

Abstract
Metazoans have evolved multiple paralogues of the TATA binding protein (TBP), adding another tunable level of gene control at core promoters. While TBP-related factor 1 (TRF1) shares extensive homology with TBP and can direct both Pol II and Pol III transcription in vitro, TRF1 target sites in vivo have remained elusive. Here, we report the genome-wide identification of TRF1-binding sites using high-resolution genome tiling microarrays. We found 354 TRF1-binding sites genome-wide with ∼78% of these sites displaying colocalization with BRF. Strikingly, the majority of TRF1 target genes are Pol III-dependent small noncoding RNAs such as tRNAs and small nonmessenger RNAs. We provide direct evidence that the TRF1/BRF complex is functionally required for the activity of two novel TRF1 targets (7SL RNA and small nucleolar RNAs). Our studies suggest that unlike most other eukaryotic organisms that rely on TBP for Pol III transcription, in Drosophila and possibly other insects the alternative TRF1/BRF complex appears responsible for the initiation of all known classes of Pol III transcription.
Keywords: ChIP-on-chip analysis, RNA polymerase III transcription, small nonmessenger RNA, TBP-related factor
Introduction
It is essential for multicellular organisms to support a complex network of gene expression patterns that are highly regulated during development and responsive to a variety of physiological stimuli. Under the current working hypothesis of eukaryotic transcriptional initiation, mechanisms that support the specificity of transcriptional activation are largely attributed to enhancer–core-promoter interactions. The protein machinery that enables these interactions includes sequence-specific enhancer DNA-binding proteins and the core-promoter recognition machinery, which is generally believed to be universal at all promoters and largely invariant. However, a number of recent studies have revealed that there are alternatives of core-promoter complexes that have evolved in multicellular organisms. It appears that mixing and matching these two classes of transcriptional factors represent a significant mechanism by which activation and repression at specific promoters are achieved. Although activators that direct specific transcriptional responses have been amply documented, functions of alternative core-promoter recognition complexes remained poorly understood. Diversified core-promoter machinery such as variant TFIID and TATA binding protein (TBP)-related factors (TRFs) are found in many metazoan species (Hochheimer and Tjian, 2003), but it remains unclear how these alternative core-promoter recognition complexes contribute to mechanisms of transcriptional regulation. Early studies addressing individual genes have been conducted largely using in vitro biochemical approaches to dissect the role of these alternative core-promoter recognition factors (Hansen et al, 1997; Holmes and Tjian, 2000; Takada et al, 2000; Hochheimer et al, 2002). However, a bottleneck to progress further has been the identification of genome-wide in vivo targets for these factors. Here, we describe the use of chromatin immunoprecipitation (ChIP) assays combined with genome tiling microarrays (ChIP-on-chip) coupled with a new computational tool to more accurately identify, in an unbiased manner, genome-wide targets of core-promoter recognition factors. To test the usefulness of this strategy, we have applied this methodology to the mapping of specific promoters targeted by the TRF1/BRF complex.
TRF1 represents a unique class of TRF found in insect species such as Drosophila and Anopheles. TRF1 is ubiquitously expressed, although it is upregulated in the central nervous system during embryogenesis and in primary spermatocytes in adults (Crowley et al, 1993; Hansen et al, 1997). Extensive sequence conservation between TBP and TRF1 is found within the core DNA-binding domains, whereas significant divergence is seen in the N-terminal domain. Previously, in vitro biochemical approaches established that TRF1 is likely involved in transcription of both Pol II and Pol III genes (Hansen et al, 1997; Holmes and Tjian, 2000). Importantly, a majority of the TRF1 in Drosophila appears to form a complex with BRF (Takada et al, 2000). In vitro transcription assays revealed that the TRF1/BRF complex plays a critical role in the transcription of several tRNA, 5S rRNA and U6 snRNA genes. Salivary gland polytene chromosome staining suggested that TRF1 can occupy a few hundred genomic sites, the majority of which are co-occupied by BRF (Takada et al, 2000). Our findings also suggested that the TRF1/BRF complex at these few promoters displayed an apparent dominance over TBP-containing complexes. To gain a more comprehensive of the transcriptional role played by TRF1, it would be advantageous to decipher the potential utilization of TRF1 on a genome-wide scale. In particular, we hoped to discover how TRF1 might be utilized for transcription mediated by different types of RNA polymerases. However, inherent limitations of resolution have precluded the analysis of polytene chromosome staining as a means to unambiguously identify specific promoters recognized and regulated by TRF1/BRF. Here, we report the higher resolution and genome-wide mapping of TRF1/BRF-binding sites using ChIP-on-chip assays. Using this experimental platform, we obtained a high-resolution (35 bp) in vivo map of TRF1- and BRF-binding sites throughout the Drosophila genome. Consistent with our previous in vitro biochemical findings, a major class of TRF1/BRF targets represents Pol III genes such as tRNAs. A small percentage of sites bound by TRF1 were mapped to Pol II promoters. In addition, we report two new classes of TRF1/BRF targets, 7SL RNA and small nucleolar RNAs (snoRNAs), which are small nonmessenger RNAs (snmRNAs). In vitro transcription assays were used to verify that the TRF1/BRF complex is functionally required for accurate transcription initiation of these new target genes. Taken together, these results strongly support a global role of the TRF1/BRF complex in Drosophila Pol III transcription.
Results
Genome-wide colocalization of TRF1 and BRF at noncoding small RNA promoters
In order to determine high-resolution in vivo target genes of the TRF1/BRF complex, we performed ChIP-on-chip analyses using Drosophila genome tiling arrays (Affymetrix). This high-density oligonucleotide array covers the entire genome of Drosophila melanogaster at 35 bp resolution with the notable exception of repeat regions such as transposons and 28S and 5S rRNA genes. We first established robust ChIP assays using affinity-purified anti-TRF1 and anti-BRF antibodies that efficiently co-precipitate specific genomic fragments such as 5S rRNA and tRNA genes. These few genes had previously been characterized as targets of the TRF1/BRF complex in vitro and are typically precipitated by the specific antibodies at a level 20- to 100-fold above nonspecific IgG controls (Figure 2A). These co-precipitated genomic fragments were amplified and subsequently hybridized to the microarrays in duplicate. The data were extensively analysed using a newly developed statistical platform (Tiling Hierarchical Gamma Mixture Model, TileHGMM). This statistical approach explicitly modeled binding of the probes in the control sample and TRF1/BRF-enriched samples (Figure 1A). The fitting of this statistical model provided us with probabilities of binding that is specific to a genomic region of interest. We then identified TRF1- and BRF-bound regions by thresholding these probabilities while controlling the false discovery rate using a false discovery rate calculation (Newton et al, 2004). Initially, 215 genomic regions (2 kb on average) were identified as bound by TRF1 and 211 by BRF. Some of the genomic regions contained multiple peaks indicative of multiple target sites, which were identified as part of postprocessing by calculating the odds of binding according to our statistical model (Supplementary data). Overall, we identified 354 binding sites for TRF1 and 359 binding sites for BRF, which appear largely colocalized and uniformly distributed on each chromosome (Figure 1B and C).
Figure 2a.
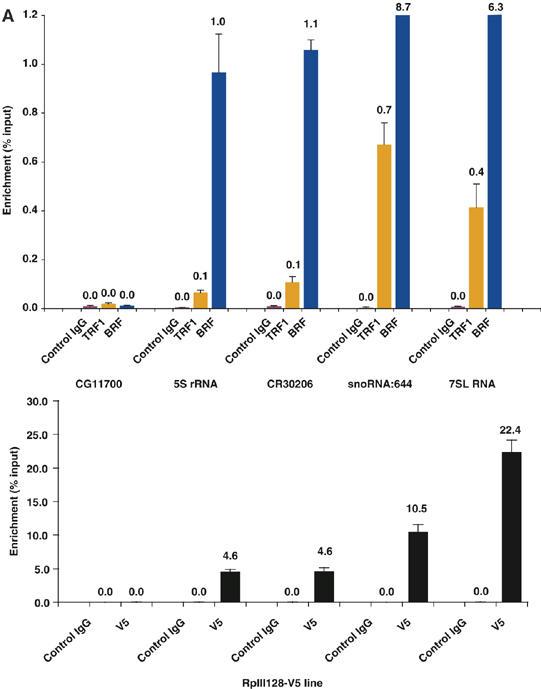
Spatial structure of enrichment by TRF1/BRF ChIP. (A) (top) Quantitative PCR detection of specific enrichment of small noncoding RNA promoters by TRF1/BRF ChIP. 5S rRNA, CR30206 (tRNA), snoRNA:644, and 7SL RNA promoter regions are significantly enriched by TRF1/BRF ChIP whereas the promoter region of a Pol II gene (CG11700) is not. (Bottom) ChIP assay using S2 cells expressing V5-tagged Pol III-specific subunit RPIII128.
