

Short abstract
We have analyzed the pattern of procoagulant and fibrinolytic gene expression in affected joints during the course of arthritis in two murine models. In both models, we found an increased expression of tissue factor, tissue factor pathway inhibitor, urokinase plasminogen activator, and plasminogen activator inhibitor 1, as well as thrombin receptor. The observed pattern of gene expression tended to favor procoagulant activity, and this pattern was confirmed by functional assays. These alterations would account for persistence of fibrin within the inflamed joint, as is seen in rheumatoid arthritis.
Keywords: arthritis, coagulation, fibrinolysis, mice, RNase protection
Abstract
Introduction:
Accumulation of fibrin in the joints remains one of the most striking histopathological features of rheumatoid arthritis (RA). Recently, we have provided evidence of the deleterious role of synovial fibrin deposition in arthritic joints in antigen-induced arthritis (AIA), a well-established murine model of RA.
A local imbalance between fibrin formation and fibrin dissolution may result in fibrin deposition in the joints.
On the one hand, fibrin formation is mainly initiated by tissue factor (TF), a transmembrane protein serving as a receptor for factor VII. Under normal conditions, TF expression and activity are tightly regulated. Constitutive TF expression is restricted to perivascular and epithelial cells, and the catalytic activity of the TF/VIIa complex can be inhibited by tissue factor pathway inhibitor (TFPI). Pathological conditions can perturb the cell-type-restricted pattern of TF expression. In particular, recent reports have shown that transcriptional activation of TF can be mediated by molecular mechanisms involving induction of the early growth response gene 1 (EGR1) or of the protease-activated receptor (PAR1) or vascular endothelial growth factor (VEGF) genes.
On the other hand, fibrin degradation is mediated primarily by plasmin, which is the active form of the zymogen plasminogen. Conversion of plasminogen to plasmin is under the control of serine protease plasminogen activators, such as the urokinase plasminogen activator (uPA), and their inhibitors, such as the plasminogen activator inhibitor (PAI-1).
Aims:
We hypothesized that the deposition of fibrin in the joints may result from an imbalance in the local expression of key genes involved in coagulation and fibrinolytic pathways. To test this hypothesis, we investigated mRNA levels in arthritic versus nonarthritic joint tissues from two murine models of RA: AIA and collagen-induced arthritis (CIA). Genes that are directly implicated in coagulation (TF, TFPI) and fibrinolysis (UPA, PAI1), and other genes that may influence the expression of TF (EGR1, PAR1, VEGF), were investigated using a novel multiprobe RNase protection assay (RPA). Furthermore, we evaluated coagulation activity in arthritic and nonarthritic mice.
Methods:
Mice with AIA or CIA were sacrificed at different time points: 2, 4, and 16 h and 3, 7, and 14 d after intra-articular antigen injection for AIA; 42 d after the first immunization for CIA. Total RNA was prepared from arthritic and nonarthritic knees for AIA, or arthritic and nonarthritic hind paws for CIA. Messenger RNA (mRNA) levels of the genes described above were determined by RPA and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels. Coagulation assays were performed on joint tissue extracts and concentrations of thrombin-antithrombin III (TAT) complex were measured in plasma.
Results:
In AIA, all the genes studied except VEGF were upmodulated as early as 2 h. PAR1, TFPI, EGR1, and UPA expression decreased to control levels by 16 h, whereas the expression of TF and PAI1 remained elevated. At later times, only TF, PAI1, and UPA showed sustained overexpression. In CIA, gene expression was assayed at only one time point (42 d after immunization) and all genes showed higher mRNA levels in the affected paws than in control paws. In AIA mice, procoagulant activity and TF activity were significantly increased in arthritic joints, and in CIA mice, plasma TAT levels were significantly enhanced.
Discussion:
Fibrin deposition in synovia is prominent in both RA and experimental arthritis, suggesting that this protein may play a role in the pathogenesis of chronic inflammation. In this study, we have tried to shed some light on the molecular mechanisms leading to extravascular fibrin deposition, using two well-established mouse models of RA: AIA and CIA. The kinetics of gene expression was first analyzed in mice with AIA, because this model allows for an accurate, temporally controlled sampling of synovial inflammation. We then extended our observations by analyzing one time point in CIA, 42 d after immunization, when chronic inflammation is present. We found that in both models, coagulation and fibrinolysis in arthritic joints were significantly increased, and that the most significant increases were in TF and PAI-1.
Although the molecular mechanism or mechanisms responsible for the transcriptional changes observed are not completely understood, the increases in TF, PAI-1, and uPA are probably due to the production of proinflammatory cytokines such as IL-1 and TGF-α. These cytokines, whose presence in the inflamed synovium is well documented, are known to induce these genes through the activation of nuclear factor κB (NF-κB), a transcription factor. TF induction is also under the control of a proximal enhancer containing a binding site for the inducible transcription factor EGR1. Indeed, the early rise of EGR1 expression in AIA is consistent with its classification as immediate-early gene and may be responsible for the induction of early expression of TF. Early TF stimulation in AIA can also be accounted for by the transient overexpression of PAR1. Contrary to what has been shown in RA, VEGF expression remained essentially unchanged throughout the progression of AIA, probably reflecting a peculiarity of this murine model.
