a standard diet and tap water ad libitum. Mice were used between the ages of 8 and 12 weeks and weighed 25–30 g.
Chemicals
Poly-L-lysine (PLL), lysozyme, 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (EDC), and zymosan A (from Saccharomyces cerevisiae) were obtained from Sigma Chemical Company, St Louis, MO, USA. N,N-dimethyl-1,3-propanediamine (DMPA) was obtained from BDH Chemicals Ltd, Poole, UK.
Lysozyme coupling to PLL
Lysozyme was coupled to PLL in accordance with the method of Danon et al [36]. As described by those authors, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide was used as an activator and PLL as a nucleophil. Free carboxyl groups of the protein were then coupled to amino groups of PLL. The molecular mass was raised whereas the isoelectric point remained high, as was determined in a 5% polyacrylamide slab gel with 0.8% ampholines (pH gradient 3.5-9.5). The molecular mass appeared to be 74 kD on SDS-PAGE.
Induction of arthritis by immune complexes, zymosan, or SCW
Specific rabbit anti-lysozyme antisera, made as described elsewhere [35] and made complement-free by heating at 56°C for 30 min, were injected (0.2 ml) intravenously into mice. ICA was then induced by injecting 3 μg of PLL-coupled lysozyme into the right knee joint. The left knee joint was injected with saline solution and used as a control. As an additional control, some mice were given normal rabbit serum instead of specific anti-lysozyme. When PLL-lysozyme was injected into the knee joint without prior administration of specific anti-lysozyme antibodies, no inflammation or cartilage damage developed, in either C57BL/6 or DBA/1 mice.
Zymosan (30 mg) was dissolved in 1 ml saline solution by heating up to 100°C twice and was then sonicated to obtain a homogeneous suspension. Arthritis was induced in other mice by injecting 180 μg zymosan into both left and right knee joints. Fcγ-chain knockout mice and the control strain (C57BL/6) were matched for age and sex.
SCW arthritis was induced by injecting 25 μg SCWs (rhamnose content), prepared as described elsewhere [37] and dissolved in 6 μl saline, into the right knee joint of C57BL/6 and DBA/1 mice.
Histology
Total knee joints of mice were isolated 3 days after induction of arthritis in Fcγ-chain deficient mice and at days 3, 7, and 14 in C57BL/6 and DBA/1 mice. For standard histology, tissue was fixed in 4% formaldehyde, decalcified in formic acid, and subsequently dehydrated and embedded in paraffin. Paraffin sections were cut at 7 μm and mounted on gelatine-coated slides. Haematoxylin/eosin (H&E) staining was performed to study the inflammatory cells.
Infiltrate and exudate were scored separately. The severity was determined by two blinded observers, using an arbitrary score (0–3): 0 = none, 1 = mild, 2 = moderate, and 3 = maximal cellularity. To study PG depletion from the cartilage matrix, sections were stained with safranin O and then coun-terstained with fast green. PG depletion from several cartilage surfaces was scored (patella + adjacent femur surface, lateral condyle femur + adjacent surface tibia plateau, and medial femoral condyle + adjacent surface tibia plateau).
The severity of depletion was scored by two blinded observers, using an arbitrary score reflecting the level of destaining: 0 = none, 1 = mild, 2 = moderate, and 3 = maximal destaining of cartilage. Chondrocyte death was scored using H&E-stained sections. The amount of empty lacunae was given as a percentage of total amount of cells (empty lacunae + viable chondrocytes). Cartilage erosion was scored by expressing the amount of eroded cartilage as a percentage of the cartilage surface.
Detection of Fcγ receptors
To compare expression of FcγRII/RIII by DBA/1 and C57BL/6 mice, we performed immunohistochemistry using a rat antibody against murine FcγRII/RIII (2.4G2, Pharmin-gen, San Diego, CA, USA) [38]. Briefly, cryostat sections were fixed in acetone vapour for 3 min and endogenous peroxidase was blocked using 1% H2O2. Sections were incubated overnight with 2.4G2 (1 μg/ml) in phosphate-buffered saline (PBS)/0.1% bovine serum albumin (BSA). Sections were washed in PBS and then incubated with rabbit anti-rat IgG coupled to biotin (Vector Laboratories, Burlingame, CA, USA). Then the sections were incubated with avidin biotin complexes (ABC; Vectastain Elite-kit, Vector Laboratories) and developed using diaminobenzidine (Sigma Chemical Company, St Louis, MO, USA).
Detection of IgG and C3c
IgG deposited in the tissue by immune-complex formation was detected on paraffin-embedded knee-joint sections. Sections were incubated with biotinylated rat anti-rabbit antibodies (Vector Laboratories) for 1 h. Sections were washed thoroughly with PBS, incubated with ABC complexes, and developed using the method described above.
C3c was detected using cryostat sections. Sections were incubated with rat anti-murine C3c (Nordic, Tilburg, The Netherlands) after acetone fixation and inhibition of endogenous peroxidase. Isotype-matched antibodies directed against an irrelevant epitope (Vector) were used as a negative control. Staining was scored using image analysis by measuring the mean amount of blue light that passed through a well-defined location in the tissue section near the cruciate ligaments.
Analysis by fluorescence-activated cell sorter (FACS)
Peritoneal macrophages of different strains were isolated from the peritoneal cavity by lavage with ice-cold DMEM/ 10% fetal calf serum (FCS)/1% pyruvate. These cells (5 × 106/100 μl) were incubated with 2.4G2 (5 μg/ml). They were then washed and incubated with mouse-adsorbed hamster anti-rat F(ab')2 fragments labelled withfluorescein isothiocyanate (FITC). FACS analysis was performed using a Coulter Epics XL/XL-MCL (Coulter Electronics Ltd, Mijdrecht, The Netherlands). Omitting the first antibody and substitution of 2.4G2 for isotype-matched irrelevant antibodies (DAKO, Glostrup, Denmark) were used as negative controls. The window was set so that >95% of cells were F4/80-positive, indicating that >95% of cells were macrophages.
Stimulation of peritoneal macrophages with heat-aggregated gamma globulin
Peritoneal macrophages were isolated by peritoneal lavage using ice-cold DMEM/10% FCS. Cells were put into 24-well plates (Costar, Acton, MA, USA) at a concentration of 1 × 106 cells/ml. After a 4-day adjustment period, the culture medium was changed to one containing heat-aggregated gamma globulin (HAGG) at 100 μg/ml, or a control medium. After 24 or 48 h, the culture medium was collected. HAGGs were obtained by heating 10 mg/ml rabbit IgG (Sigma Chemicals, St Louis, MO, USA) to 63°C for 30 min. After heating, the solution was centrifuged at 12 000 g for 10 min. The concentration of the HAGG in the supernatant was determined by reading the absorbance at 280 nm.
Production of inflammatory mediators by synovial tissue
Synovial tissue was isolated by dissection of patellar tendon and patellar plate containing the patella, tendons, and synovium, as described earlier [39]. Mediators were obtained by elution from synovial specimens derived from six knee joints in 2 ml Roswell Park Memorial Institute (RPMI) medium for 1 h at room temperature. Washouts were tested for their bioactive IL-1 levels in an IL-1-sensitive bioassay (NOB-1 assay) and in a radioimmunoassay (RIA) to determine the total protein levels.
Bioassay for IL-1
IL-1 activity was measured in the one-stage bioassay for IL-1 as described by Gearing et al [40]. The assay was performed as a culture of the IL-1-specific thymoma cell EL-4, designated NOB-1, which produces IL-2 and IL-4 in response to IL-1, in co-culture with the lymphokine-responder CTLL line. Briefly, EL-4 cells were washed twice and resuspended at 2.5 × 105 cells per ml RPMI medium containing 5% FCS. The cells were distributed into 96-well microtitre plates at 2.5 × 104 cells per well in 100-μl volumes. CTLL cells (4 × 103) suspended in 50 μl RPMI medium were added, followed by appropriate dilutions of test sample to a final volume of 200 μl. After 20 h, 0.5 μCi 3H-thymidine (specific activity 20 Ci/mmol; Amersham Nederland BV, 's Hertogenbosch, The Netherlands) was added to each well. After a 3-h incubation, the contents were harvested, and incorporated activity was determined. The EL-4 line, from which NOB-1 was derived, does not incorporate thymidine, because it is deficient in thymidine kinase, and therefore the incorporation of 3H-thymidine is a measure of only CTLL proliferation. Maximal 3H-thymidine incorporation in the bioassay in the presence of IL-1 was between 15 000 and 20 000 cpm. Culture media contained only minor concentrations of IL-2 or IL-4, as was assessed by testing samples with CTLL alone.