Figure 1.

Overview of ChIP-on-chip data analysis. (A) TileHGMM: a statistical framework for the analysis of Chip-on-chip data. Our pipeline for the Chip-on-chip data analysis involves: (1) preprocessing and normalization; (2) performing diagnostic checks for validating statistical model assumptions; (3) partitioning each chromosome into genomic regions of approximately 2000 base pairs; (4) fitting a hierarchical gamma mixture model that models probe-level occupancy measures while allowing information sharing across probes to accommodate small sample sizes; (5) identifying a final set of bound regions by thresholding posterior probability of binding estimated by the statistical model fit; (6) annotation of the genomic regions. In (4), TileHGMM assumes that each genomic partition has at most one peak. Within each unbound genomic region, probe-specific control and IP-enriched observations follow different Gamma distributions conditional on latent (unobserved) mean binding measure. Let μj1(i) and μj2(i) represent the latent control and IP-enriched means for probe j in genomic region i. The model assumes that control binding measurements for probe j form a random sample from a Gamma distribution with scale and shape parameters equal to a1 and μj1(i)/a1, respectively. This ensures probe-specific control binding distributions with mean μj1 while avoiding overparameterization through a common scale parameter. Similarly, IP-enriched binding measurements for probe j form a random sample from a Gamma distribution with scale and shape parameters equal to a2 and μj2(i)/a2, respectively. If region i is unbound, μj1(i)=μj2(i). Otherwise, we have μj2(i)>μj1(i), reflecting transcription factor–DNA interactions, as we expect the IP-hybridizations to be greater than the control hybridizations. (B) Genome-wide colocalization of TRF1/BRF-binding sites. For each chromosome, the top graph represents TRF1, and the bottom graph represents BRF. Chromosome 4 was omitted as no binding sites were observed. The X-axis represents the genomic location in Mbp, and the Y-axis represents the binding efficiency indicated by likelihood ratio score. (C) Distribution of TRF1/BRF bound regions across the genome. Number of total TRF/BRF-binding sites per each chromosome is plotted on the graph.
In order to visualize the spatial topology of the enrichment revealed by TRF1/BRF immunoprecipitations, the signal intensity of each probe was plotted over the chromosomal locations for the selection of genomic regions. Figure 2B displays a region on the X chromosome that encodes three tRNA genes (CR30208, CR30206, and CR30207) displaying equally strong hybridization signals, indicating that all three sites are bound by TRF1 and BRF with comparable efficiency. The second region represents chromosome 3R with multiple TRF1/BRF occupancies at five tRNA genes (CR31485, CR31490, CR31487, CR31489, and CR31486) and two noncoding RNA genes corresponding to 7SL RNA (Figure 2C). The third example illustrates snoRNA:644 gene on chromosome 2R (Figure 2D). In all three cases, it is evident that the hybridization signals peak precisely in register with these noncoding RNA genes. We further verified these microarray results by conventional quantitative PCR detection of co-immunoprecipitated fragments. All sites except for CG11700, a control locus that is not bound by TRF1 and BRF, displayed a significant enrichment with antibodies against TRF1 and BRF, but not with control IgG (Figure 2A). These results confirm that our microarray detection methodology faithfully recapitulates conventional ChIP detected by quantitative PCR.
Figure 2b.
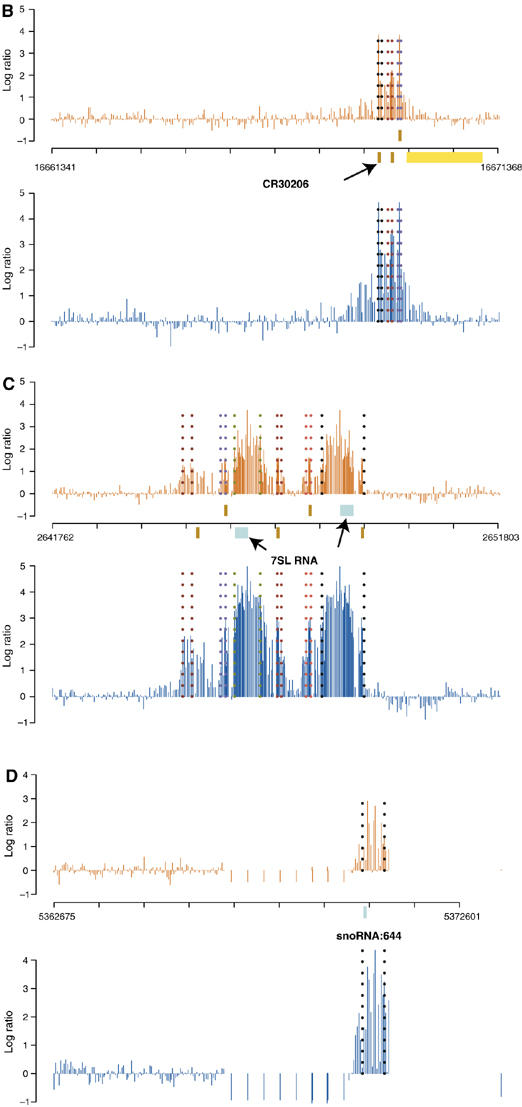
(B–D) High-resolution detection of TRF1/BRF ChIP by genome tiling microarrays. Top plot is the averaged log ratios of ChIP and control binding intensities over two TRF1 replicate experiments, bottom plot is the same for BRF1. Dashed lines mark the predicted peak start and end positions. All the annotations are based on the 4.2.1 version of the D. melanogaster genome. Brown bars indicate tRNA genes, and light blue bars indicate snmRNA genes. (B) Cytosolic tRNA genes (CR30208 on the + strand; tRNAs CR30206, CR30207) on chromosome 2R are bound by the TRF1/BRF complex. The yellow bar indicates the Pol II gene CG4266 on the − strand. (C) 7SL RNA locus (from left to right, CR31490, CR31489, 7SL RNA, CR32864 on the + strand; CR31485, 7SL RNA, CR31487, CR31486 on the − strand) on chromosome 3R is bound by the TRF1/BRF complex. We identified the second 7SL RNA on the − strand by a BLAST search. (D) snoRNA:644 locus on chromosome X is bound by the TRF1/BRF complex.
We next annotated the identified TRF1/BRF-binding sites using the 4.2.1 version of the D. melanogaster genome from FlyBase. We classified the target genes of the TRF1/BRF complex into several categories based on the available annotation (Figure 3 and Table I, full lists of the identified targets are in Supplementary Tables). Strikingly, we found that 77.7% of all the identified sites are shared by TRF1 and BRF, consistent with our previous biochemical observation that the majority of TRF1 in Drosophila cells appears to be in a complex with BRF (Takada et al, 2000). The major class of these colocalized sites corresponded to tRNA genes. Importantly, we found that 93% of all known tRNA genes in Drosophila are bound by both TRF1 and BRF. In addition, a minor fraction (4%) is bound only by BRF. For the 20 tRNA genes that are not identified as TRF1-bound, two corresponded to regions that are not tiled on the array, and six can be identified with a less stringent threshold on the binding probabilities. Likewise, for the seven tRNA genes not identified as BRF-bound, two corresponded to regions not tiled on the array, and three can be identified using a lower probability threshold (Supplementary data). Therefore, our methods not only identified conventional targets of the TRF1/BRF complex with good sensitivity but also provided a clear correlation between tRNA genes and the TRF1/BRF complex throughout the entire genome.
Figure 3.