The alteration of the patterns of gene expression was accompanied by increased functional coagulation activity, which was more marked in AIA than in CIA.
Conclusion:
Prominent fibrin deposition in two different animal models of RA – AIA and CIA – can be attributed to modulations in key regulatory genes for coagulation and fibrinolysis.
Introduction
Rheumatoid arthritis (RA) is a common autoimmune disease of unknown etiology, characterized by chronic synovial inflammation that leads to progressive destruction of cartilage and bone [1]. Immunological mechanisms are thought to initiate synovial inflammation, which becomes persistent with the disease progression. Among the many histopathological features described, one of the most striking is the accumulation of fibrin [2,3]. We have recently provided evidence that synovial deposition of this protein plays a deleterious role in arthritic joints in antigen-induced arthritis (AIA), a well-established model of RA [4]. This accumulation of fibrin could result from a local imbalance between its formation and dissolution. Previous studies have revealed enhanced coagulation activity in rheumatoid synovial fluid and membrane [3,5] as well as increased activity of synovial urokinase plasminogen activator (uPA) in rheumatoid synovial membrane [6]. Little is known about the expression of procoagulant molecules in the arthritic synovial membrane, and the molecular events that tip the natural balance between synovial procoagulant and fibrinolysis in favor of coagulation remain to be elucidated.
Synovial fibrin deposition is mediated principally by tissue factor (TF), an activator of the extrinsic pathway of coagulation. TF is a transmembrane protein that initiates coagulation by serving as a cofactor for activated factor VII [7]. TF is constitutively expressed in perivascular and epithelial cells, but its expression can be induced on endothelial cells and monocytes by inflammation [8] and hypoxia [9]. Increased expression of TF during hypoxia is mediated by the transcription factor early growth response gene 1 (EGR1) product and leads to pulmonary fibrin deposition [9]. Tissue factor pathway inhibitor (TFPI), a naturally occuring Kunitz-type inhibitor, regulates coagulation by inhibiting the catalytic activity of the TF/VIIa complex [10]. Although little is known about its role in pathological processes, by virtue of its anticoagulant effect, under- or over-expression of this gene could alter the procoagulant/ anticoagulant balance.
Besides modulating fibrin deposition, activation of the coagulation pathway can also modulate genes that play an accessory role in inflammation by mediating cellular activation, for example thrombin receptor, protease-activated receptor 1 (PAR-1), and vascular endothelial growth factor (VEGF). PAR-1 is a G-protein-coupled receptor that binds thrombin [11] and is abundantly expressed in inflamed rheumatoid synovia [12]. Activation of PAR1 by thrombin can lead to proliferation of synovial fibroblasts and rapidly induces the transcription of TF mRNA [13]. Since PAR1 mRNA is itself upregulated by thrombin [14], PAR1 may be part of a positive-feedback loop that potentiates the coagulation cascade. VEGF stimulates endothelial-cell proliferation in vitro and induces neovascularization in vivo [15]. Significant amounts of antigenic VEGF have been detected in synovial fluids and tissues from RA patients [16], and VEGF mRNA is abundantly expressed in highly vascularized areas of the RA synovial tissue [17]. A TF-dependent production of VEGF by human fibroblasts in response to activated factor VII binding has been reported [18]. Conversely, VEGF can induce the expression of TF [19] and of UPA [15], thereby influencing the coagulation/fibrinolysis equilibrium.
Degradation of extravascular fibrin is primarily mediated by plasmin, which is formed upon cleavage of plasminogen by plasminogen activators [20]. This fibrin degradation is supported by increased synovial uPA activity evidenced in human and experimental arthritis and by the observation that lack of uPA leads to increased fibrin deposition in the joint [4]. The activity of uPA is regulated by plasminogen activator inhibitors (PAIs), in particular PAI-1 [20]. Both UPA and PAI1 expression are induced by cytokines that are abundant within the inflamed joint. [21].
In this study, we have analyzed the modulation of the mRNA levels of TF, TFPI, PAR1, EGR1, VEGF, UPA, and PAI1 in the inflamed joint in two murine models of arthritis, namely AIA and collagen-induced arthritis (CIA). These two models recapitulate many features of RA, especially fibrin deposition in arthritic joints (for AIA, see [4]; for CIA, Marty et al, submitted). We have used a novel multiprobe RNase protection assay that was developed to facilitate the simultaneous analysis of all the genes studied from small amounts of tissue. Functional assays of procoagulant activity (PCA) and TF activity were also performed on these tissues. Evidence of ongoing coagulation was sought in arthritic mice by measuring the plasma concentration of TAT.