Measurement of IL-1α and IL-1β protein levels
IL-1 culture supernatants were measured in duplicate by a nonequilibrium RIA as described elsewhere [41]. Briefly, 100 μl polyclonal rabbit anti-murine IL-1α and IL-1β (diluted in RIA buffer, pH 7.4) was added to 100 μl of samples and standards and kept on ice. After vortexing, the tubes were incubated at 4°C. After 24 h, 100 μl of the appropriate 125I-labelled IL-1α and β containing approximately 10 000 cpm was added to each tube, and incubation was continued for a further 24 h at 4°C. RIA buffer (750 μl) containing 9% (w/v) polyethylene glycol 6000 (Merck Diagnostica, Darmstadt, Germany) and 3% (w/v) goat anti-rabbit serum was added, to separate bound and free tracer. The tubes were incubated for 20 min at room temperature. After centrifugation at 1500 × g for 15 min, supernatants were quickly drained on adsorbent paper. A gamma-counter was used to count the radioactivity remaining on the paper. The radioactivity in control tubes (the nonspecific binding activity) was subtracted from samples and standards. The detection limit of the assay was 20 pg/ml for both IL-1α and IL-1β.
Measurement of IL-1 receptor antagonist
Protein levels of IL-1 receptor antagonist (IL-1Ra) in synovial washout specimens were detected using a specific sandwich enzyme-linked immunosorbent assay (ELISA). Briefly, microtitre plates were coated with unconjugated first antibody (anti-IL1Ra: MAB480, R&D Systems, Abingdon, UK) overnight at 4°C. Subsequently, wells were blocked using 1% BSA followed by a 3-h incubation with patella washouts at 37°C. Wells were then incubated with a biotinylated specific antibody (IL-1Ra: BAF480, R&D Systems) and a signal enhancement step was performed by incubating with PolyHRP (CLB, Amsterdam, The Netherlands). Microtitre plates were developed using o-phenylene-diamine and optical density was read at 492 nm. Between all steps, a washing episode was included using PBS/0.1% Tween 20.
Semiquantitative detection of mRNA using reverse-transcription polymerase chain reaction
Levels of mRNA for four different cytokines/chemokines were detected using a semiquantitative method. In brief, mice were killed by cervical dislocation, immediately followed by dissection of the patella with adjacent synovial tissue. Six patellae per group were obtained. Two biopsies with a diameter of 3 mm were punched from the synovial tissue, using a biopsy punch (Stiefel, Wachtersbach, Germany): one from the medial and one from the lateral aspect of the patella. Three lateral and three medial biopsies were pooled (six total), to obtain two samples per group. The samples were immediately frozen in liquid nitrogen. Thereafter samples were ground to powder using a Micro-dismembrator II (B Braun, Melsungen, Germany) and total RNA was extracted using 1 ml TRIzol reagent.
One microgram of RNA was used for reverse-transcription polymerase chain reaction (RT-PCR). Reverse transcription of mRNA was done using oligoDT primers, and 1/20 of the cDNA was used in one amplification by polymerase chain reaction (PCR). PCR was performed at a final concentration of 200 μM dNTPs, each primer at 0.1 μM, and 1unit Taq polymerase (Life Technologies, Breda, The Netherlands) in standard PCR buffer. Message for glycer-aldehyde-3-phosphate dehydrogenase (GAPDH), IL-1β, IL-1Ra, MCP-1, and MIP-2 was amplified using specific primers. The primer sequences for IL-1β were: 5'-TTGACGGACCCCAAAAGATG-3' (sense) and 5'-AGAAGGTGCTCATGTCCTCA-3' (antisense), for IL-1ra: 5'-TGCTGGGGACCCTACAGTCAC-3' (sense) and 5'-GCAAGTGCATCATCGTTGTTC-3' (antisense), for MCP-1: 5'-CTCACCTGCTGCTACTCATTC-3' (sense) and 5'-GCATGAGGTGGTTGTGAAAA-3' (antisense) and for MIP-2: 5'-GCTGGCCACCAACCACCAGG-3' (sense) and 5'-AGCGAGGCACATCAGGTACG-3' (antisense). The PCR-reaction was paused after 20, 23, 26, 29, 32, 35, 38, 41, and 44 cycles at the very end of the extension phase (72°C) and 5-μl samples were taken. PCR products were separated on 1.6% agarose gel and stained with ethidium bromide. The results are presented as cycle number in which the first detectable amount of DNA appears on the agarose gel. Samples were compared after correction for GAPDH content for each individual sample, to rule out confounding by variation of cellularity in the biopsies.
Statistical analysis
Differences between experimental groups were tested for significance using the Wilcoxon rank test. P values <0.05 were considered significant.
Results
Fcγ receptors are essential for synovial inflammation during immune-complex-mediated arthritis
To investigate the involvement of Fcγ receptors in locally induced ICA, we used FcR γ-chain-/- C57BL/6 mice, which lack functional FcγRI and RIII. ICA was induced in knee joints of FcR γ-chain-/- mice and control C57BL/6 mice. Histology of total knee joints showed florid inflammation in C57BL/6 mice 3 days after induction of ICA (Fig. 1b). In knee joints of FcR γ-chain-/- mice, however, virtually no inflammatory cells were observed at day 3 of ICA (Fig. 1a). Joint inflammation, defined as cells present in the joint cavity (exudate) and synovium (infiltrate), was scored. Both exudate and infiltrate were substantial in C57BL/6 mice whereas, in FcR γ-chain-/- mice, cell influx was virtually absent (Table 1).
Figure 1.
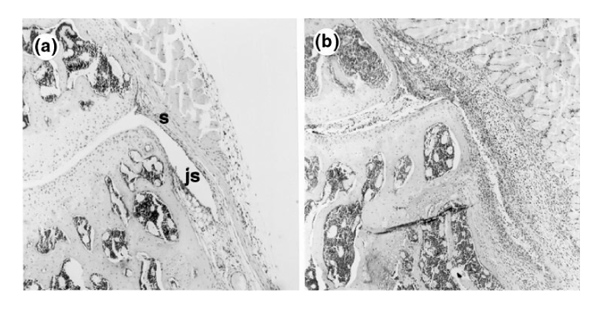
Inflammation in H&E-stained sections of mouse knee joints after induction of immune-mediated arthritis (ICA). (a) Section from FcR γ-chain-/- mouse 3 days after ICA induction. No inflammatory cells are visible in the joint space (js) or synovium (s). (b) Section from C57BL/6 (control) mouse 3 days after ICA induction. Florid inflammation is visible both in the joint space (exudate) and in the synovium (infiltrate) (orginal magnification 100×).
Table 1.