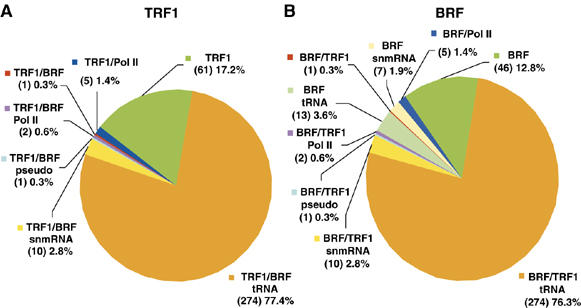
Classification of the TRF1 and BRF occupied regions. TRF1/BRF-binding sites are annotated using version 4.2.1 of the D. melanogaster genome and classified according to the type of noncoding RNAs. Percentage and number of target sites are indicated. (A) TRF1/BRF-tRNA: tRNA promoter regions bound by both TRF1 and BRF (77.4%); TRF1/BRF/snmRNA: snmRNA promoters bound by TRF1 and BRF (2.8%); TRF1/BRF/pseudo: pseudo-gene promoters bound by TRF1 and BRF (0.3%); TRF1/BRF/Pol II: Pol II promoters bound by TRF1 and BRF (0.6%); TRF1/BRF: unannotated regions bound by both TRF1 and BRF (0.3%); TRF1/Pol II: Pol II promoters bound only by TRF1 (1.4%); TRF1: unannotated regions bound only by TRF1 (17.2%). (B) BRF/TRF1-tRNA: tRNA promoter regions bound both by BRF and TRF1 (76.3%); BRF/TRF1/snmRNA: snmRNA promoters bound by BRF and TRF1 (2.8%); BRF/TRF1/pseudo: pseudo gene promoters bound by BRF and TRF1 (0.3%); BRF/TRF1/Pol II: Pol II promoters bound by BRF and TRF1 (0.6%); BRF/tRNA: tRNA promoters bound only by BRF (3.6%); BRF/TRF1: unannotated regions bound by both TRF1 and BRF (0.3%); BRF/snmRNA: snmRNA promoters bound only by BRF (1.9%); BRF/Pol II: Pol II promoters bound only by BRF (1.4%); BRF: unannotated regions bound only by BRF (12.8%).
Table 1.
Representative genomic regions with significant TRF1/BRF binding
| Rank | Start | End | Chr | Score | Class | Genes |
|---|---|---|---|---|---|---|
| 1/1 | 8952366 | 8954214 | chr2R | 41.8 | I | CR30509 |
| 2/2 | 16687498 | 16691515 | chr2R | 34.3 | I | CR33539, CR30210, CR30209 |
| 3/3 | 14928678 | 14930667 | chr2R | 33.1 | I | CR30224, CR30225, CR30326 |
| 4/5 | 7167448 | 7169438 | chr2R | 32.5 | I | CR30255, CR30254 |
| 11/6 | 8944250 | 8950296 | chr2R | 31.2 | I | CR30244, CR32842, CR32841, CR30521, CR30246, CR30247, CR30508 |
| 15/7 | 18578516 | 18580486 | chr2R | 29.0 | I | CR30202, CR30201 |
| 24/9 | 14637600 | 14641600 | chr3L | 28.3 | I | CR32144, CR32142 |
| 5/10 | 3077634 | 3081648 | chr3L | 27.4 | I | CR32288, CR32289, CR32287, CR32285, CR32286, CR32272 |
| 10/11 | 10498592 | 10500568 | chr2R | 27.2 | I | CR30241 |
| 14/12 | 22781067 | 22783043 | chr3L | 26.7 | I | CR32460 |
| 13/21 | 2581411 | 2583397 | chr2R | 24.9 | I | CR30298, CR30299 |
| 14/12 | 1362366 | 1364341 | chr3L | 24.9 | I | CR32330, CR32328, CR32329 |
| 15/23 | 8039304 | 8043250 | chr3L | 24.7 | I | CR32361, CR32362, CR32363 |
| 17/22 | 7175511 | 7177469 | chr2R | 24.4 | I | CR32844 |
| 18/27 | 21041337 | 21043328 | chrX | 24.0 | I | CR32526, CR32518 |
| 19/13 | 16667371 | 16669351 | chr2R | 23.9 | I | CR30208, CR30206, CR30207 |
| 20/7 | 752349 | 756360 | chr3L | 23.7 | I | CR32480, CR32481 |
| 32/16 | 1647601 | 1651588 | chr2R | 14.0 | I | CR30304, CR32837 |
| 22/17 | 20582213 | 20586210 | chr2R | 13.8 | I | CR30198, CR30199, CR30200 |
| 35/19 | 8000971 | 8004963 | chr3L | 12.7 | I | CR32370 |
| 42/20 | 14153176 | 14155175 | chr2R | 12.6 | I | CR30227, CR30228 |
| 21/8 | 12657949 | 12662132 | chr2R | 16.3 | I, II | CR30234, CR30235, CR33921 (snoRNA:U3:54Aa), CR33628 (snoRNA:U3:54Ab) |
| 85/87 | 20379116 | 20383094 | chr3R | 13.1 | I, II | CR31540, CR31379** (snRNA:U6:96Aa), CR32867** (snRNA:U6:96Ab), CR31539** (snRNA:U6:96Ac) |
| 109/108 | 2643770 | 2649796 | chr3R | 8.8 | I, II | CR31490, CR31485, CR31489, CR31487, CR31486, CR32864 (7SLRNA) |
| 143/142 | 3297494 | 3303497 | chr3R | 1.7 | I, II | CR31500, CR31502, CR33925** (snmRNA:331) |
| 162/167 | 15238196 | 15244198 | chr2R | 2.4 | I, II | CR30218, CR30452, CR30451, CR30455, CR30453, CR30454, CR30220, CR33930** (snoRNA:185) |
| 23/41 | 5369112 | 5370107 | chrX | 22.9 | II | CR33787 (snoRNA:644) |
| 24/34 | 1470403 | 1473835 | chr3L | 22.9 | II | CR33656 (snoRNA:3) |
| 34/— | 19555572 | 19557535 | chr3R | 20.7 | II | CR33682** (snmRNA:342) |
| 92/129 | 18860853 | 18862812 | chr3L | 11.8 | II | CR33686 (snoRNA:269) |
| 93/141 | 7106831 | 7108826 | chr2R | 11.7 | II | CR33661 (snoRNA:535) |
| 102/— | 8389072 | 8391062 | chr2L | 10.0 | II | CR32989** (snRNA:U6atac:29B) |
| 111/138 | 3877139 | 3879109 | chr3L | 8.5 | II | CR33708 (snmRNA:149) |
| 119/127 | 19345596 | 19349590 | chr2R | 7.2 | II | CR33913 (snoRNA:314) |
| 121/109 | 10192815 | 10194807 | chrX | 7.0 | II | CR33662 (snoRNA:U3:9B) |
| 6/4 | 6919489 | 6923502 | chr2R | 32.6 | III | CG7759 |
| 16/30 | 12370895 | 12372879 | chr2R | 24.5 | III | CG5935 |
| 18/8 | 13130719 | 13132695 | chr2R | 28.7 | IV | 7th intron of CG10936 |
| —/9 | 4236361 | 4238331 | chr2R | 16.0 | IV | 1st intron of CG8411-RA |
| Highly statistically significant TRF1/BRF binding regions were categorized into four classes: I, tRNA, II, snmRNA, III, Pol II genes, IV, unannotated regions. A statistical score was used to list the top binding regions, and the coordinates for start and end sites of the binding regions are indicated. Double asterisks indicate that the binding was observed only in the BRF dataset. | ||||||
Approximately 20% of the binding sites corresponded to non-tRNA sites. These non-tRNA target regions of the TRF1/BRF complex contained promoters of snmRNA genes (such as 7SL RNA and snoRNAs) and Pol II genes, in addition to genomic loci with no current annotation. Interestingly, we observed some snmRNA genes that are only bound by BRF under our experimental conditions. This could be due to the differences in antibody affinity, or it could indicate that some BRF can associate with subsets of promoters in the absence of TRF1, possibly in association with other, as yet, uncharacterized partners.
Functional characterization of novel TRF1/BRF targets
Our ChIP-on-chip analysis has provided not only a comprehensive high-resolution profile of TRF1/BRF target sites but also revealed several potentially novel target genes. We were particularly intrigued by the finding that the TRF1/BRF complex was associated with 7SL RNA and snoRNA genes. Therefore, we wanted to determine whether the binding of TRF1/BRF to these sites has functional relevance for the activity of these genes. We selected several representative promoters from these newly identified, putative targets of the TRF1/BRF complex to further characterize by direct in vitro transcription reactions. Figure 4 shows in vitro transcribed RNA products using S2 cell extracts directed by templates containing promoters for tRNA (CR30206), 7SL RNA, and snoRNA genes. As expected, transcription from all three of these classes of genes was resistant to low concentrations of α-amanitin (25 ng/μl) typically used to inhibit Pol II transcription (Takada et al, 2000). By contrast, addition of tagetin (0.5 U/μl), a Pol III-specific inhibitor, completely abolished transcription from all three templates. tRNA and 7SL RNA have previously been established as Pol III genes in Drosophila, human, and plants (Ullu and Weiner, 1985; Takada et al, 2000; Yukawa et al, 2005). Interestingly, snoRNAs have been reported to rely on either Pol II or Pol III for transcription (Antal et al, 2000; Kiss, 2002; Harismendy et al, 2003; Roberts et al, 2003; Moqtaderi and Struhl, 2004). Here, we found that snoRNA:314 and snoRNA:644 are specifically targeted by the TRF1/BRF complex and appear to depend exclusively on the Pol III machinery. This finding is consistent with the observation that these two genes are localized to intergenic regions, rather than as part of introns of host Pol II genes (Yuan et al, 2003), suggesting that at least this class of snoRNAs may consist of functionally independent Pol III transcription units. Indeed, our bioinformatic analysis of novel snoRNA targets revealed conserved B-box sequences, which serve as binding sites for TFIIIC (Figure 4B).