Materials and methods
Induction of AIA
C57Bl/6 mice (Iffa-Credo, L'Arbresle, France) between 8 and 10 weeks old were immunized at day 0 and 7 with 100 μg of methylated bovine serum albumin (Sigma Chemical Company, Buchs, Switzerland) emulsified in 0.1 ml Freund's complete adjuvant containing 200 μg mycobacterial strain H37RA (Difco, Basel, Switzerland) by intradermal injection at the base of the tail. On the same day, 2 ×109 heat-killed Bordetella pertussis organisms (Berna, Bern, Switzerland) were injected intraperitoneally as additional adjuvant. Arthritis was induced at day 21 by intra-articular injection of 100 μg of methylated bovine serum albumin in 10 μl sterile phosphate-buffered saline solution (PBS) into the right knee, the left knee being injected with sterile PBS alone. Institutional approval was obtained for these experiments.
Induction of CIA
Male DBA/1J mice between 8 and 10 weeks of age were obtained from BRL/RCC Biotechnology & Animal Breeding (Füllinsdorf, Switzerland). Native chicken type II collagen (MM Griffith, Salt Lake City, UT, USA) was dissolved in 0.1 M acetic acid overnight at 2 mg/ml. Collagen (100 μg) emulsified in Freund's complete adjuvant containing 5 mg/ml Mycobacterium tuberculosis was injected intradermally at the base of the tail. At day 24 after the first injection, a booster injection of 100 μg of native chicken collagen in Freund's incomplete adjuvant was given intradermally at the base of the tail. All immunization reagents were purchased from DIFCO (Basel, Switzerland). Clinical assesment of arthritis was performed in accordance with established protocols [22].
RNA extraction
Cryostat sections of synovial tissues from knee joints (AIA model) or cryostat sections of total paws (CIA model) were homogenized in Trizol reagent (Gibco BRL, Berne, Switzerland) and total RNA extractions were performed in accordance with the manufacturer's instructions.
DNA template set preparation for RNase protection assay (RPA)
Complementary DNA (cDNA) fragments of a different size for each of the chosen genes were recovered by reverse transcriptase-polymerase chain reaction (RT-PCR) and subsequently subcloned into the pGEM-T plasmid (Promega, Wallisen, Switzerland). Positive clones with antisense orientation with respect to the T7 promoter were identified by colony polymerase chain reaction (PCR) and then their complete sequence was verified by DNA sequencing. Table 1 summarizes the details for each probe.
Table 1.
Probes used in the multiprobe RPA set
| Gene | Gene-bank | Sequence | Protected |
| template | accession number | chosen, nt | probe, nt |
| PAR1 | L03529 | 881-1280 | 400 |
| TFPI | AF004833 | 781-1090 | 310 |
| TF | M57896 | 655-940 | 286 |
| EGR1 | M20157 | 1581-1840 | 260 |
| VEGF | M95200 | 436-650 | 215 |
| UPA | X02389 | 1154-1354 | 201 |
| PAI1 | M33960 | 1151-1330 | 180 |
| GAPDH | M32599 | 944-1050 | 107 |
nt, nucleotides.
Probe labeling and RPA
Radioactive multiriboprobe preparation and RPA were performed in accordance with standard protocols. Briefly, anti-sense a32P-UTP-labeled riboprobes were synthesized by transcription in vitro of the DNA template set, which contained all the linearized plasmid cDNAs pooled in equimolar amounts. DNase I treatment was performed to remove the DNA templates and riboprobes were purified by phenol/chloroform extraction followed by ethanol precipitation using glycogen as carrier. For each sample, about 5 μg of total RNA was hybridized overnight at 52°C with 3 × 105 cpm of the labeled multiprobe. Samples were first treated with an RNase cocktail (Ambion, Austin, TX, USA); RNase was then removed by treatment with proteinase K and samples were purified by extraction in phenol/chloroform followed by ethanol precipitation using glycogen as carrier. Protected fragments were resolved through a 5% sequencing gel. Precise quantification of all the experimental samples for all time points studied was determined by analyzing the gels with an InstantImager apparatus and software (Packard Instruments, Berne, Switzerland). For each protected band analyzed, a cpm/mm2 value was obtained. This value was corrected for sample-loading errors by normalizing with the respective cpm/mm2 value calculated for the constitutively expressed glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, which was also included in the template set. Ratio values between arthritic/nonarthritic joints were then calculated for each time point.
Preparation of tissue extracts
Tissue extracts were prepared from cryostat sections (see RNA preparation above) as described before [4].
Measurement of procoagulant activity and TF activity
PCA was measured in tissue extracts from knee joints of mice with AIA or from paws of mice with CIA by a modified one-stage clotting assay. Briefly, tissue extracts (25 μl) were mixed with an equal volume of phospholipids (Dade-Behring, Düdingen, Switzerland). After adding 50 μl rabbit citrated plasma and 50 μl 0.02 M calcium chloride, time to thrombus formation was recorded using a micro-coagulometer (Dialine, Itingen, Switzerland). TF activity in tissue extracts was measured using a commercially available kit (American Diagnostica, Greenwich, CT, USA).