Joint inflammation in FcR γ-chain-/-, wild-type C57BL/6, and DBA/1 mice
| Days after induction of ICA | ||||||||
| 1 | 3 | 7 | 14 | |||||
| Mouse strain | Infiltrate | Exudate | Infiltrate | Exudate | Infiltrate | Exudate | Infiltrate | Exudate |
| FcRγ-/- | 0.4 ± 0.1 | 0.3 ± 0.2 | 0.1 ± 0.1* | 0.1 ± 0.1* | ND | ND | ND | ND |
| C57BL/6 | 1.4 ± 0.1 | 0.8 ± 0.2 | 1.4 ± 0.3 | 1.5 ± 0.4 | 1.2 ± 1.2 | 0.5 ± 0.6 | 0.4 ± 0.3 | 0 |
| DBA/1 | ND | ND | 2.1 ± 0.2* | 1.8 ± 0.3 | 3.0 ± 0* | 2.5 ± 0.4* | 2.1 ± 0.3* | 1.6 ± 0.2* |
Cellular score in knee joints 3, 7, and 14 days after induction of ICA in knee joints of FcR γ-chain-/-, C57BL/6, and DBA/1 mice. The amount of inflammatory cells in the joint cavity (exudate) and in the synovial layer (infiltrate) were scored blind by two independent observers, on an arbitrary scale from 0-3 (0 = no; 1 = minor; 2 = moderate; 3 = maximal). Note that FcR γ-chain-/- mice show markedly less inflammation and that knee joints of DBA/1 mice were significantly more inflamed at all time points in comparison with C57BL/6 mice. Data are means of scores for six mice, and significance (FcR γ-chain-/- versus C57BL/6, and DBA/1 versus C57BL/6) was evaluated using the Mann-Whitney U test (*P < 0.05). ND, not done.
To investigate whether FcγRs are involved in the upregulation of inflammatory mediators seen during the first phase of this arthritis, RT-PCR was performed on synovial specimens from arthritic knee joints. Levels of mRNA of inflammatory mediators (IL-1β, IL-1Ra, MCP-1, MIP-2) were detected semiquantitatively in knockout and control mice, 6 and 24 h after ICA induction (Fig. 2). IL-1β and IL-1Ra mRNA levels were high in control arthritis and appeared to be decreased (down to 28) in FcR γ-chain-/- mice. MCP-1 and MIP-2 mRNA levels were also diminished in the early phase of ICA (6 h) relative to the levels of controls (respectively down to 26 and 24) suggesting downregula-tion of both polymorphonuclear neutrophils (PMNs) and monocyte-specific chemokines.
Figure 2.
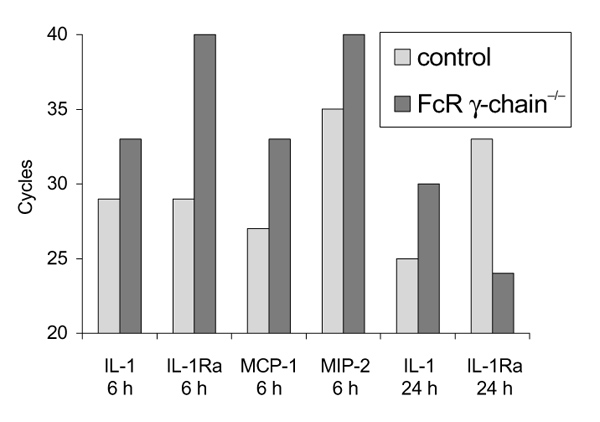
Semiquantitative mRNA measurements in synovial tissue of FcR γ-chain-/- and C57BL/6 (control) mice. Synovial mRNA levels of IL-1, IL-1Ra, MCP-1, and MIP-2, 6 and 24 h after induction of ICA. At certain points during the PCR reaction, samples were put on agarose gel and electrophoresis was performed. The number of the cycle in which the first band appeared was found: thus, a low cycle number means a higher mRNA content. Data were corrected for GAPDH signal. (Each group: n = 6.)
Subsequently, intra-articular production of IL-1 and IL-1Ra protein during arthritis was measured. Washouts of synovial specimens from joints of control C57BL/6 mice contained considerable amounts of IL-1 (240 pg/ml) at 6 h after induction of arthritis; at 24 h, IL-1 levels were reduced to 70 pg/ml (Table 2). Washouts of arthritic FcR γ-chain-/- joints contained 200 ng/ml IL-1 at 6 h but no detected IL-1 at 24 h after induction, indicating that in the absence of functional FcγRI and RIII, a rapid downregulation of IL-1 production is found. IL-1Ra production was comparable at 6 h in the two strains (respectively 180 and 215 pg/ml) and below the detection limit at 24 h after ICA induction in both strains (Table 2).
Table 2.
IL-1 and IL-1Ra production (pg/ml) in knee joints of mice after induction of immune-complex-mediated arthritis (ICA) and zymosan-induced arthritis (ZIA)
| During ICA | During ZIA | ||||
| IL-1Ra | IL-1β | IL-1β | |||
| Mouse strain | At 6 h | At 6 h | At 24 h | At 6 h | At 24 h |
| FcRγ-/- | 180 ± 15 | 200 ± 23 | <20* | ND | ND |
| C57BL/6 | 215 ± 20 | 240 ± 21 | 70 ± 12 | >1000 | 550 ± 30 |
| DBA/1 | ND | 390 ± 43* | 120 ± 15* | >1000 | 565 ± 5 |
Production of IL-1Ra at 6 h and IL-1β at 6 and 24 h after induction of ICA and ZIA in knee joints of three mouse strains. IL-1β was measured using a radioimmunoassay. Data are means ± SD of at least 5 mice and were evaluated using the Mann-Whitney U test (*P < 0.05). ICA, immune-complex-mediated arthritis; IL-1Ra, IL-1 receptor antagonist; ND = not done; ZIA, zymosan-induced arthritis.
To exclude the possibility that the strong reduction of inflammation during ICA in FcR γ-chain-/- mice is caused by a lower level of immune complexes which are formed or retained within the knee joint, deposition of IgG in the knee joint was detected using biotinylated goat anti-rabbit IgG. As Table 3 shows, at 6 and 24 h after ICA induction, no difference was found between FcR γ-chain-/- and C57BL/6 mice in the amount of immune complexes deposited within the knee joints. We compared deposition of complement (C3c) in the synovial tissue of both strains to rule out the possibility that a difference in complement deposition caused differences in inflammation. Control and knockout mice showed the same amounts of C3c in the tissue as was determined by immunohistochemistry (Table 3).
Table 3.
Deposition of IgG and complement factor C3c in knee joints of mice at various times after induction of immune-complex- mediated arthritis
| IgG | C3c | |||
| Mouse strain | 6 h | 24 h | 24 h | |
| FcRγ-/- | 128 ± 6 | 139 ± 4 | 152 ± 9 | |
| C57Bl/6 (control) | 126 ± 4 | 140 ± 9 | 155 ± 7 | |
IgG and C3c were studied immunohistochemically. Deposition was assessed using an image analyser to measure the mean amount of blue light, as a measure of staining intensity, passing through a defined area in the tissue. Data are means ± SD for six mice. Significance was evaluated using Student's t test.
Expression of FcγRII/III is elevated in resident macrophages of naïve CIA-sensitive mice that are hypersensitive to immune complexes
In a recent study [36], we noticed that certain mouse strains that are prone to develop collagen-type-II autoimmune arthritis are also particularly susceptible to immune-complex-induced inflammation. Induction of ICA in the DBA/1 knee joint resulted in a more severe arthritis than that in C57BL/6 mice, and this became chronic (Table 1). Local production of IL-1 was also significantly higher and more prolonged than in C57BL/6 mice, both at 6 and 24 h after ICA induction (Table 2). To investigate whether the enhanced inflammation in DBA/1 mice during ICA was still mediated by FcRs, FcR γ-chain-/- mice were backcrossed to DBA/1 (DBAγ-/-) mice. ICA was induced in these mice and the inflammation was compared with that in DBAγ+/+ littermates. We found that at day 3 after arthritis induction, arthritis was full-blown in DBAγ+/+mice (arbitrary scores of 1.0 ± 0.4 for exudate and 1.6 ± 0.3 for infiltrate) but was completely absent in γ-chain-deficient DBA/1 mice (0.1 ± 0.2 and 0.1 ± 0.1, respectively; not shown).
To examine whether the different reactions in knee joints of C57BL/6 and DBA/1 mice to immune complexes might be due to variable local expression of FcγR, expression in situ of FcγRs in naïve knee joints was determined immunohistochemically, using the monoclonal antibody (mAb) 2.4G2 (FcγRII/III). Synovial macrophage-like type A lining cells and deeper-lying synovial macrophages were the dominant cell types expressing these receptors, as was seen in double-labeling studies with F4/80 (data not shown). Interestingly, synovial macrophages of naïve DBA/1 mice stained more intensely than those of C57BL/6 mice (Fig. 3), indicating a higher constitutive expression of FcγRs in lining macrophages of these autoimmune-prone mice.