Figure 4.
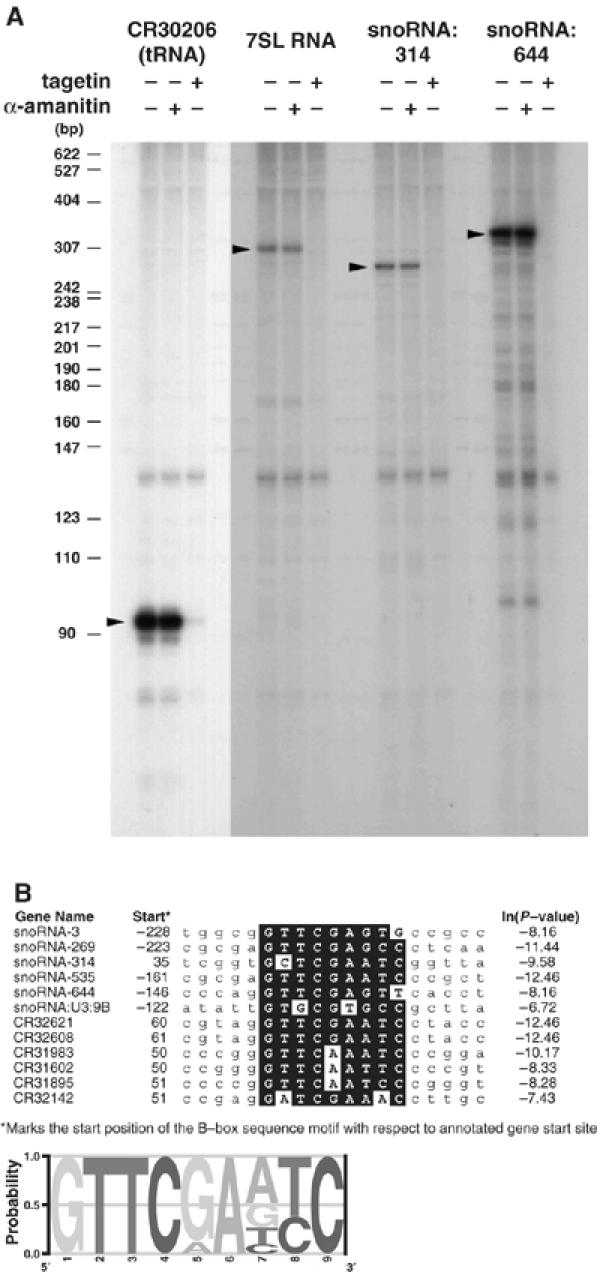
In vitro transcription of tRNA, 7SL RNA, and snoRNA genes. (A) In vitro transcription assays were carried out using templates for the putative targets of the TRF1/BRF complex, CR30206 (tRNA), 7SL RNA, snoRNA:314, and snoRNA:644. Transcription from these promoters gave template-dependent transcription products that are resistant to a low concentration of α-amanitin (25 ng/μl), which is sufficient to inhibit Pol II transcription, but displayed sensitivity to tagetin (0.5 U/μl), a Pol III-specific inhibitor. (B) Alignment of conserved B-box sequences found in snoRNAs and tRNAs bound by TRF1 and BRF. The conserved sequences are boxed in black. The consensus B-box sequence is represented as a logo at the bottom. The start sites indicated are the distances from the annotated gene start sites in FlyBase. As most annotated snoRNAs appear to be derived from processed transcripts, the identified B-boxes typically reside upstream of the mature snoRNA sequences.
To further establish that these novel TRF1/BRF target genes are indeed bona fide Pol III transcribed genes, we conducted ChIP assays probing directly for the presence of RNA polymerase III at these novel TRF1/BRF targets in vivo. Using S2 cells expressing a V5-tagged RpIII128, the second largest subunit of Drosophila RNA Pol III that is a unique class III subunit, we found that 5S rRNA, tRNA, snoRNA, and 7SL RNA genes are all specifically precipitated via anti-V5 antibody whereas CG11700, a Pol II gene, failed to exhibit any enrichment (Figure 2A, bottom). This result strongly corroborates our finding that the TRF1/BRF complex is responsible for regulating the transcription of both of these novel target genes (7SL and snoRNA) and that this diversified TRF1 containing initiation complex indeed works in conjunction with RNA Pol III in Drosophila.
snoRNA:644 is transcribed as a longer precursor
Although we observed robust, template-dependent as well as Pol III-dependent transcription from snoRNA templates, we noticed that the size of the in vitro products were significantly longer than what had been reported previously. For example, the snoRNA:314 template produced a ∼250 bp RNA product in vitro whereas stable transcripts detected by Northern blot were only 130 bp in length. Likewise, snoRNA:644 transcribed in vitro produced a ∼300 bp transcription product instead of the 170 bp RNA observed in cells (Yuan et al, 2003). We hypothesized that this discrepancy may be due to RNA processing events that occur at the 5′ and 3′ ends of putative precursor-snoRNAs (Kiss, 2002). Therefore, we tested whether these in vitro products accurately reflect primary transcripts of pre-snoRNAs. To address this, we performed primer extension analysis to compare the in vitro transcription products with the primary in vivo transcripts present in total RNA extracted from S2 cells (Figure 5). Primer extension products using two different primers complementary to different segments of the snoRNA:644 transcript confirmed that the vast majority of snoRNA:644 transcripts exists as two distinct processed RNA species (Figure 5A). Importantly, however, a fraction of the in vivo snoRNA:644 contains a start site that matches perfectly with the long in vitro transcription products (Figure 5B). Similarly, the in vitro transcription start site of snoRNA:314 gene matched exactly the in vivo start site (data not shown). Thus, we conclude that the in vitro products likely represent the unprocessed ‘primary transcript' and that the in vitro transcription start site we observe accurately reflects the in vivo transcription start site.
Figure 5.
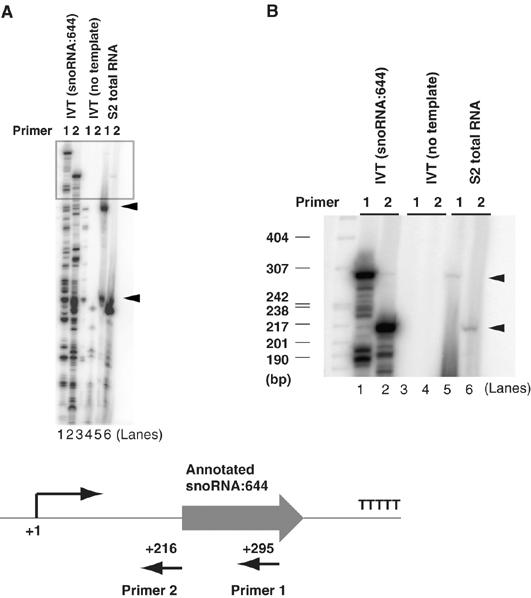
Mapping the in vivo transcriptional start site of snoRNA:644. (A, B) In vitro transcription products directed by the snoRNA:644 template were subject to primer extension analysis using two independent primers (lanes 1 and 2) hybridizing to mature and precursor snoRNA:644 transcripts. Control mock transcription reactions without the template did not yield any primer extension products (lanes 3 and 4). The primer extension products with identical sizes with the in vitro products were obtained using S2 total RNA (lanes 5 and 6). Arrowheads represent two major processed forms of snoRNA:644 in lane 5 using Primer 1. (B) Longer exposure of (A) in top region of the gel. Arrowheads represent unprocessed nascent transcripts. (Bottom) Schematic diagram of primers used for the primer extension analysis.