Determination of thrombin-antithrombin III
Citrated plasmas were obtained from arthritic and nonarthritic mice. Thrombin–antithrombin III (TAT) concentration in plasma was measured using a commercially available ELISA (enzyme-linked immunosorbent assay) kit designed for human TAT (Enzygnost TAT, Dade-Behring, Düdingen, Switzerland), which cross-reacts also with murine TAT. The concentration of murine TAT was calculated according to the human TAT standard curve.
Statistical analysis
The unpaired Student's t-test was used to compare means for normally distributed values. A level of P < 0.05 was considered statistically significant.
Results
Detection of gene expression by multiprobe RNAse protection assay
A typical autoradiograph obtained with the multiprobe set is shown in Fig. 1a. It reveals the expression of eight probe-protected bands in RNA from mouse skin. The actual size of each of the protected fragments corresponded with their expected size (compare with Table 1).
Figure 1.
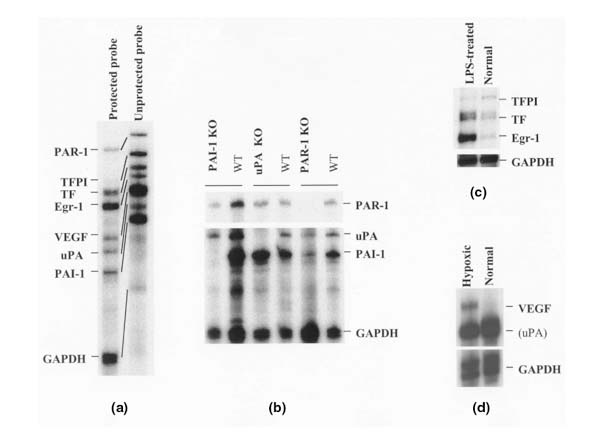
RNase protection assay with the set of coagulation/fibrinolytic genes. (a) Typical eight-band protection pattern resulting from the analysis of 5 μg of mouse-skin total RNA. (b) Kidney RNA prepared from various knockout (KO) mice (PAI1-, UPA-, or PAR1-deficient mice) and from wild-type (WT) mice, analyzed using RPA. (c) Kidney RNA prepared from lipopolysaccharide (LPS)-treated or untreated mice, analyzed using RPA. (d) RNA from murine hypoxic or normal endothelial cells, analyzed using RPA. In all these RPA experiments, 5 μg of total RNA/lane was analyzed.
Confirmation of multiprobe specificity
To confirm the specificity of the bands detected, we analyzed RNA samples that either lacked a particular mRNA species or over- or under-expressed it.
In Fig. 1b, the patterns of the protected fragments detected in RNAs prepared from PAI1, UPA, and PAR1 knockout mice were compared with those prepared from wild-type mice. As expected, no PAI1, UPA, or PAR1 protected bands were detected in kidney RNA from the respective deficient mice. For TF, TFPI, and EGR1, we analyzed kidney RNA from lipopolysaccharide-treated mice (Fig. 1c), which confirmed the upregulation of TF and EGR1 upon lipopolysaccharide treatment, while TFPI was downregulated [23]. For VEGF, we observed its striking upregulation in hypoxic endothelial cells using the multi-probe assay, whereas the mRNA was not detected in normoxic cells (Fig. 1d).
Synovial expression of procoagulant and fibrinolytic genes in AIA
Total RNAs were extracted from dissected synovial tissues of arthritic and nonarthritic joints (injected with PBS) at various time points (2, 4, and 16 h, and 3, 7, and 14 d) after AIA induction. A representative multiprobe assay autoradiograph showing the mRNA expression at 4 h and at 7 d in the arthritic and nonarthritic knees is shown in Fig. 2. As early as 4 h after AIA induction, there was a clear-cut upregulation of all the genes except VEGF in the arthritic (right) knee as compared with the uninvolved (left) knee. At 7 d after AIA induction, although increased expression of TF and PAI1 mRNA was still evident in the arthritic joint, the increased expression of the other mRNA species was less marked than before. Analysis of the whole time course of expression yielded four major observations (Fig. 3):
Figure 2.