Figure 3.
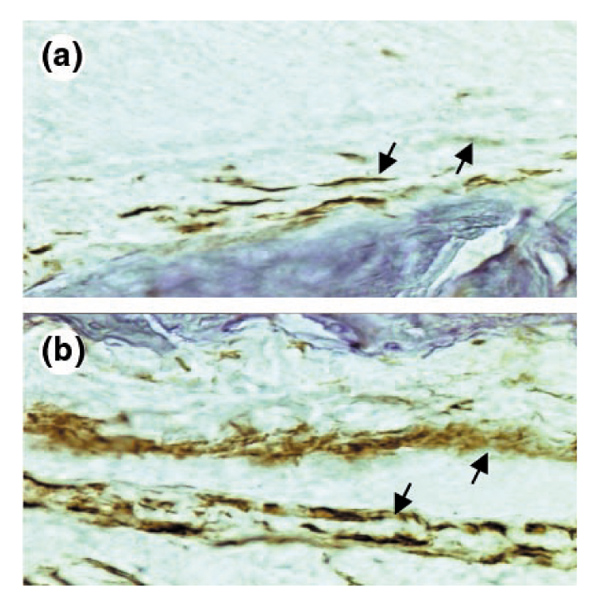
Expression of FcγRs in naïve knee joints of C57BL/6 and DBA/1 mice as detected by immunohistochemical staining using specific anti-FcγRII/III antibodies (mAb: 2.4G2) and subsequent development using di-aminobenzidine. (a) Naïve C57BL/6 mice. Very light staining of both synovial lining layer (arrows) and deeper layer. (b) Naïve DBA/1. Note the markedly higher staining intensity, especially of the cells of the synovial lining (arrows) (original magnification 200×).
To quantify the difference in FcγR expression by macrophages of the two strains (DBA/1 and C57BL/6), we further investigated receptor expression on peritoneal macrophages, using FACS analysis. Macrophages of DBA/1 mice (mean fluorescence 440 ± 50 intensity per cell) displayed almost twice the mean fluorescence with mAb 2.4G2 as those of C57BL/6 mice (240 ± 30 intensity per cell) (Fig. 4). These findings also indicate that FcγRII/III is significantly upregulated on the membrane of macrophage populations present in various body compartments of naïve DBA/1 mice.
Figure 4.
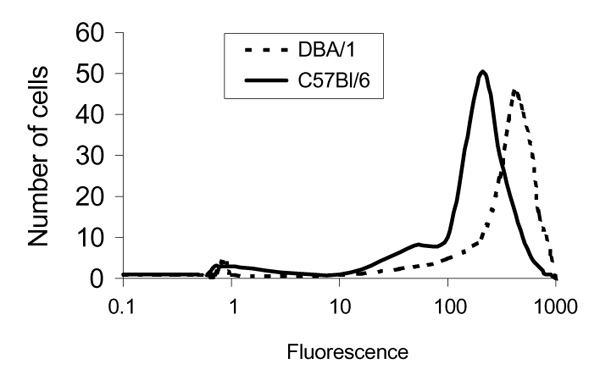
Expression of FcγRs by resident peritoneal macrophages of C57BL/6 and DBA/1 mice as determined by fluorescence in FACS analysis. Almost twice as many receptors were expressed in DBA/1 as in C57BL/6 (control) mice. Cells were isolated from naïve mice using ice-cold medium and thereafter incubated with an anti-FcγR antibody (2.4G2) that recognises both FcγRII and RIII. Incubation with FITC-labeled secondary antibody followed.
To further investigate whether a higher basal FcγR expression on macrophages has consequences for the activation of cells upon interaction with immune complexes, peritoneal macrophages of both strains were incubated with aggregated IgG and production of IL-1 was determined by bioassay. Incubation of C57BL/6 cells for 24 or 48 h caused the release of considerable amounts of bioactive IL-1 (45 pg/ml at 24 h and 50 pg/ml at 48 h) (Fig. 5). Interestingly, DBA/1 macrophages produced significantly higher amounts of IL-1 (120 and 135 pg/ml, respectively) than C57BL/6 mice.
Figure 5.
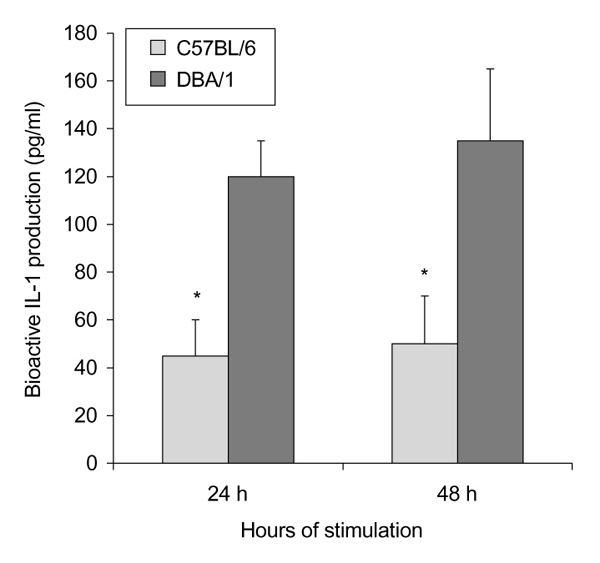
IL-1 production by peritoneal macrophages. Production of bioactive IL-1 (pg/ml) by peritoneal macrophages of C57BL/6 and DBA/1 mice 24 and 48 h after stimulation with HAGGs (100 μg/ml). IL-1 production was measured using an IL-1-specific bioassay (NOB assay). The higher production by macrophages of DBA/1 than of C57BL/6 mice was significant (*P < 0.02).
Arthritogenic triggers other than immune complexes cause comparable inflammation in knee joints of mice that differ in FcγR expression levels
To substantiate the hypothesis that differences in FcR expression underlie the variation seen during immune-complex-induced joint inflammation, we injected various arthritogenic triggers (zymosan, SCWs) that do not act via FcγRs into the knee joints of three strains of mice (FcR γ-chain-/-, C57BL/6, and DBA/1). A previous study had shown that when these triggers were used, there was no difference between DBA/1 and C57BL/6 mice in joint inflammation [34]. In the present study, when zymosan was injected into knee joints of FcR γ-chain-/- mice, the inflammation was similar to that in control C57BL/6 mice. At day 3 after injection, inflammation in FcR γ-chain-/-mice was scored as 1.0 ± 0.4 for infiltrate and 0.9 ± 0.6 for exudate, versus 0.9 ± 0.3 and 0.9 ± 0.5, respectively, in control C57BL/6 mice. These findings indicate that knee joints of the three strains examined develop comparable, marked inflammation after injection of zymosan or SCWs. Moreover, IL-1β production in the knee joints during zymosan-induced arthritis was comparable in DBA/1 and C57BL/6 mice (Table 2).
FcγR expression on macrophages is correlated with cartilage destruction during immune-complex-mediated arthritis
Apart from inflammation, absence or overexpression of FcγRs by local macrophages may also have serious consequences for cartilage destruction. We investigated several parameters of cartilage destruction (depletion of proteogly-cans [PGs], chondrocyte death, and erosion of the cartilage matrix). Arthritic knee joints of the three strains were compared at different time points after ICA induction. At day 3, control C57BL/6 mice showed marked PG depletion in various cartilage surfaces (patella, femur adjacent to patella, lateral and medial condyle of the femur, and lateral and medial condyle of the tibia). Strikingly, PGs were not depleted in arthritic knee joints of the FcR γ-chain-/- mice (Fig. 6a,b; Table 4). In contrast, arthritic DBA/1 knee joints showed markedly greater depletion of PGs than those of C57BL/6 mice. Moreover, PG depletion was still severe at days 7 and 14 in the DBA/1 mice, whereas in the C57BL/6 mice it was almost fully restored by day 14 (Fig. 6b-e; Table 4). When ICA was induced in FcR γ-/- mice with a DBA/1 background, PG depletion at day 3 was scored 2.6 ± 0.3 in the Fcγ-chain+/+ mice and was totally absent in the FcR γ-chain-/- ones.