TRF1/BRF complex is required for transcription of novel targets
Having established an efficient in vitro transcription system that accurately reflects in vivo transcription start sites, we next asked whether the TRF1/BRF complex is required to potentiate activation of these novel target promoters. To test this, we first prepared transcription extracts depleted of the TRF1/BRF complex by preincubating S2 extracts with protein A beads conjugated with affinity-purified anti-BRF antibody (Figure 6A). Importantly, we confirmed that the levels of other proteins such as TBP and α-tubulin remain unaffected in the depleted extracts. Using these immunodepleted transcription extracts, we performed in vitro transcription reactions directed by tRNA (CR30206), 7SL RNA, and snoRNA:644 templates. As expected, we detected very low levels of transcription from these templates after immunodepletion of the TRF1/BRF complex (Figure 6C, lanes 1 and 2). To ascertain the specificity of the immunodepletion, we performed add-back experiments using a series of recombinant purified TRF1/BRF complexes (Figure 6B). Transcription from these three templates was efficiently restored to levels comparable to the control transcription using mock-depleted extracts (Figure 6C, lanes 1–5). Thus, these studies indicate that the TRF1/BRF complex is likely an essential component of the Pol III-mediated transcription of tRNA, 7SL RNA, and snoRNA genes in vitro.
Figure 6.
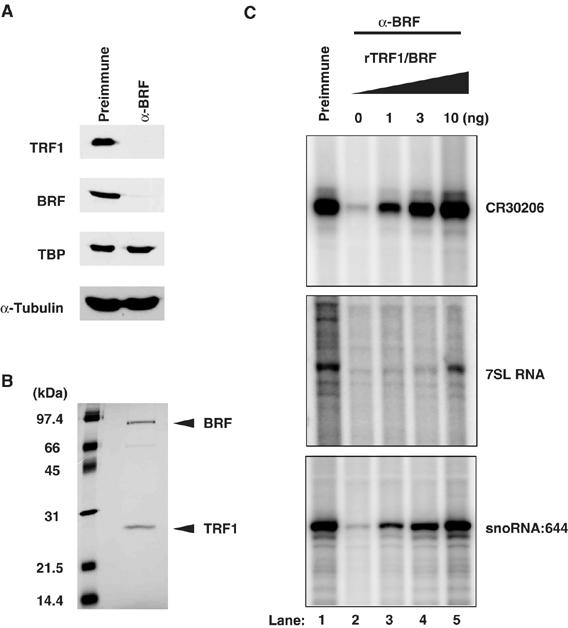
TRF1/BRF complex is required for transcription of tRNA, 7SL RNA, and snoRNA genes. (A) Immunodepletion of the TRF1/BRF complex. The S2 extract depleted of the TRF1/BRF complex was used for the immunoblot to examine the level of TRF1, BRF, and TBP. α-BRF antibody selectively depletes TRF1 and BRF from the S2 extract, but not TBP. α-Tubulin is used to show that approximately equal amount of total proteins was loaded in each lane. (B) Highly purified recombinant TRF1/BRF complex used for the add-back experiments. Arrows indicate bands corresponding to recombinant TRF1 and BRF. (C) In vitro transcription using TRF1/BRF depleted extracts. The transcription from tRNA (CR30206), 7SL RNA, and snoRNA:644 templates was significantly diminished using the extracts depleted of the TRF1/BRF complex (lanes 1 and 2). The transcriptional activity was recovered by the addition of purified recombinant TRF1/BRF complex (lanes 3–5).
TRF1/BRF complex regulates snoRNA transcription via promoter proximal binding sites
Although we have mapped genome-wide locations of TRF1 and BRF at high resolution (35 bp), determining the exact binding sites at ±1 nucleotide resolution would be required in order to begin deciphering the molecular basis of Pol III promoter recognition that has evolved to replace the more conventional TBP containing TFIIIB core promoter recognition complex. To address this issue, we have used DNase I footprint assays to determine which segment of the snoRNA:644 promoter is specifically recognized by the TRF1/BRF complex. Surprisingly, we observed a reproducible footprint spanning from ∼−19 to ∼+6 relative to the transcription start site (Figure 7A), indicating that the snoRNA promoter recognition site for TRF1/BRF is at least partly internal to the gene.
Figure 7.
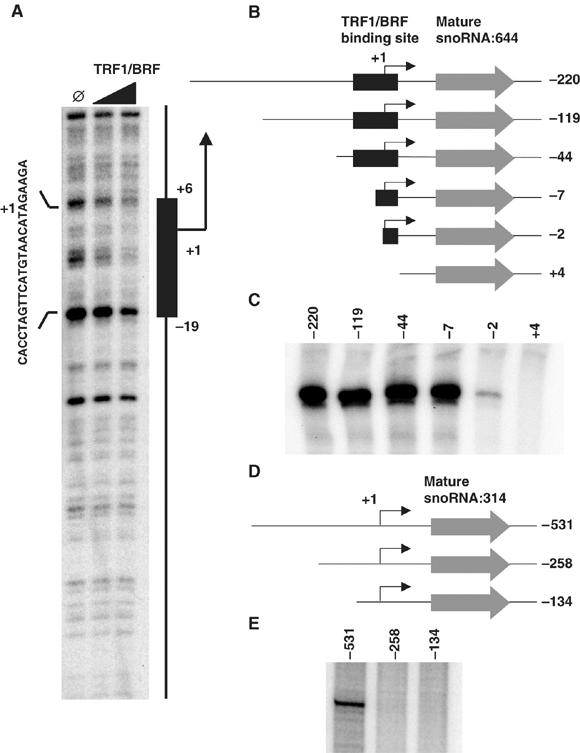
TRF1/BRF complex binding to proximal promoter of snoRNA:644 gene is essential for the gene activity. (A) DNase I footprinting assay utilizing a snoRNA:644 template and recombinant TRF1/BRF complex. The footprint region spanning approximately 25 bp is indicated by a black box and sequences on the forward strand of the gene. (B) Schematic representation of the snoRNA:644 templates used for the promoter deletion experiments. pCR4 vector backbone is omitted from the diagram. The numbers represent the position of the promoter fragment relative to the transcription start site determined by primer extension analysis. (C) In vitro transcription of the snoRNA:644 templates described above. The snoRNA:644 promoter retains activity even when the majority of gene external sequence is removed. (D) Schematic representation of the snoRNA:314 templates used for the promoter deletion experiments. pCR4 vector backbone is omitted from the diagram. The numbers represent the position of the promoter fragment relative to the transcription start site determined by primer extension analysis. (E) In vitro transcription of the snoRNA:314 templates described in (D).
To determine if this footprint region is functionally required for snoRNA:644 promoter activity, we used in vitro transcription to test promoter activity from a deletion series of gene constructs (Figure 7B and C). Consistent with the notion of a gene-internal promoter, a deletion up to −2 did not obliterate the activity, although some decrease in activity was observed after removing −7 to −2 region. In contrast, further 5 bp deletion at the transcriptional start site completely abolished the promoter activity. Taken together, the binding and transcription data suggest that DNA sequences spanning the transcriptional start site of the snoRNA:644 gene, which is recognized by the TRF1/BRF complex, serves as a critical target site for the Pol III machinery to assemble and form an active initiation complex. We also conducted promoter deletion experiments with another snoRNA gene, snoRNA:314. Unlike the partially gene-internal snoRNA:644 promoter, an essential element for snoRNA:314 promoter activity appears to reside in the gene-external region as constructs retaining up to least ∼250 bp upstream of the transcription start site nevertheless failed to produce any transcripts whereas that contains 531 bp upstream of the transcription start site directed normal levels of transcription (Figures 4A, 7D and E). This surprising observation reveals that Pol III transcribed snoRNA promoters have substantially different structural elements suggesting that perhaps these promoters can employ different mechanisms involving the TRF1/BRF complex and Pol III machinery.