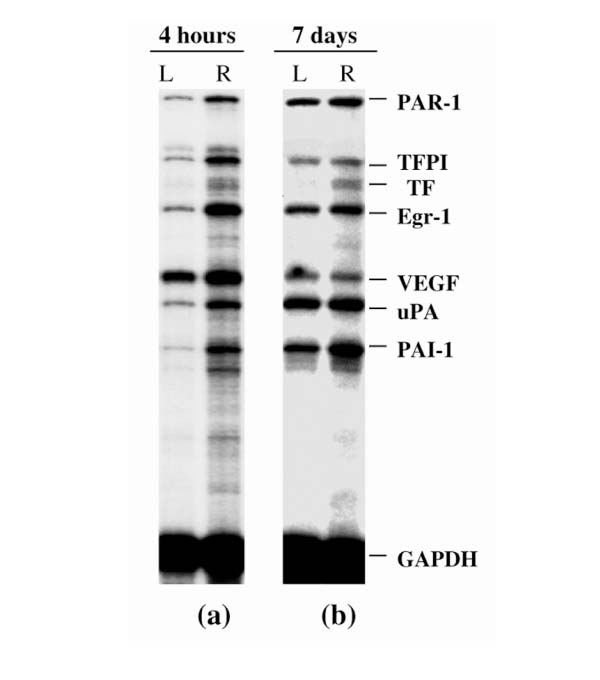
Synovial levels of mRNA from procoagulant and fibrinolytic genes in AIA, as analyzed by RPA. (a) Autoradiographic signals of synovial RNA samples from left (L; control) and right (R) knees, 4 h after arthritis induction. The single sample shown for each condition shows the typical, reproducible pattern obtained from the RPA analysis of at least three different RNAs, each from different animals. (b) Same as (a), but 7 d after arthritis induction.
Figure 3.
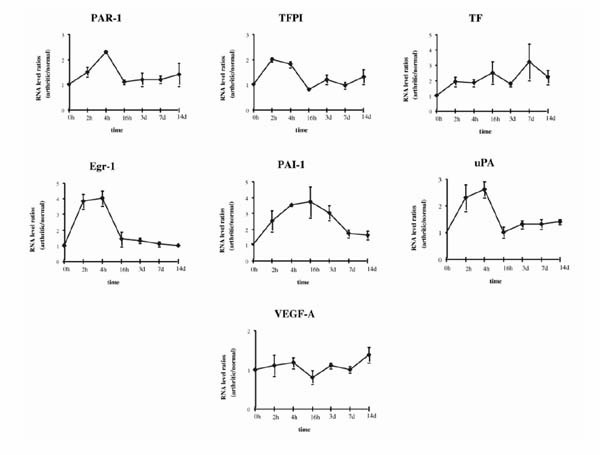
Time course of accumulation of mRNA from procoagulant and fibrinolytic genes during AIA. RNA levels are expressed as arthritic/normal knee value ratios, after correcting for GAPDH signal. The 0 time point is given the value 1. Values are the mean ± SEM of at least three different mRNA values from three different mice.
1) PAR1, TFPI, EGR1, and UPA showed a rapid initial increase of mRNA levels, which peaked between 2 and 4 h, followed by a sharp decrease to near basal levels at 16 h.
2) The kinetics of PAI1 and TF induction differed from that of the other genes studied. PAI1 reached its peak level (a 3.5-fold increase in comparison with the control knee) at 16 h, followed by a slow decrease, so that the level was twofold by day 7 and 1.5-fold by day 14. TF expression increased gradually, peaking (to more than threefold) by day 7, and remaining at more than twofold by day 14.
3) At day 14, expression of TF, PAI1, and UPA was still increased (to about 1.4-fold each).
4) VEGF expression hardly changed during the experiment.
Increased expression of procoagulant and fibrinolytic genes changes in collagen-induced arthritis
The expression of the same genes during CIA was analyzed using the same multiprobe set. RNA samples from arthritic paws of collagen-immunized mice were compared with samples from nonimmunized control mice. For this experiment, we used only one time point. Animals were sacrified 42 d after the first immunization, when the incidence of arthritis reaches >80% and the severity, as evaluated by clinical scores, is maximal.
Figure 4a shows a representative autoradiograph demonstrating the upregulation of all the genes assayed in the arthritic paw in comparison with the control paw. TF and PAI1 had the highest increases – about fourfold and about threefold, respectively (Fig. 4b). All the other genes showed an approximately twofold increase over controls.
Figure 4.
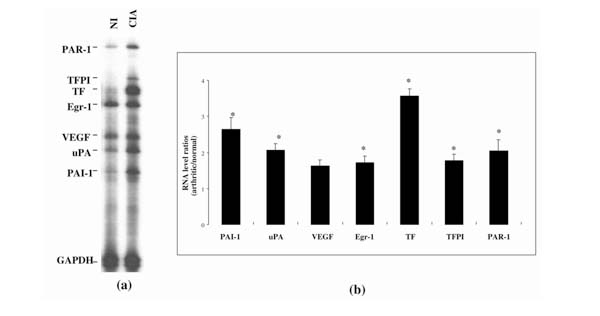
RNA from procoagulant and fibrinolytic genes in paws of mice with CIA, as analyzed by RPA. (a) Autoradiographic signals of RNA samples from eitherarthritic paws (CIA) or normal paws from nonimmunized (NI) animals. The single sample shown for each condition represents the typical, reproducible pattern obtained from the RPA analysis of four RNAs, each from a different animal. (b) Expression of genes studied during CIA, shown as the ratio of mRNA in arthritic to that in normal paws (CIA/NI), after correction for GAPDH signal. Values are means ± SEM of at least four different mRNA values from four different mice. *P < 0.05, CIA versus NI values.