Figure 6.
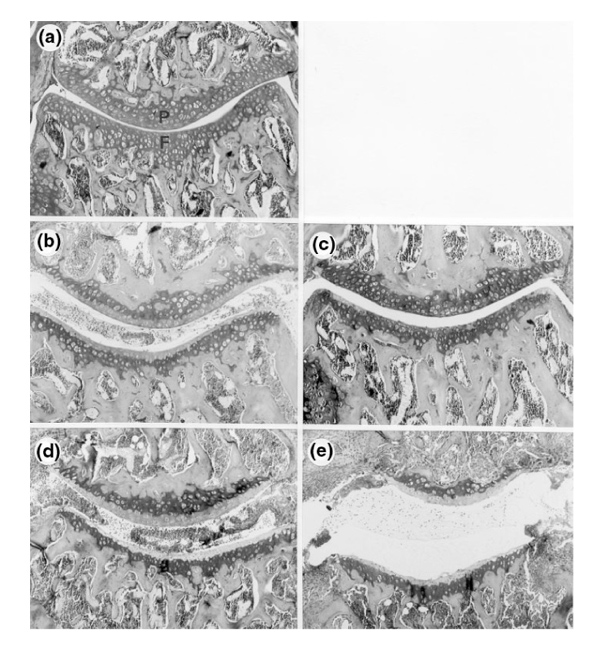
Cartilage damage during ICA. Safranin-O-stained knee-joint sections of FcR γ-chain-/-, C57BL/6, and DBA/1 mice 3 and 7 days after induction of ICA. Proteoglycan depletion was correlated with destaining of the superficial layer of the cartilage matrix, which is normally stained red. Both patellar (P) and femoral (F) cartilage are shown. (a) FcR γ-chain-/- 3 days after ICA induction. No proteoglycan depletion is seen. (b) C57BL/6 mouse 3 days after ICA induction. Marked destaining of the matrix is found. (c) C57BL/6 mouse 7 days after ICA induction. The absence of depleted areas suggests that the matrix has been completely restored. (d) DBA/1 mouse 3 days after ICA induction. The cartilage matrix is completely depleted, indicating considerable PG loss. (e) Same mouse strain (DBA/1) as in (d), 7 days after ICA induction. The cartilage matrix seems still devoid of PG, suggesting that no repair has taken place. Moreover, marked erosion of the matrix is visible, mainly of the femoral cartilage.
Table 4.
Fc receptors and cartilage damage in knee joints of mice during immune-complex-mediated arthritis
| Mouse strain | ||||
| Index of cartilage damage | Day | FcRγ-/- | C57BL/6 | DBA/1 |
| PG depletion† | 1 | 0 | 0.2 ± 0.1 | ND |
| 3 | 0.1 ± 0.1* | 2.2 ± 0.5 | 2.8 ± 0.3* | |
| 7 | ND | 1.5 ± 0.4 | 3.0 ± 0* | |
| 14 | ND | 0.6 ± 0.4 | 3.0 ± 0* | |
| Chondrocyte death (%)‡ | 1 | 0 | 0 | ND |
| 3 | 0 | 18 ± 6 | 35 ± 12 | |
| 7 | ND | 20 ± 10 | 68 ± 24* | |
| 14 | ND | 20 ± 9 | 98 ± 5* | |
| Matrix erosion§ | 1 | 0 | 0 | ND |
| 3 | 0 | 0 | 0.4 ± 0.3 | |
| 7 | ND | 0.2 ± 0.2 | 1.5 ± 0.7* | |
| 14 | ND | 0.2 ± 0.2 | 2.3 ± 0.6* | |
Cartilage damage was assed using total knee-joint sections stained with safranin O (for PG depletion and matrix erosion) or H&E (for chondrocyte death). PG depletion was absent in FcRγ-/-, moderate in C57BL/6, and high in DBA/1 mice. Irreversible cartilage damage, chondrocyte death, and erosion of the cartilage matrix, were almost absent in both FcRγ-/- and their control C57BL/6 mice, whereas erosion in DBA/1 mice was massive. To evaluate significance, the Mann-Whitney U-test (*P < 0.05) was used. †Scored by two blinded, independent observers. 1 = mild, 2 = moderate, 3 = maximal destaining of cartilage. ‡ Scored by two blinded, independent observers, as the percentage of total cartilage area in the surface layer (up to the tide mark) containing empty lacunae. §Measured by determining the area in the cartilage surface layer (up to the tide mark) that had been lost during arthritis by two blinded observers, and expressed using an arbitrary score from 0–3 (0 = no; 1 = minor (0-10%); 2 = moderate (>10-50%); 3 = maximal (>50-100%). ND = Not done.
Chondrocyte death was measured by determining empty lacunae in the cartilage matrix and breaks in DNA strands as evaluated using an in situ cell detection kit (TUNEL method; data not shown). In knee joints of C57BL/6 mice, chondrocyte death at day 3 was 5% in the patella and 30% in the adjacent femur. In the knee joints of knockout mice, chondrocyte death was completely absent (Table 4), whereas in the DBA/1 mice, it was strikingly higher in both the patella (50%) and the femur (90%) than in the control mice (C57BL/6).
Finally, erosion of the cartilage matrix in knee joints was measured. Whereas no erosion was found in either FcR γ-chain-/- or C57BL/6 mice at day 3 after arthritis induction, in DBA/1 mice erosion was already considerable by day 3, and complete loss of the cartilage surface layer up to the tidemark was observed at days 7 and day 14 (Fig. 6d,e; Table 4).
Severe cartilage destruction in DBA/1 mice is specific for immune-complex-mediated arthritis
To further substantiate the interpretation that immune complexes and FcγRs, rather than inflammatory cells, caused the observed severe joint cartilage destruction, various concentrations of zymosan were injected into the knee joints of all three mouse strains (FcR γ-chain-/-, C57BL/6, and DBA/1). Even large amounts of zymosan (up to 180 μg), although inducing a tremendous influx of inflammatory cells, failed to induce chondrocyte death or cartilage erosion. PG depletion at day 3, however, was scored as 3.6 ± 0.4 in FcR γ-/- mice, versus 0.6 ± 0.3 in C57BL/6 mice. At day 7, mean PG depletion was scored as 1.8 ± 0.5 in C57BL/6 mice and 1.7 ± 0.8 in DBA/1. PG depletion in the cartilage matrix in response to zymosan, therefore, was marked but did not differ between the three strains.
Discussion
We have found that inflammation and subsequent cartilage destruction caused by immune complexes may be related to the expression of FcγRs on macrophages. Three strains differing in levels of expression of FcγRs were compared. Absence of functional FcγRI and RIII prevents synovial inflammation and cartilage destruction after induction of immune complex arthritis, whereas elevated expression of FcγR on resident joint macrophages of mice susceptible for development of autoimmune arthritis is correlated to markedly more synovial inflammation and cartilage destruction after induction of ICA. The difference in joint inflammation seen within the three strains was not due to genetic differences resulting in a different susceptibility to inflammation, since injection of zymosan or SCWs caused comparable inflammation in the three strains.
Mice lacking the FcR γ-chain were used to investigate the role of FcγRs in ICA. The FcR γ subunit is essential not only for functional FcRI and RIII IgG receptors but also for the high-affinity IgE receptor FcεRI and TcR-CD3 complex of γδT cells. In this passively induced arthritis, B and T cells do not play a role [35]. Even during the chronic phase of ICA as seen in the DBA/1 knee joint, no effect on synovial inflammation was found after giving anti-αβTCR antibodies (data not shown). The high-affinity receptor FcγRI, which binds IgE, is found on mast cells. A previous study showed, however, that experimentally induced ICA in knee joints of mice that are deficient for mast cells (WBB6F1-W/Wv) [42] was not significantly different from that in control mice, indicating that mast cells do not play an important role in ICA.