Discussion
Genome-wide mapping of TRF1- and BRF-binding sites
In this paper, we sought to identify at relatively high-resolution specific genome-wide binding sites for the TRF1/BRF core promoter recognition machinery in Drosophila. Previously, our laboratory used in vitro biochemical methods to identify a few TRF1 target genes and found that this TRF can mediate transcription from both Pol II and Pol III promoters. This observation suggested that at least in Drosophila some of the key promoter recognition functions of TBP are carried out by an alternative core promoter recognition factor TRF1. However, our previous studies were hampered by technical limitations that prevented us from directly comparing the in vivo role of TRF1 in Drosophila cells with our in vitro observations. One problem was the resolution of TRF1 localization on polytene chromosomes that did not allow us to map accurately (10–100 kb) TRF1 target promoters in vivo. Another problem was the finding that TRF1 can drive both Pol II and Pol III-mediated transcription, thus complicating our analysis of identifying bona fide promoters subject to regulation by TRF1. Indeed, given the blunt resolution of polytene sites, one could not distinguish between multiple tRNA sites from adjacent Pol II genes with potential TRF1 target sites. In this report, we have employed a range of in vivo and in vitro assays including genome-wide ChIP-on-chip assays to obtain a more accurate and global picture of how the TRF1 factor directs promoter recognition. Our present study identified ∼350 sites in the Drosophila genome that are specifically targeted by TRF1, BRF or both. These data revealed that, in S2 cells, TRF1 as well as BRF are found in a majority of known Pol III gene promoters whereas Pol II promoters appear to constitute a minor proportion of TRF1 targets. It should also be noted that these classes of small noncoding RNA genes we identified here generally pose a particularly difficult challenge in determining the exact binding sites using existing lower resolution tiling arrays as they are on average much smaller than Pol II transcripts. High-resolution (35 bp) oligonucleotide microarrays such as the ones used in this study provide a much more accurate mapping of protein-binding sites than ones that have been typically employed in previous studies.
Analysing vast amounts of data from high-resolution (35 bp), high-density (3.1 × 106 probes) genome tiling microarrays poses a significant challenge especially if the objective is to identify, in an unbiased manner, bona fide functional transcription factor binding sites. Most often, these studies are compromised by a large number of false-positive target sites. A significant advance would be the development of automated and statistically motivated methods for de novo prediction of binding regions. Using TRF1/BRF ChIP-on-chip data as a case study, we present here a newly developed statistical framework (TileHGMM) that provides a powerful computational analysis platform. The strength of TileHGMM is its ability to allow information sharing across probes using a hierarchical model which, in turn, provides more power and accuracy than simple sliding window testing approaches (Cawley et al, 2004) especially when there are limits to the number of replicate samples that can be obtained (Keleş, 2006). Furthermore, by allowing probe-specific distributions of binding, TileHGMM accommodates probe-specific hybridization efficiencies. TileHGMM was indeed shown to provide advantages over more recent hidden Markov methods (Ji and Wong, 2005; discussed in Keleş et al, 2006). Thus, this algorithm provides a significant advantage to molecular geneticists performing microarray analysis using limited sources of materials. Importantly, we found that peak predictions by TileHGMM are highly accurate and extremely sensitive, judging from the striking correlation between tiling array results and traditional quantitative PCR detection of ChIP signals. Therefore, this new framework should be highly useful for de novo unbiased identification of genome-wide transcription factor binding sites not only for the TRF1/BRF data sets but should also be generally applicable to a broad range of other ChIP-on-chip experiments performed using high-density genome tiling microarrays.
Genome-wide evidence for TRF1/BRF as alternative TFIIIB in Drosophila
Surveying all the genomic sites identified by this study, we observed a striking degree (77.7%) of colocalization between TRF1 and BRF. This is entirely consistent with but also significantly extending our previous biochemical study indicating that most of the TRF1 protein in Drosophila S2 cells appears to be in a complex with BRF (Takada et al, 2000). Among the colocalized sites, we found that by far the most dominant class represents tRNA genes, which is consistent with our in vitro studies. Remarkably, 93% of known tRNA genes in the Drosophila genome scored as TRF1/BRF targets. This result indicates that the TRF1/BRF complex in Drosophila is tightly linked to Pol III transcription, in contrast to most other eukaryotes where TBP is the core component of the TFIIIB complex. In addition, our recent ChIP-on-chip analysis of Drosophila TBP confirmed that less than 1% of the Pol III genomic sites that are bound by either TRF1 or BRF are also bound by TBP, further supporting the role of TRF1, but not TBP, in Pol III transcription (Y Isogai, R Tjian and S Keleş, unpublished data). Importantly, several of the other mapped sites corresponded to genes that had not been previously described as TRF1/BRF targets, including 7SL RNA, snoRNAs, and various functionally uncharacterized snmRNAs. We also found that approximately 19% of the identified sites are occupied only by TRF1 or BRF, but not by both. These ‘single-hit' sites could be due to differences in the sensitivity of the assays (i.e., variability in antibody strength) or they could reflect some aspect of TRF1 and BRF functional specificity that we do not yet understand. Polytene chromosome staining conducted previously (Hansen et al, 1997; Takada et al, 2000) was consistent with our finding that not all TRF1 sites are also BRF sites. One possibility is that TBP or some other as yet unidentified TRFs could play a role in the recognition of these non-TRF1-associated, BRF sites. Interestingly, these ‘single-hit' sites are found most frequently in potential promoter regions of Pol II genes or in regions where no gene annotations are found. This suggests that both TRF1 and BRF may be involved in transcriptional specificity possibly involving Pol II that remains to be characterized. For example, we detected TRF1/BRF binding at the 5′ upstream region of the tudor gene, which had previously been biochemically characterized as an in vitro Pol II gene target of TRF1 (Holmes and Tjian, 2000). However, the presence of several tRNA sites proximal to this genomic locus made it difficult for us to determine whether these binding sites are utilized for directing transcription of tudor, tRNA genes or possibly both.
Identification of a snoRNA transcription unit and potential processing events
To date, there have been relatively few studies characterizing snoRNA transcription in Drosophila (Tycowski and Steitz, 2001; Yuan et al, 2003). In yeast, it has been reported that the majority of snoRNAs are transcribed by Pol II, and only one snoRNA gene (snR52) has been identified as a Pol III target (Harismendy et al, 2003; Roberts et al, 2003; Moqtaderi and Struhl, 2004; Guffanti et al, 2006). We report here at least two snoRNA genes that are transcribed by the Pol III machinery in Drosophila, snoRNA:314 and snoRNA:644, possess independent transcriptional units that are localized to intergenic regions. By examining other snoRNA targets of the TRF1/BRF complex, we found that five are localized to intergenic regions whereas three are embedded in the introns of Pol II genes. Therefore, it is likely that at least some of these other uncharacterized intergenic snoRNA targets are also Pol III genes. Moreover, of the two different classes of snoRNAs (box C/D type and box H/ACA type), we did not find any bias in our list of snoRNA targets. Thus, the chromosomal location and promoter structures, rather than specific types of snoRNAs, may be key determinants for designating the class of transcriptional machinery (Pol II or III) utilized for snoRNA genes. Indeed, these snoRNA targets contain the conserved B-box sequence, underscoring the regulation of these promoters by the Pol III transcription machinery.
Another observation regarding snoRNA transcriptional units revealed by these studies is the apparent production of a larger primary transcript precursor that is then most likely subject to processing at its 5′ end. In the latest annotation of the Drosophila genome, snoRNAs are mapped according to the size of the mature forms and therefore may not reflect their true transcriptional start sites. The in vitro transcription assays used in this study provide a powerful complementary approach to mapping the promoter regions of these snoRNAs as this assay correctly predicted the transcriptional start sites that were then confirmed in vivo by primer extension.
Pol III promoters have been subdivided into at least two classes, gene internal (5S rRNA and tRNAs) and gene external (U6 snRNA) promoters (Schramm and Hernandez, 2002). What then is the common structure of snoRNA gene promoters? In the yeast snR52 gene, potential A/B boxes have been mapped suggesting that a gene-internal promoter may be important (Harismendy et al, 2003; Guffanti et al, 2006). Consistent with this observation, the snoRNA:644 gene in Drosophila we identified here also appears to require gene-internal elements. In addition, we found that the bulk of the TRF1/BRF complex binds a region overlapping the transcription start site and extending well into the gene (+6), which is reminiscent of a gene-internal promoter element. Importantly, this element has substantially diverged from the typical upstream TATA box. This finding is also consistent with the observation that, unlike fungi, plants, and mammals, Drosophila Pol III genes generally lack conspicuous TATA box sequences. Thus, the core promoter recognition apparatus consisting of TRF1/BRF in insects has apparently evolved to accommodate a more diversified Pol III promoter structure utilized by Drosophila.