Increased procoagulant and TF activity in arthritic joints
Since the pattern of mRNA modulation observed in AIA and CIA strongly suggested a shift of the coagulation/fibrinolytic balance in favor of coagulation during joint inflammation, we next tried to confirm this prediction at a functional level. We first compared PCA in tissue extracts prepared from arthritic and nonarthritic joints (Fig. 5a). Time to reach thrombus formation was decreased in arthritic joints in both models (by 24% in CIA, and by 72% in AIA), although only the decrease in AIA was statistically significant. Because increased PCA in arthritic joints could result from increased TF activity, we also studied TF activity in these samples (Fig. 5b). It was increased in both models (by 78% in CIA, and by 365% in AIA), but only the increase in AIA was statistically significant.
Figure 5.
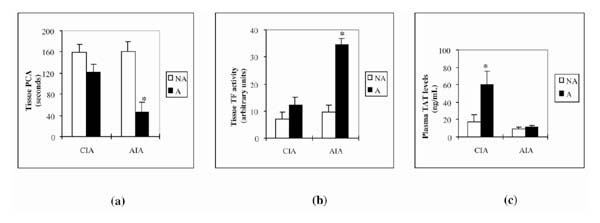
Activity of procoagulant and TF and concentrations of plasma TAT in mice with induced arthritis (CIA or AIA) and controls. (a) PCA (expressed in seconds taken for thrombus formation) in tissue extracts. Arthritic tissue extracts (A) were prepared from arthritic, swollen paws at day 42 after the first immunization (CIA model) or from arthritic right knee joints at day 3 after arthritis induction (AIA model). Nonarthritic (NA) tissue extracts were from paws of normal, nonimmunized mice (CIA model) or normal left knees (contralateral to induced arthritis; AIA model). (b) TF activity in arthritic (A) and nonarthritic (NA) tissue extracts (the same extracts as were used for PCA), measured using a chromogenic assay. (c) Plasma concentrations of TAT complexes as determined in arthritic (A) mice in comparison with nonimmunized, nonarthritic (NA) controls. Citrated plasmas from arthritic mice were prepared at day 42 after the first immunization (CIA model) or at day 3 after arthritis induction (AIA model). Values are means ± SEM of at least four samples from four mice. –*P < 0.05, arthritic versus nonarthritic values.
Increased plasma TAT levels in arthritic mice
Because TAT is considered to be an indicator of ongoing activation of the clotting system, we measured its concentration in the plasma of arthritic mice. We found a statistically significant increase of TAT in the plasma of CIA animals (3.5 times its concentration in nonarthritic mice) but only a very modest increase (by 27%; not statistically significant) in AIA (Fig. 5c).
Discussion
Excessive fibrin deposition has been shown to be deleterious in inflammation associated with various diseases such as hypoxia, sepsis, and RA [24,25]. Because the molecular mechanisms underlying fibrin deposition in RA have not been elucidated, we have characterized the alteration in coagulation and fibrinolytic gene expression in two murine models of RA. By analyzing the kinetics of gene expression in the AIA model, we were able to sample the very early phases of synovial inflammation. To extend our observations, we then analyzed total paw RNA prepared from CIA mice. In this model, synovial fibrin is also present and has a deleterious role in inflammation (Marty et al, submitted). In CIA, we studied gene expression at 42 d after immunization, when inflammation is clinically active. One possible drawback of using total paw RNA is that paws contain tissues in addition to synovia. Nevertheless, the use of total paw RNA has been shown to accurately reflect synovial RNA changes of cytokine levels in CIA [26].
An upregulation of both procoagulant and fibrinolytic genes was observed in both models. The pattern of expression in CIA at day 42 approximated that at the early time points in AIA, with TF and PAI1 showing the most significant increases. If the protein expression of these molecules parallels their transcription, then the combined increase of TF and PAI-1 would favor fibrin formation and its persistence in the joint.
The transcriptional regulation of the changes noted are not completely understood. The increases in TF, PAI-1, and uPA are most likely to be due to the increased secretion of cytokines such as IL-1 and TNF-α, which are known inducers of these genes and whose presence in the synovium in AIA has been documented by other workers [27]. The transcriptional regulation of the TF gene probably involves activation of the transcription factor nuclear factor κB (NF-κB). Indeed, an enhancer in the TF gene containing an NF-κB site that is activated by endotoxin, TNF-α, and IL-1 has been found [28]. Moreover, findings in a recent study [29] suggest that induced TF and PAI1 expression by vascular smooth muscle and endothelial cells may be simultaneously mediated by activation of NF-κB. In addition, TF expression is under the control of another proximal enhancer [28], containing overlapping binding sites for the constitutively expressed SP1 and the inducible EGR1 transcription factor. The early rise of EGR1 expression in AIA, at 2 to 4 h, is consistent with the immediate-early gene expression pattern of this gene and may be responsible for the induction of early expression of TF. However, by day 3, EGR1 expression had declined to control levels, while TF expression remained elevated, an observation suggesting that EGR1 does not contribute to the sustained overexpression of TF in AIA synovial tissues. Transient PAR1 overexpression can also contribute to the early stimulation of TF [13]. VEGF expression remained essentially unchanged throughout the progression of AIA, with only a slight increase at the late time points. In CIA there was only a slight, not significant, increase of VEGF mRNA levels. This is somewhat surprising, in view of data showing increased expression in RA [17].