The findings described above suggest that the observed joint inflammation is locally regulated by synovial cells. Earlier studies revealed that macrophage-like type A cells, which form the dominant population (more than 80%) of the lining layer of diarthrodial joints in mice, regulate the onset and chronicity [19,20,21,22] as well as the exacerbation [23] of experimental arthritis. Selective removal of type A cells by clodronate-containing liposomes prior to induction of several experimental arthritides prevented the onset of arthritis and also the larger part of cartilage destruction [21,43]. These studies suggested that synovial macrophages form an important source of chemotactic factors and that activation by immune complexes results in production of chemokines by these cells. It was also found that chronicity of synovial inflammation is mostly seen in models in which immune complexes are present [44]. Immune complexes communicate with macrophages via FcRs, especially the FcγRs, which bind IgG. IgG represents the most dominant immunoglobulin class in the blood and might be involved in activation of macrophages. Coordinate expression of activating FcγRI and RIII and the inhibiting receptor FcγRIIb exposed on joint macrophages probably determines the reaction to immune complexes [15].
Indeed, we found that the absence of functional FcγRI and RIII completely abrogated the onset of ICA, as was seen in knee joints of FcR γ-chain-/- mice. The proinflammatory cytokine IL-1, which has been shown to regulate synovial inflammation within this model [34,35], was significantly decreased at protein level 24 h after induction. Semiquantitative determination of mRNA levels also suggested downregulation of IL-1. At 6 h after induction of ICA, no difference in cytokine production was found between FcR γ-chain-/- mice and controls. It is therefore possible that the first hours of ICA are mediated by a component other than FcγRs. This component may very well be complement. In addition to IL-1, complement split products have been shown to be important within this model. Depletion of complement generation by cobra venom factor treatment blocked ICA for the most part [35]. In the present study, complement C3c deposition did not differ in knee-joint structures of FcR γ-chain-/- and control mice, suggesting that complement activation does not differ between the two strains; this observation confirms earlier findings [8]. To further investigate whether complement or other mediators play a role in the enhanced inflammation in DBA/1 mice, we compared FcRγ-chain-/- mice in a DBA/1 background with FcR γ-chain+/+ mice. ICA did not develop in FcR γ-chain deficient mice with a DBA/1 background, indicating that in the DBA/1 strain, also, FcRs are crucial mediators. Another difference may be the removal of immune complexes from the joint. Earlier studies revealed that absence of functional FcγRI and RIII retarded the removal of insoluble immune complexes from the body [6]. We found, however, no differences in IgG deposition in the knee joints of the two mouse strains during the first days. The reason for this may be that the arthritogenic molecule we use to elicit ICA has an excellent retention, because of its positive charge. Cationic antigen will be bound electrostatically to the negatively charged structures of the joint [35,39]. Moreover, inflammation in the knee joint may be differently regulated than in other body compartments. The knee-joint cavity is covered mainly with macrophages, which are the first cells that come into contact with the immune complexes during ICA, whereas the kidney and skin contain a broader range of cell types handling immune complexes.
Next, we investigated ICA in knee joints of DBA/1 mice and found that the severity and the chronicity of arthritis corresponded with a higher level of expression of FcγRs on macrophages. In contrast, ICA in C57BL/6 mice was only mild, without a chronic phase. Macrophages in the knee-joint lining of normal DBA/1 mice stained more intensely than those of normal C57BL/6 mice, suggesting a higher basal level of FcγRII/III expression on DBA/1 mice. Furthermore, higher FcγR expression was not limited to joint macrophages. Peritoneal macrophages showed a twofold higher expression of FcγRs, suggesting that the higher basal expression of FcγRII/III is found on all resident macrophages. Other cell types were also tested for differences in FcγR expression. No significant differences of FcγR expression were found on peripheral blood monocytes nor on a mixed B- and T-cell population, indicating that the differences in expression of FcγRs between C57BL/6 and DBA/1 are not found on all cells of the haematopoietic lineage (unpublished results). Antibodies to FcγRI or FcγRIII are not available and therefore we could not determine the different classes of FcγR separately. Genetic differences in DBA/1 may be responsible for altered FcγR expression, as Holmdahl et aI have pointed out [45]. This would imply that this mouse strain is at higher risk for immune-complex diseases in body compartments that contain macrophages. This is in line with findings in other studies [46]. The finding that FcR γ-chain-/-DBA/1 mice did not show any inflammation when ICA was induced strongly suggests that, in this strain, full-blown ICA is also mediated by FcγRs. Thus the differences in the severity and the chronicity of inflammation during ICA and in the severity of subsequent cartilage damage are likely to be caused by differences in FcγR function or expression. The elevated expression of FcγRs on macrophages was also functional, as was found in experiments in vitro in which IL-1 production was measured after stimulation of macrophages with HAGGs. ICA induction caused much higher levels and longer-lasting production of IL-1 in the knee joints of DBA/1 mice than in the control C57BL/6 mice. IL-1 may well be the factor responsible for the chronicity of the synovial inflammation. Although IL-1 levels at day 3 were found to be below the detection limit of the RIA, blocking of IL-1 with anti-IL-1 antibodies starting at day 3 after ICA induction abrogated the chronic phase completely [34].
In addition to inflammation, cartilage destruction was also investigated. In earlier studies, we found that only in those experimental-arthritis models in which immune complexes are present was severe, irreparable cartilage damage found [44]. Mice injected with zymosan or SCW did not suffer irreparable cartilage damage, although a tremendous influx of inflammatory cells in the joint was observed.
In FcR γ-chain-/- mice, depletion of PG was entirely absent during ICA. In knee joints of DBA/1 mice, loss of PG from the cartilage matrix was more severe than in C57BL/6 mice. This finding is in line with the respective expression of FcγR on macrophages in these strains and suggests that activation of FcγR may be one of the mechanisms mediating PG loss. Activation of macrophages by FcγRI and RIII leads to the release of IL-1 [47,48], which is the dominant cytokine involved in inhibition of PG synthesis [49,50] and production of aggrecan-degrading enzymes [51,52]. Depletion of PG within this model is probably mediated by aggrecanase [44], an enzyme that recently was identified as a metalloproteinase of the ADAM (A disintegrin and metalloproteinase) family [53]. Aggrecanase neoepitopes were abundantly found during the first phase of this arthritis [44]. Injection of zymosan into the knee joints of the three strains we studied was followed by a comparable depletion of PG; this finding suggests that the three strains have similar potencies to produce aggrecanase.
Severe cartilage destruction, like chondrocyte death and cartilage erosion, was found only in DBA/1 mice. During the acute phase, PMNs constitute the dominant cell type present in the joint, whereas during the chronic phase, macrophages are the predominant cell type present in the synovium and synovial cavity. FcγR activation on both PMNs and macrophages may lead to the release of oxygen and nitrogen radicals, which in high concentrations can kill chondrocytes either by necrosis or apoptosis [54,55] or may activate latent enzymes in the matrix. Activated macrophages form an important source of latent metalloproteinases such as stromelysin and collagenase; after activation, these enzymes are involved in degradation of the collagen matrix, resulting in severe cartilage destruction [56,57]. Immune complexes activate macrophages in the synovium, causing the release of enzymes that are hindered in their activity by large amounts of inhibitors present in the synovial fluid. Immune complexes present on the cartilage surface may cause PMNs or macrophages to attach to this surface, as we have seen by clear pannus formation on the cartilage during chronic arthritis in the DBA/1 joint. This attachment to the cartilage surface may cause release of enzymes that directly penetrate the cartilage matrix thus escaping from their inhibitors (manuscript in preparation).
The present findings partly underline the findings in our previous study, in which an antigen-induced arthritis was produced in FcR γ-/-mice [58], in the sense that stimulation of FcγRs by immune complexes seems necessary in order to induce irreversible cartilage damage. However, in contrast to our present findings, antigen-induced arthritis in FcR γ-chain-/- mice did not significantly reduce joint inflammation. This indicates that within that model, FcRs do not play any role in the chronic phase of inflammation, but are of utmost importance in the induction of severe cartilage damage.
To provide conclusive evidence of the role of various classes of FcγR during ICA, experiments are needed in which FcγRs are blocked with specific antibodies, or in which knockout mice are used that lack one of the specific classes of FcγR. However, specific antibodies to FcγRs (2.4G2), once bound to the receptor, have a stimulatory effect and therefore cannot be used in blocking experiments. Experiments using specific knockout mice are therefore now being performed in our laboratory.
Macrophages are the dominant type of cell present in chronic inflammation during RA and their number has been shown to correlate well with severe cartilage destruction [16,17,18]. Apart from that, in humans, these macrophages express activating Fc receptors, mainly FcγIIIa [26], which may lead to activation of these cells by IgG-containing immune complexes. Fc receptor expression on the surface of these cells may have important implications for joint inflammation and severe cartilage destruction and may form new targets for therapeutic intervention.
References
- Winchester RJ, Agnello V, Kunkel HG. Gamma globulin complexes in synovial fluids of patients with rheumatoid arthritis. Partial characterization and relationship to lowered complement levels. . Clin Exp Immunol. 1970;6:689–706. [PMC free article] [PubMed] [Google Scholar]
- Cooke TD, Richer S, Hurd E, Jasin HE. Localization of antigen-antibody complexes in intraarticular collagenous tissues. . Ann N Y Acad Sci. 1975;256:10–24. doi: 10.1111/j.1749-6632.1975.tb36032.x. [DOI] [PubMed] [Google Scholar]
- Henson PM, Johnson HB, Spiegelberg HL. The release of granule enzymes from human neutrophils stimulated by aggregated immunoglobulins of different classes and subclasses. J Immunol. 1972;109:1182–1192. [PubMed] [Google Scholar]
- Deo YM, Graziano RF, Repp R, van de Winkel JG. Clinical significance of IgG Fc receptors and Fc gamma R-directed immunotherapies. . Immunol Today. 1997;18:127–135. doi: 10.1016/s0167-5699(97)01007-4. [DOI] [PubMed] [Google Scholar]
- Ravetch JV. Fc receptors. Curr Opin Immunol. 1997;9:121–125. doi: 10.1016/s0952-7915(97)80168-9. [DOI] [PubMed] [Google Scholar]
- Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Sylvestre D, Clynes R, Ma M, Warren H, Carroll MC, Ravetch JV. Immunoglobulin G-mediated inflammatory responses develop normally in complement-deficient mice. J Exp Med. 1996;184:2385–2392. doi: 10.1084/jem.184.6.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. . Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, Ra C. Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int. 1998;54:1166–1174. doi: 10.1046/j.1523-1755.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Park SY, Ueda S, Ohno H, Hamano Y, Tanaka M, Shiratori T, Yamazaki T, Arase H, Arase N, Karasawa A, Sato S, Ledermann B, Kondo Y, Okumura K, Ra C, Saito T. Resistance of Fc receptor-deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102:1229–1238. doi: 10.1172/JCI3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakov N. Immunoreceptor tyrosine-based activation motif (ITAM), a unique module linking antigen and Fc receptors to their signaling cascades. J Leukoc Biol. 1997;61:6–16. doi: 10.1002/jlb.61.1.6. [DOI] [PubMed] [Google Scholar]
- Cambier JC. Antigen and Fc receptor signaling. The awesome power of the immunoreceptor tyrosine-based activation motif (ITAM). J Immunol. 1995;155:3281–3285. [PubMed] [Google Scholar]
- Daeron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, Fridman WH. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. 1995;3:635–646. doi: 10.1016/1074-7613(95)90134-5. [DOI] [PubMed] [Google Scholar]
- Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, Ravetch JV, Takai T. Deletion of Fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J Exp Med. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J Exp Med. 1999;189:179–185. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanni G, Whelan A, Feighery C, Bresnihan B. Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann Rheum Dis. 1994;53:39–44. doi: 10.1136/ard.53.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. . Arthritis Rheum. 1996;39:115–124. doi: 10.1002/art.1780390116. [DOI] [PubMed] [Google Scholar]
- Bresnihan B. Pathogenesis of joint damage in rheumatoid arthritis. J Rheumatol. 1999;26:717–719. [PubMed] [Google Scholar]
- van Lent PL, van den Hoek AE, van den Bersselaar LA, Spanjaards MF, van Rooijen N, Dijkstra CD, van de Putte LB, van den Berg WB. In vivo role of phagocytic synovial lining cells in onset of experimental arthritis. . Am J Pathol. 1993;143:1226–1237. [PMC free article] [PubMed] [Google Scholar]
- van Lent PL, Holthuysen AE, van den Bersselaar LA, van Rooijen N, Joosten LA, van de Loo FA, van de Putte LB, van den Berg WB. Phagocytic lining cells determine local expression of inflammation in type II collagen-induced arthritis. Arthritis Rheum. 1996;39:1545–1555. doi: 10.1002/art.1780390915. [DOI] [PubMed] [Google Scholar]
- van Lent PL, Holthuysen AE, van Rooijen N, van de Putte LB, van den Berg WB. Local removal of phagocytic synovial lining cells by clodronate-liposomes decreases cartilage destruction during collagen type II arthritis. Ann Rheum Dis. 1998;57:408–413. doi: 10.1136/ard.57.7.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lent PLEM, van den Bersselaar LAM, Holthuysen AEM, van Rooijen N, van de Putte LB, van den Berg WB. Phagocytic synovial lining cells in experimentally induced chronic arthritis: down-regulation of synovitis by CL2MDP-liposomes. Rheumatol Int. 1994;13:221–228. [Google Scholar]
- van Lent PL, Holthuysen AE, van Rooijen N, van de Loo FA, van de Putte LB, van den Berg WB. Phagocytic synovial lining cells regulate acute and chronic joint inflammation after antigenic exacerbation of smouldering experimental murine arthritis. J Rheumatol. 1998;25:1135–1145. [PubMed] [Google Scholar]
- Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Ann Hematol. 1998;76:231–248. doi: 10.1007/s002770050396. [DOI] [PubMed] [Google Scholar]
- Broker BM, Edwards JC, Fanger MW, Lydyard PM. The prevalence and distribution of macrophages bearing Fc gamma RI, Fc gamma RII, and Fc gamma RIII in synovium. Scand J Rheumatol. 1990;19:123–135. doi: 10.3109/03009749009102116. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Blades S, Cambridge G. Restricted expression of Fc gammaRIII (CD16) in synovium and dermis: implications for tissue targeting in rheumatoid arthritis (RA). Clin Exp Immunol. 1997;108:401–406. doi: 10.1046/j.1365-2249.1997.3941286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizaki T, Ookawara T, Oh IS, Itoh Y, Iwabuchi K, Onoe K, Day NK, Good RA, Ohno H. An increase in basal glucocorticoid concentration with age induces suppressor macrophages with high-density Fc gamma RII/III. . Immunology. 1998;93:409–414. doi: 10.1046/j.1365-2567.1998.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago A, Mori T, Satriano J, Schlondorff D. Regulation of Fc receptors for IgG on cultured rat mesangial cells. Kidney Int . 1991;39:87–94. doi: 10.1038/ki.1991.11. [DOI] [PubMed] [Google Scholar]
- de Andres B, Cardaba B, del Pozo V, Orozco ME, Gallardo S, Tramon P, Palomino P, Lahoz C. Modulation of the Fc gamma RII and Fc gamma RIII induced by GM-CSF, IFN-gamma and IL-4 on murine eosinophils. . Immunology. 1994;83:155–160. [PMC free article] [PubMed] [Google Scholar]