Although the snoRNA:644 gene represents one type of snoRNA promoter structure, we found that not all the snoRNAs regulated by TRF1/BRF exhibit the same type of promoter structure. In the case of snoRNA:314 gene, it appears that significant gene-external sequences and promoter elements may be necessary for transcriptional initiation as in vitro transcription experiments with promoter deletions of the snoRNA:314 template revealed that at least ∼250 bp upstream of the putative transcription start site are essential for efficient initiation. This suggests that the promoter structure of snoRNA:644 gene may resemble tRNAs whereas that of snoRNA:314 is more similar to the 7SL RNA gene in plants wherein both gene-external and -internal sequence elements play a role in directing transcriptional initiation (Yukawa et al, 2005). Interestingly, under our in vitro transcription system, the snoRNA:644 template produced larger amounts of transcripts than the snoRNA:314 template. This observation appears well correlated with our ChIP-on-chip results in which the occupancy score of TRF1/BRF at the snoRNA:644 promoter is significantly higher than at the snoRNA:314 promoter, indicating that the recruitment of the TRF1/BRF complex may be a crucial step for successful initiation of transcription by Pol III. Therefore, the snoRNA:314 promoter may represent a case where the Pol III transcription machinery may be potentially directed by yet unknown DNA binding factors, allowing tight transcriptional control of these snoRNAs.
Small non-messenger RNAs comprise a novel class of TRF1/BRF targets
Small nonmessenger RNAs are abundantly expressed in eukaryotic cells and thought to participate in critical cellular functions. For example, 7SL RNA is part of the signal recognition particle and snoRNAs plays an important role in guiding modification (such as pseudouridylation) of ribosomal RNAs (Kiss, 2002). However, the functional roles of the majority of other snmRNAs remain to be characterized. For example, the putative TRF1/BRF target snmRNA:149 gene appears to be transcribed in the antisense direction to a protein coding gene, CG1079. One proposal is that this class of snmRNAs may play a role in the regulation of the corresponding Pol II genes via splicing or potential RNAi-like mechanisms (Yuan et al, 2003).
We have not yet determined the localization of the TRF1/BRF complexes in different cell types or in different Drosophila tissues. It may be particularly interesting to examine neural tissues where TRF1 was found to be prominently upregulated (Crowley et al, 1993; Hansen et al, 1997). It is possible that TRF1 mediates cell-type-specific transcription in these tissues. Recently, an snoRNA in humans was specifically expressed in the brain and was implicated in alternative splicing of the serotonin receptor (Kishore and Stamm, 2005). Such post-transcriptional RNA modification events may also occur in the central nervous system of Drosophila. Therefore, the role of the TRF1/BRF complex in snmRNA expression in S2 cells may point to a potential link between the TRF1/BRF complex and the regulation of yet to be identified brain-specific snmRNAs. It is thus tempting to speculate that the TRF1/BRF complex may have broad implications for gene regulation in the Drosophila neural system. Our finding that some snoRNA promoters rely on gene-external promoter elements supports a potential tissue or developmental stage-specific expression of these snmRNA by employing additional upstream transcription factors in conjunction with TRF1/BRF.
At least in S2 cells, the majority of the TRF1/BRF complex is found to direct the regulation of small non-coding RNA genes, most of which are transcribed by Pol III. Apparently in Drosophila and other insects, TRF1 has evolved to be responsible for initiating all the known classes of Pol III genes. This presents an interesting functional diversification in insects between TBP and TRF1 that may have implications in other organisms.
Materials and methods
Antibodies
Affinity-purified anti-BRF and anti-TRF1 antibodies have been described (Hansen et al, 1997; Takada et al, 2000). Rabbit anti-V5 antibody was obtained from Sigma.
ChIP assay and quantitative PCR
ChIP assays were conducted as described (Puig et al, 2003) except a formaldehyde concentration of 0.5% was used for crosslinking. For the immunoprecipitation experiments, preimmune rabbit IgG and normal mouse IgG (Sigma) were used as negative controls. Quantitative PCR was conducted with Opticon (MJ Research) using iQ SYBR Green Supermix (Bio-Rad) at a primer concentration of 150 nM. Primer sequences used to amplify 5S, 7SL, snoRNA:644, CG11700 and CR30206 genomic regions are provided in the Supplementary data. For ChIP assays probing Pol III occupancy, we generated S2 cells stably expressing a Pol III-specific subunit RPIII128, tagged with V5 at the C-terminus. We used 1% formaldehyde for crosslinking and conducted subsequent steps as described above.
Probe preparation and hybridization
The materials from ChIP assays were first amplified (Bohlander et al, 1992). Two micrograms of amplified DNA were treated with DNase I (Sigma) and sheared to 50–100 bp fragments. The sheared DNA probes were labeled with biotin-N6-ddATP (Enzo) using terminal deoxytransferase (Promega) and hybridized to two replicates of Drosophila Tiling Forward Array (Affymetrix) per antibody. Hybridization cocktail (200 μl) contained the following components: 2 μg of biotinylated DNA probe, 3 M trimethylammonium chloride (Sigma), 30 pM of biotinylated oligo B2 (Affymetrix), 0.1 mg/ml herring sperm DNA (Invitrogen), and 0.02% Triton X-100 (Sigma). Post-hybridization washes and signal detection were carried out using the protocol described in the GeneChip Expression Analysis Technical Manual (Affymetrix).
In vitro transcription
Templates for transcription were prepared by inserting tRNA (CR30206), snoRNA:314, snoRNA:644, and 7SL RNA gene regions to pCR4-TOPO vector (Invitrogen). Cell extracts from Drosophila Schneider line 2 (S2) cells were prepared as described (Dingermann et al, 1981) with one modification: after the cell lysis, 1/10 volume of buffer B (50 mM HEPES, pH 7.6, 2 M KCl, 50% glycerol, 30 mM MgCl2, 0.1 mM EDTA) was added before ultracentrifugation. The recombinant TRF1/BRF complex was prepared as described (Takada et al, 2000). Immunodepletion of the TRF1/BRF complex was carried out by mixing 100 μl of protein A sepharose prebound with either preimmune rabbit serum or affinity-purified BRF-antibody and 1 ml of S2 extract and incubating for 4 h at 4°C. The in vitro transcription carried out in Figure 4 contained 20 μl of S2 extract (∼120 μg), 1 μg of template DNA, 0.5 mM of ATP, CTP, and UTP, and 0.1 mM of GTP, 1 μl of 3000 Ci/mmol α-32P-GTP in a buffer containing 30 mM HEPES-KOH, pH 7.6, 100 mM KCl, 5 mM MgCl2, 3 mM DTT, 5% glycerol in 42 μl reaction. α-Amanitin (Sigma, at 25 ng/μl) and tagetin (Epicentre, at 0.5 U/μl) were added for select reactions. The rescue experiments were carried out using the following mixture: for CR30206 and snoRNA:644 templates, 12 μl of depleted extract, 1 μl of template DNA, 0.5 mM of ATP, CTP, and UTP, 1 μl of 3000 Ci/mmol α-32P-GTP, recombinant TRF1/BRF (in TGED buffer: 0.5 M NaCl, 20 mM Tris, 10% glycerol/0.25 mM EDTA, 1 mM DTT), in the reaction buffer above. For the 7SL RNA template, 20 μl of depleted extract was used. Primer extension assays were conducted as described (Takada et al, 2000) using the primers hybridizing to snoRNA:644 gene. All the primer sequences used for vector construction and primer extension assays are provided in the Supplementary data.
DNase I footprinting
DNase I footprinting experiments were conducted essentially as described (Ziegelbauer et al, 2001) using a 243-bp (spanning +124 to −119 relative to the transcription start site) probe labeled at the sense strand of the snoRNA:644 gene.
Statistical data analysis
Preprocessing of the data. We carried out the analysis of the data from each chromosome separately. For each chromosome, the IP-enriched and control samples were quantile normalized (Bolstad et al, 2003) within two replicates and median scaled across the two groups. Log-normalized intensities were used as the final measurements of occupancy.