We found significantly increased functional coagulation activity in the AIA model but only a slight, nonsignificant increase in the CIA model. TF activity in AIA can be totally accounted for by changes in TF mRNA. By contrast, the discrepancy between TF activity and TF mRNA levels in CIA needs to be clarified. Furthermore, we were able to demonstrate increased plasma TAT levels in both models, though the difference was more striking in CIA than in AIA. Plasma TAT levels result most probably from the spillover into the systemic circulation of TAT that originated in arthritic joints. As multiple joints are affected in CIA, but only one in AIA, one could expect TAT values to be more elevated in CIA than in AIA. Altogether, these findings support the conclusion that articular inflammation is accompanied by significant upregulation of molecules that favor increased extravascular coagulation activity and fibrin deposition.
In conclusion, we have found that experimental arthritis is associated with a pronounced upregulation of both pro-coagulation and fibrinolytic genes, that in its later stages tends to favor the formation and persistence of fibrin. Workers using mice with targeted disruptions of either the fibrinogen gene and/or genes of the fibrinolytic pathway have observed that fibrin persistence may delay wound healing [30] and exacerbate progression of atherosclerosis [31], glomerulonephritis [32], and arthritis [4]. In the latter case, if efficient fibrinolytic mechanisms are not sufficiently activated in the course of joint inflammation, the persistence of joint fibrin may contribute to continued synovitis and subsequent joint damage.
Acknowledgments
Acknowledgements
This work was supported by a grant from the Fonds national suisse de la recherche scientifique (number 32-56710.99).
Contributor Information
Roberto Salvi, Email: Nathalie.Busso@chuv.hospvd.ch.
Nathalie Busso, Email: natalie.busso@chuv.hospvd.ch.
References
- Firestein GS. Etiology and pathogenesis of rheumatoid arthritis. . In Textbook of Rheumatology Edited by Kelley WN, Harris ED, Ruddy S, Sledge CB Philadelphia: W B Saunders, 1997. pp. 851–897.
- Clemmensen I, Holund B, Andersen RB. Fibrin and fibronectin in rheumatoid synovial membrane and rheumatoid synovial fluid. Arthritis Rheum. 1983;26:479–485. doi: 10.1002/art.1780260405. [DOI] [PubMed] [Google Scholar]
- Weinberg JB, Pippen AM, Greenberg CS. Extravascular fibrin formation and dissolution in synovial tissue of patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheum. 1991;34:996–1005. doi: 10.1002/art.1780340809. [DOI] [PubMed] [Google Scholar]
- Busso N, Péclat V, Van Ness K, Kolodziesczyk E, Degen J, Bugge T, So A. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J Clin Invest. 1998;102:41–50. doi: 10.1172/JCI2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmassi F, De Negri F, Morale M, Puccetti R, Chung SI. Elastase- and plasmin-mediated fibrinolysis in rheumatoid arthritis. . Int J Tissue React. 1994;16:89–93. [PubMed] [Google Scholar]
- Busso N, Péclat V, So A, Sappino AP. Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann Rheum Dis. 1997;56:550–557. doi: 10.1136/ard.56.9.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerer E, Kolsto AB, Prydz H. Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res. 1996;81:1–4. doi: 10.1016/0049-3848(95)00209-x. [DOI] [PubMed] [Google Scholar]
- Mackman N, Sawdey MS, Keeton MR, Loskutoff DJ. Murine tissue factor gene expression in vivo: tissue and cell specificity and regulation by lipopolysaccharide. Am J Pathol. 1993;143:76–84. [PMC free article] [PubMed] [Google Scholar]
- Yan SF, Zou YS, Gao Y, Zhai C, Mackman N, Lee SL, Milbrandt J, Pinsky D, Kisiel W, Stern D. Tissue factor transcription driven by Egr-1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxia. . Proc Natl Acad Sci USA. 1998;95:8298–8303. doi: 10.1073/pnas.95.14.8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broze GJ., Jr Tissue factor pathway inhibitor and the revised theory of coagulation. Annu Rev Med. 1995;46:103. doi: 10.1146/annurev.med.46.1.103. [DOI] [PubMed] [Google Scholar]
- Vu T-KH, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Morris R, Winyard PG, Brass LF, Blake DR, Morris CJ. Thrombin receptor expression in rheumatoid and osteoarthritic synovial tissue. . Ann Rheum Dis. 1996;11:841–843. doi: 10.1136/ard.55.11.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm AK, Norstrom E, Sundelin J, Nysted S. Stimulation of proteinase activated receptor-2 causes endothelial cells to promote blood coagulation in vitro. Thromb Haemost. 1999;81:984–988. [PubMed] [Google Scholar]
- Shin H, Nakajima T, Kitajima I, Shigeta K, Abeyama K, Imamura T, Okano T, Kawahara K, Nakamura T, Maruyama I. Thrombin receptor-mediated synovial proliferation in patients with rheumatoid arthritis. Clin Immunol Immunopathol. 1995;76:225–233. doi: 10.1006/clin.1995.1120. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, Jackman RW, Senger DR, Dvorak HF, Brown LF. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med . 1994;180:341–346. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekanecz Z, Szegedi G, Koch AE. Angiogenesis in rheumatoid arthritis: pathogenic and clinical significance. J Invest Med . 1998;46:27–41. [PubMed] [Google Scholar]
- Olivier V, Bentolila S, Chabbat J, Hakim J, de Prost D. Tissue factor-dependent vascular endothelial growth factor production by human fibroblasts in response to activated factor VII. Blood. 1998;91:2698–2703. [PubMed] [Google Scholar]
- Mechtcheriakova D, Wlachos A, Holzmüller H, Binder BR, Hoefer E. Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by Egr-1. Blood. 1999;11:3811–3823. [PubMed] [Google Scholar]
- Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest. 1991;88:1067–1072. doi: 10.1172/JCI115405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA, Campbell IK, Wojta J, Cheung D. Plasminogen activators and their inhibitors in arthritic disease. Ann NY Acad Sci. 1992;667:87–100. doi: 10.1111/j.1749-6632.1992.tb51602.x. [DOI] [PubMed] [Google Scholar]
- Bakker AC, Joosten LA, Arntz OJ, Helsen MM, Bendele AM, van de Loo FA, van den Berg WB. Prevention of murine collagen-induced arthritis in the knee and ipsilateral paw by local expression of human interleukin-1 receptor antagonist protein in the knee. Arthritis Rheum. 1997;40:893–900. doi: 10.1002/art.1780400517. [DOI] [PubMed] [Google Scholar]
- Hara S, Asada Y, Hatakeyama K, Marutsuka K, Sato Y, Kisanuki A, Sumiyoshi A. Expression of tissue factor and tissue factor pathway inhibitor in rats lungs with lipopolysaccharide-induced disseminated intravascular coagulation. Lab Invest. 1997;77:581–589. [PubMed] [Google Scholar]
- Idell S, James KK, Levine EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest. 1989;84:695–705. doi: 10.1172/JCI114217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coalson JJ. Pathology of sepsis, septic shock, and multiple organ failure. In Perspective on Sepsis and Septic Shock Edited by the Society of Critical Care Medicine Fullerton, CA, 1986. pp. 27–59.
- Thornton S, Duwel LE, Boivin GP, Ma Y, Hirsch R. Association of the course of collagen-induced arthritis with distinct patterns of cytokine and chemokine messenger RNA expression. Arthritis Rheum. 1999;42:1109–1118. doi: 10.1002/1529-0131(199906)42:6<1109::AID-ANR7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- van de Loo FA, Joosten LA, van Lent PL, Arntz OJ, van den Berg WB. Role of interleukin-1, tumor necrosis factor alpha, and interleukin-6 in cartilage proteoglycan metabolism and destruction. Effect of in situ blocking in murine antigen- and zymosan-induced arthritis. . Arthritis Rheum. 1995;38:164–172. doi: 10.1002/art.1780380204. [DOI] [PubMed] [Google Scholar]
- Mackman N. Regulation of the tissue factor gene. FASEB J. 1995;9:883–889. doi: 10.1096/fasebj.9.10.7615158. [DOI] [PubMed] [Google Scholar]
- Dechend R, Maass M, Gieffers J, Dietz R, Scheidereit C, Leutz A, Gulba DC. Chlamydia pneumoniae infection of vascular smooth muscle and endothelial cells activates NF-κB and induces tissue factor and PAI-1 expression. Circulation. 1999;100:1369–1373. doi: 10.1161/01.cir.100.13.1369. [DOI] [PubMed] [Google Scholar]
- Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–719. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- Lou XJ, Boonmark NW, Horrigan FT, Degen JL, Lawn RM. Fibrinogen deficiency reduces vascular accumulation of apolipoprotein(a) and development of atherosclerosis in apolipoprotein(a) transgenic mice. . Proc Natl Acad Sci USA. 1998;95:12591–12595. doi: 10.1073/pnas.95.21.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitching AR, Holdsworth SR, Ploplis VA, Plow EF, Collen D, Carmeliet P, Tipping PG. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med. 1997;185:963–968. doi: 10.1084/jem.185.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]