- Wahl SM, Allen JB, Welch GR, Wong HL. Transforming growth factor-beta in synovial fluids modulates Fc gamma RII (CD16) expression on mononuclear phagocytes. J Immunol. 1992;148:485–490. [PubMed] [Google Scholar]
- Welch GR, Wong HL, Wahl SM. Selective induction of Fc gamma RIII on human monocytes by transforming growth factor-beta. J Immunol . 1990;144:3444–3448. [PubMed] [Google Scholar]
- Conrad DH, Waldschmidt TJ, Lee WT, Rao M, Keegan AD, Noelle RJ, Lynch RG, Kehry MR. Effect of B cell stimulatory factor-1 (interleukin 4) on Fc epsilon and Fc gamma receptor expression on murine B lymphocytes and B cell lines. J Immunol. 1987;139:2290–2296. [PubMed] [Google Scholar]
- Isomaki P, Luukkainen R, Toivanen P, Punnonen J. The presence of interleukin-13 in rheumatoid synovium and its antiinflammatory effects on synovial fluid macrophages from patients with rheumatoid arthritis. . Arthritis Rheum. 1996;39:1693–1702. doi: 10.1002/art.1780391012. [DOI] [PubMed] [Google Scholar]
- Blom AB, van Lent PLEM, Holthuysen AEM, van den Berg WB. Immune complexes, but not streptococcal cell walls or zymosan, cause chronic inflammation in mouse strains susceptible for collagen type II autoimmune arthritis. Cytokine. 1999;11:1046–1056. doi: 10.1006/cyto.1999.0503. [DOI] [PubMed] [Google Scholar]
- van Lent PL, van den Bersselaar LA, van den Hoek AE, van de Loo AA, van den Berg WB. Cationic immune complex arthritis in mice - a new model. Synergistic effect of complement and interleukin-1. Am J Pathol. 1992;140:1451–1461. [PMC free article] [PubMed] [Google Scholar]
- Danon D, Goldstein L, Marikovsky Y, Skutelsky E. Use of cationized ferritin as a label of negative charges on cell surfaces. . J Ultrastruct Res. 1972;38:500–510. doi: 10.1016/0022-5320(72)90087-1. [DOI] [PubMed] [Google Scholar]
- van den Broek MF, van den Berg WB, van de Putte LB, Severijnen AJ. Streptococcal cell wall-induced arthritis and flare-up reaction in mice induced by homologous or heterologous cell walls. Am J Pathol . 1988;133:139–149. [PMC free article] [PubMed] [Google Scholar]
- Unkeless JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med . 1979;150:580–596. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg WB, van de Putte LB, Zwarts WA, Joosten LA. Electrical charge of the antigen determines intraarticular antigen handling and chronicity of arthritis in mice. J Clin Invest. 1984;74:1850–1859. doi: 10.1172/JCI111604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing AJ, Bird CR, Bristow A, Poole S, Thorpe R. A simple sensitive bioassay for interleukin-1 which is unresponsive to 10(3) U/ml of interleukin-2. J Immunol Methods. 1987;99:7–11. doi: 10.1016/0022-1759(87)90025-1. [DOI] [PubMed] [Google Scholar]
- Drenth JP, van Uum SH, van Deuren M, Pesman GJ, van der Ven-Jongekrijg , van der Meer JW. Endurance run increases circulating IL-6 and IL-1ra but downregulates ex vivo TNF-alpha and IL-1 beta production. J Appl Physiol. 1995;79:1497–1503. doi: 10.1152/jappl.1995.79.5.1497. [DOI] [PubMed] [Google Scholar]
- van den Broek MF, van den Berg WB, van de Putte LB. The role of mast cells in antigen induced arthritis in mice. J Rheumatol. 1988;15:544–551. [PubMed] [Google Scholar]
- van Lent PL, van den Hoek AE, van den Bersselaar LA, van Rooijen N, van den Berg WB. Role of phagocytic synovial lining cells in experimental arthritis. Agents Actions. 1993;38 Spec No:C92–C94. doi: 10.1007/BF01991148. [DOI] [PubMed] [Google Scholar]
- van Meurs JB, van Lent PL, Holthuysen AE, Singer II, Bayne EK, van den Berg WB. Kinetics of aggrecanase- and metalloproteinase-induced neoepitopes in various stages of cartilage destruction in murine arthritis. . Arthritis Rheum. 1999;42:1128–1139. doi: 10.1002/1529-0131(199906)42:6<1128::AID-ANR9>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Jirholt J, Cook A, Emahazion T, Sundvall M, Jansson L, Nordquist N, Pettersson U, Holmdahl R. Genetic linkage analysis of collagen-induced arthritis in the mouse. Eur J Immunol. 1998;28:3321–3328. doi: 10.1002/(SICI)1521-4141(199810)28:10<3321::AID-IMMU3321>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Raine CS, Barnett LB, Brown A, Behar T, McFarlin DE. Neuropathology of experimental allergic encephalomyelitis in inbred strains of mice. Lab Invest. 1980;43:150–157. [PubMed] [Google Scholar]
- Simms HH, Gaither TA, Fries LF, Frank MM. Monokines released during short-term Fc gamma receptor phagocytosis up-regulate polymorphonuclear leukocytes and monocyte-phagocytic function. J Immunol. 1991;147:265–272. [PubMed] [Google Scholar]
- Marsh CB, Pope HA, Wewers MD. Fc gamma receptor cross-linking down-regulates IL-1 receptor antagonist and induces IL-1 beta in mononuclear phagocytes stimulated with endotoxin or Staphylococcus aureus. J Immunol. 1994;152:4604–4611. [PubMed] [Google Scholar]
- Arner EC, Pratta MA. Independent effects of interleukin-1 on proteoglycan breakdown, proteoglycan synthesis, and prostaglandin E2 release from cartilage in organ culture. Arthritis Rheum. 1989;32:288–297. doi: 10.1002/anr.1780320310. [DOI] [PubMed] [Google Scholar]
- van de Loo FA, Arntz OJ, Otterness IG, van den Berg WB. Protection against cartilage proteoglycan synthesis inhibition by anti-interleukin 1 antibodies in experimental arthritis. J Rheumatol. 1992;19:348–356. [PubMed] [Google Scholar]
- Chubinskaya S, Huch K, Mikecz K, Szabo G, Hasty KA, Kuettner KE, Cole AA. Chondrocyte matrix metalloproteinase-8: up-regulation of neutrophil collagenase by interleukin-1 beta in human cartilage from knee and ankle joints. Lab Invest. 1996;74:232–240. [PubMed] [Google Scholar]
- Sandy JD, Gamett D, Thompson V, Verscharen C. Chondrocyte-mediated catabolism of aggrecan: aggrecanase-dependent cleavage induced by interleukin-1 or retinoic acid can be inhibited by glucosamine. Biochem J. 1998;335:59–66. doi: 10.1042/bj3350059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner EC, Pratta MA, Trzaskos JM, Decicco CP, Tortorella MD. Generation and characterization of aggrecanase. A soluble, cartilage-derived aggrecan-degrading activity. J Biol Chem. 1999;274:6594–6601. doi: 10.1074/jbc.274.10.6594. [DOI] [PubMed] [Google Scholar]
- Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995;146:75–85. [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Song YW. Apoptotic chondrocyte death in rheumatoid arthritis. Arthritis Rheum. 1999;42:1528–1537. doi: 10.1002/1529-0131(199907)42:7<1528::AID-ANR28>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Williams RJ, Smith RL, Schurman DJ. Purified staphylococcal culture medium stimulates neutral metalloprotease secretion from human articular cartilage. J Orthop Res. 1991;9:258–265. doi: 10.1002/jor.1100090214. [DOI] [PubMed] [Google Scholar]
- Arner EC, Tortorella MD. Signal transduction through chondrocyte integrin receptors induces matrix metalloproteinase synthesis and synergizes with interleukin-1. Arthritis Rheum. 1995;38:1304–1314. doi: 10.1002/art.1780380919. [DOI] [PubMed] [Google Scholar]
- van Lent PL, van Vuuren AJ, Blom AB, Holthuysen AE, van de Putte LB, van de Winkel JG, van de Berg WB. Role of Fc Receptor γ chain in inflammation and cartilage damage during experimental antigen-induced arthritis. Arthritis Rheum. 2000;43:740–752. doi: 10.1002/1529-0131(200004)43:4<740::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]