Identification of bound regions. We performed a higher level analysis of the ChIP-on-chip data using the Tiling Hierarchical Gamma Mixture Model (TileHGMM) of Keleş et al (2006). We first partitioned each chromosome into regions utilizing gaps derived mainly from repeat masking and masking of regions with low hybridization quality probes. Regions longer than 2 kb were further partitioned so that each genomic region is on the average 2 kb. The resulting total number of regions was represented by N. We utilized a hierarchical gamma model adapted from Kendziorski et al (2003) and Newton et al (2004) to model the hybridization intensities of the probes as this method had been shown to be more powerful than simple testing approaches with a small number of replicate microarray experiments (Newton et al, 2004). Underlying features of the statistical pipeline are summarized in Figure 1A. The method assumes that each genomic region has at most one peak and such a peak can have a variable size, that is, variable number of probes. Probe-specific IP-enriched and control hybridizations follow different Gamma distributions. The mean values of these distributions would be equal for ‘unbound' probes whereas the IP-enriched sample distribution would exhibit larger mean values for the ‘bound' probes (Keleş, 2006 for mathematical details). The TileHGMM package processes control and IP-enriched ChIP-on-chip data and computes the partitioning of chromosomes, and outputs the coordinates of peaks as well as peak sizes. The model parameters including the shape parameters for the underlying Gamma distributions and proportion of regions with a peak are estimated by maximum likelihood using the expectation-maximization (EM) algorithm. This algorithm outputs two important posterior probabilities (‘probabilities' in the main text for simplicity): ηi, i=1, …, N, that are region-specific posterior probabilities representing the probability of ith region having a peak as well as ξij, j=1, …, Li (Li is the total number of probes in the ith genomic region), i=1, …, N, which represents the posterior probability that the peak in ith region starts at jth probe. Using these posterior probabilities, we identified genomic regions with peaks and subsequently defined peak start and end positions. As discussed in Keleş et al (2006), the average peak size was calculated to be approximately 15 probes using the average fragment size of the sheared chromatin and the array parameters such as the length of oligonucleotides and average spacing of the tiling path. In addition, TileHGMM allows variable peak sizes (Supplementary data). We allowed both a fixed peak size of 15 probes (fixed peak, FP, approach) and a peak size distribution estimated using an agarose image gel of the sheared genomic DNA (variable peak, VP, approach). VP approach identified 10–18% more bound regions and included about 96% of the regions identified by the FP approach. As most of the additional regions identified by the VP approach corresponded to unannotated regions or Pol II gene targets, we focused on the results obtained using the FP approach at a false discovery rate of 0.01.
Annotation of binding regions. We annotated the identified peaks based on the 4.2.1 version of the D. melanogaster genome in FlyBase. For each peak, tRNA, pseudogenes, snmRNAs and Pol II genes with transcription start sites within 500 bps downstream of the peak boundaries were reported. Furthermore, Pol II targets with the peaks starting or ending within (+500, −100) bps of the annotated transcription start site were curated (Supplementary data).
De novo motif finding. We ran the motif finding program MEME (Bailey and Elkan, 1995) for a set of sequences mapping to peaks from 43 tRNAs and 6 snoRNAs. These regions had the region specific posterior probability of binding to be 1. The de novo motif finding was performed in a systematic fashion focusing on +/50, ±75, ±100, ±125, +/150 flanking base pairs of the mid-point of the peaks. The sequence logo of the B-box was generated using the enoLOGOS server (Workman et al, 2005).
Supplementary Material
Supplementary Materials
Acknowledgments
We thank D Nix and V Semetchenko for providing map files for microarray data processing, Y Fong, P Hu, R Losick, and M Marr for critical reading of the manuscript.
References
- Antal M, Mougin A, Kis M, Boros E, Steger G, Jakab G, Solymosy F, Branlant C (2000) Molecular characterization at the RNA and gene levels of U3 snoRNA from a unicellular green alga, Chlamydomonas reinhardtii. Nucleic Acids Res 28: 2959–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C (1995) Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine Learning 21: 51–80 [Google Scholar]
- Bohlander SK, Espinosa R III, Le Beau MM, Rowley JD, Diaz MO (1992) A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genomics 13: 1322–1324 [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116: 499–509 [DOI] [PubMed] [Google Scholar]
- Crowley TE, Hoey T, Liu JK, Jan YN, Jan LY, Tjian R (1993) A new factor related to TATA-binding protein has highly restricted expression patterns in Drosophila. Nature 361: 557–561 [DOI] [PubMed] [Google Scholar]
- Dingermann T, Sharp S, Appel B, DeFranco D, Mount S, Heiermann R, Pongs O, Soll D (1981) Transcription of cloned tRNA and 5S RNA genes in a Drosophila cell free extract. Nucleic Acids Res 9: 3907–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guffanti E, Ferrari R, Preti M, Forloni M, Harismendy O, Lefebvre O, Dieci G (2006) A minimal promoter for TFIIIC-dependent in vitro transcription of snoRNA and tRNA genes by RNA polymerase III. J Biol Chem 281: 23945–23957 [DOI] [PubMed] [Google Scholar]
- Hansen SK, Takada S, Jacobson RH, Lis JT, Tjian R (1997) Transcription properties of a cell type-specific TATA-binding protein, TRF. Cell 91: 71–83 [DOI] [PubMed] [Google Scholar]
- Harismendy O, Gendrel CG, Soularue P, Gidrol X, Sentenac A, Werner M, Lefebvre O (2003) Genome-wide location of yeast RNA polymerase III transcription machinery. EMBO J 22: 4738–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochheimer A, Tjian R (2003) Diversified transcription initiation complexes expand promoter selectivity and tissue-specific gene expression. Genes Dev 17: 1309–1320 [DOI] [PubMed] [Google Scholar]
- Hochheimer A, Zhou S, Zheng S, Holmes MC, Tjian R (2002) TRF2 associates with DREF and directs promoter-selective gene expression in Drosophila. Nature 420: 439–445 [DOI] [PubMed] [Google Scholar]
- Holmes MC, Tjian R (2000) Promoter-selective properties of the TBP-related factor TRF1. Science 288: 867–870 [DOI] [PubMed] [Google Scholar]
- Ji H, Wong WH (2005) TileMap: create chromosomal map of tiling array hybridizations. Bioinformatics 21: 3629–3636 [DOI] [PubMed] [Google Scholar]
- Kendziorski CM, Newton MA, Lan H, Gould MN (2003) On parametric empirical Bayes methods for comparing multiple groups using replicated gene expression profiles. Stat Med 22: 3899–3914 [DOI] [PubMed] [Google Scholar]
- Keleş S (2006) Mixture modeling for genome-wide localization of transcription factors. Published online: 16 November 2006. doi:10.1111/j.1541-0420.2005.00659.x, http://www.blackwell-synergy.com/d oi/full/10.1111/j.1541-0420.2005.00 659.x [DOI] [PubMed]
- Keleş S, van der Laan MJ, Dudoit S, Cawley SE (2006) Multiple testing methods for ChIP-Chip high density oligonucleotide array data. J Comp Biol 13: 579–613 [DOI] [PubMed] [Google Scholar]
- Kishore S, Stamm S (2005) The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 5758: 230–232 [DOI] [PubMed] [Google Scholar]
- Kiss T (2002) Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell 109: 145–148 [DOI] [PubMed] [Google Scholar]
- Moqtaderi Z, Struhl K (2004) Genome-wide occupancy profile of the RNA polymerase III machinery in Saccharomyces cerevisiae reveals loci with incomplete transcription complexes. Mol Cell Biol 24: 4118–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, Noueiry A, Sarkar D, Ahlquist P (2004) Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics 5: 155–176 [DOI] [PubMed] [Google Scholar]
- Puig O, Marr MT, Ruhf ML, Tjian R (2003) Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev 17: 2006–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DN, Stewart AJ, Huff JT, Cairns BR (2003) The RNA polymerase III transcriptome revealed by genome-wide localization and activity-occupancy relationships. Proc Natl Acad Sci USA 100: 14695–14700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L, Hernandez N (2002) Recruitment of RNA polymerase III to its target promoters. Genes Dev 16: 2593–2620 [DOI] [PubMed] [Google Scholar]
- Takada S, Lis JT, Zhou S, Tjian R (2000) A TRF1:BRF complex directs Drosophila RNA polymerase III transcription. Cell 101: 459–469 [DOI] [PubMed] [Google Scholar]
- Tycowski KT, Steitz JA (2001) Non-coding snoRNA host genes in Drosophila: expression strategies for modification guide snoRNAs. Eur J Cell Biol 80: 119–125 [DOI] [PubMed] [Google Scholar]
- Ullu E, Weiner AM (1985) Upstream sequences modulate the internal promoter of the human 7SL RNA gene. Nature 318: 371–374 [DOI] [PubMed] [Google Scholar]
- Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV (2005) enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res 33: W389–W392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Klambt C, Bachellerie JP, Brosius J, Huttenhofer A (2003) RNomics in Drosophila melanogaster: identification of 66 candidates for novel non-messenger RNAs. Nucleic Acids Res 31: 2495–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa Y, Felis M, Englert M, Stojanov M, Matousek J, Beier H, Sugiura M (2005) Plant 7SL RNA genes belong to type 4 of RNA polymerase III- dependent genes that are composed of mixed promoters. Plant J 43: 97–106 [DOI] [PubMed] [Google Scholar]
- Ziegelbauer J, Shan B, Yager D, Larabell C, Hoffmann B, Tjian R (2001) Transcription factor MIZ-1 is regulated via microtubule association. Mol Cell 8: 339–349 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials