

Abstract
Stress in the endoplasmic reticulum (ER stress) and its cellular response, the unfolded protein response (UPR), are implicated in a wide variety of diseases, but its significance in many disorders remains to be validated in vivo. Here, we analyzed a branch of the UPR mediated by xbp1 in Drosophila to establish its role in neurodegenerative diseases. The Drosophila xbp1 mRNA undergoes ire-1-mediated unconventional splicing in response to ER stress, and this property was used to develop a specific UPR marker, xbp1-EGFP, in which EGFP is expressed in frame only after ER stress. xbp1-EGFP responds specifically to ER stress, but not to proteins that form cytoplasmic aggregates. The ire-1/xbp1 pathway regulates heat shock cognate protein 3 (hsc3), an ER chaperone. xbp1 splicing and hsc3 induction occur in the retina of ninaEG69D−/+, a Drosophila model for autosomal dominant retinitis pigmentosa (ADRP), and reduction of xbp1 gene dosage accelerates retinal degeneration of these animals. These results demonstrate the role of the UPR in the Drosophila ADRP model and open new opportunities for examining the UPR in other Drosophila disease models.
Keywords: apoptosis, Drosophila, retinal degeneration, unfolded protein response, xbp1
Introduction
The endoplasmic reticulum (ER) is the cellular organelle where proteins destined for secretion or for diverse subcellular localizations are synthesized and acquire their correct conformation. Perturbations of the environment normally required for protein folding in the ER, or production of large amounts of misfolded proteins exceeding the functional capacity of the organelle, trigger a physiological response in the cell, collectively known as the unfolded protein response (UPR) (Patil and Walter, 2001; Harding et al, 2002; Schroder and Kaufman, 2005). The UPR serves to cope up with ER stress by transcriptionally regulating ER chaperones and other ER-resident proteins, attenuating the overall translation rate and increasing the degradation of misfolded ER proteins.
The mechanisms leading to the UPR activation and its short-term response in regulating gene expression are well characterized in various organisms. In Saccharomyces cerevisiae, unfolded proteins in the ER cause oligomerization of the ER transmembrane protein Ire-1. Upon oligomerization, the endoribonuclease activity of Ire-1 is activated, which catalyzes the unconventional splicing of hac-1 mRNA. Splicing of hac-1 mRNA allows its translation (Ruegsegger et al, 2001) and its protein product acts as a transcription factor, by binding to DNA motifs collectively called UPRE. This leads to the induction of proteins that help to alleviate ER stress (Mori et al, 1998). Ire-1 in Caenorhabditis elegans and mammals plays a similar role, by splicing the mRNA of xbp1 (Shen et al, 2001; Yoshida et al, 2001; Calfon et al, 2002), the functional homolog of hac-1. Other branches of the UPR include transcription factors ATF4 and ATF6 (Haze et al, 1999; Harding et al, 2000), as well as nontranscriptional mechanisms that reduce the overall amount of misfolded proteins in the ER. This occurs in part through the attenuation of protein translation through PERK, an ER transmembrane kinase (Harding and Ron, 1999), and enhanced rate of protein degradation, also known as ERAD (ER-associated degradation). PERK and the components of the ERAD machinery are activated in response to ER stress (Friedlander et al, 2000; Travers et al, 2000).
Whereas transient ER stress can be alleviated by the UPR, a prolonged condition of ER stress can trigger apoptosis. In a mouse model of Pelizaeus–Merzbacher disease (PMD), misfolded proteolipid protein in the ER triggers apoptosis of oligodendrocytes, and several components of the UPR have been shown to play a protective role against the progression of the disease (Southwood et al, 2002). On the other hand, certain components of the UPR, including the PERK/ATF4/CHOP branch, are implicated to play a proapoptotic role (Zinszner et al, 1998). Additionally, numerous cell culture studies implicate the UPR in many disorders caused by misfolded proteins in the cytoplasm, such as Huntington's and Parkinson's diseases (Kouroku et al, 2002; Nishitoh et al, 2002; Ryu et al, 2002; Takahashi et al, 2003). However, the links between the UPR and a wide variety of neurodegenerative disorders remain indirect and controversial, in part, owing to limitations of existing animal model systems (Forman et al, 2003).
To better understand the role of the UPR in disease, we focused on a set of alleles in Drosophila ninaE (or rhodopsin-1 (Rh-1)), which have molecular and phenotypic characteristics identical to those found in class III autosomal dominant retinitis pigmentosa (ADRP) (Leonard et al, 1992; Colley et al, 1995; Kurada and O'Tousa, 1995). These alleles have mutations either in transmembrane domains or in extracellular loops that are predicted to disrupt Rh-1 folding properties (Colley et al, 1995; Kurada and O'Tousa, 1995). ADRP-afflicted individuals, in both humans and Drosophila, show late-onset retinal degeneration, which occurs through the activation of apoptosis (Davidson and Steller, 1998; Galy et al, 2005). Inhibiting caspases blocks retinal degeneration and blindness in the Drosophila model, demonstrating that apoptosis is the main cause of the disease (Davidson and Steller, 1998).
Here, we examined whether the UPR contributes to the progression of retinal degeneration in class III ADRP model of Drosophila. We show that xbp1 splicing and the induction of an ER chaperone, heat shock cognate protein 3 (hsc3), occur in response to ER stress. Using this knowledge, we have devised an xbp1-EGFP fusion transgene as an in vivo marker of ER stress, designed to have EGFP expressed in frame with xbp1 only after Ire-1-mediated splicing occurs. xbp1-EGFP splicing occurs after mutant Rh-1 expression, but is not detectable in response to polyglutamine repeats or tau R406W, which cause neurodegenerative disorders in humans by forming cytoplasmic protein aggregates. xbp1-EGFP splicing also occurs in the Drosophila model of ADRP, and xbp1 has a protective role against retinal degeneration. These results demonstrate that ER stress occurs in the Drosophila ADRP model and suggest possible mechanisms by which apoptosis is activated in response to mutant Rh-1 molecules.
Results
Drosophila xbp1 gene structure
The Drosophila genome contains a gene (CG 9415) encoding a protein homologous to the mammalian xbp1 (Hollien and Weissman, 2006). In situ hybridization against xbp1 transcripts revealed that this gene is expressed at higher levels in cells specialized in protein secretion, such as the embryonic salivary gland cells (Figure 1A and B). The analysis of EST database revealed two xbp1 isoforms: the RA isoform containing an extra 23-base sequence compared to the RB isoform. Upon closer inspection, we found in the RA transcript a predicted double stem-loop structure similar to that found in mammalian xbp1 and yeast hac1 mRNAs (Figure 1C) (Shen et al, 2001; Yoshida et al, 2001; Calfon et al, 2002). These loop sequences show conservation in the bases important for Ire-1-mediated splicing, suggesting that Drosophpila xbp1 may also be spliced by Ire-1. In fact, the Drosophila genome contains a single homolog of ire-1 (Hollien and Weissman, 2006). The splicing of this 23-nucleotide sequence by Ire-1 is predicted to cause a frame shift during protein translation. As a result, the putative RA transcript encodes a 307-amino-acid protein whereas the RB transcript encodes a 498-amino-acid protein (Figure 1D).
Figure 1.
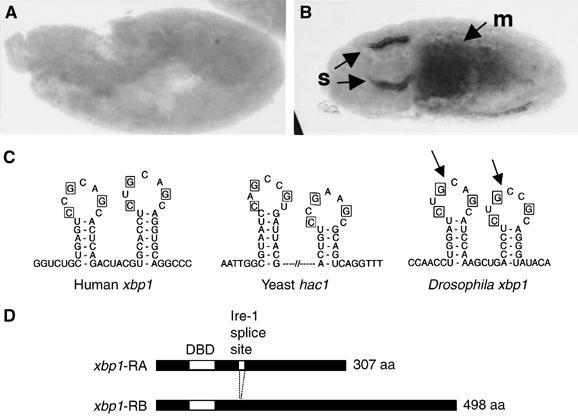
Structure and expression of Drosophila xbp1. (A, B) In situ hybridization against xbp1 mRNA in embryos. (A) A 6 h-old embryo shows little xbp1 mRNA. (B) A 12 h embryo with arrows pointing to high xbp1 mRNA in salivary glands (s) and the midgut (m). (C) A sequence comparison between the xbp1 genes of humans, Drosophila and the yeast hac1 shows a conserved stem–loop structure that is a target of Ire-1-mediated unconventional splicing. The bases required for Ire-1 recognition (in boxes) are conserved in the Drosophila xbp1 gene. The Ire-1 cleavage sites are marked with arrows. (D) The two predicted xbp1 isoforms. After Ire-1-mediated splicing, a frameshift in xbp1 translation occurs, converting a 307 aa protein into a 498 aa protein. The first white box indicates the DNA-binding domain (DBD). The second white box (smaller one) indicates the putative Ire-1 splice site.
ER stress triggers xbp1 mRNA splicing
To test whether xbp1 mRNA is spliced within the double stem–loop structure, we conducted RT–PCR for xbp1 transcripts from various Drosophila tissues. The examined tissues were adult heads, larval salivary glands and gut that normally secrete large amounts of proteins. After cloning of the PCR products and sequencing of a number of clones (n=13), we were not able to find any spliced (RB) form of xbp1, indicating that the spliced form of xbp1 is rare, if at all expressed, in the examined tissues of Drosophila. However, at least a small fraction of xbp1 mRNA is likely to be spliced, as several ESTs of the RB form from S2 cells, adult testis and mixed staged developing tissues are reported in the Berkeley Drosophila Genome Project (BDGP) database (http://www.fruitfly.org; see EST database therein). To test if xbp1 splicing is enhanced in response to ER stress, we used DTT, which blocks disulfide bond formation necessary for the folding of many ER proteins and can experimentally cause ER stress. After DTT treatment, 83% of the sequenced clones encoded the RB form of xbp1 (n=12). In contrast, no RB form clones were found in the mock-incubated tissues (n=17) (Figure 2A).
Figure 2.
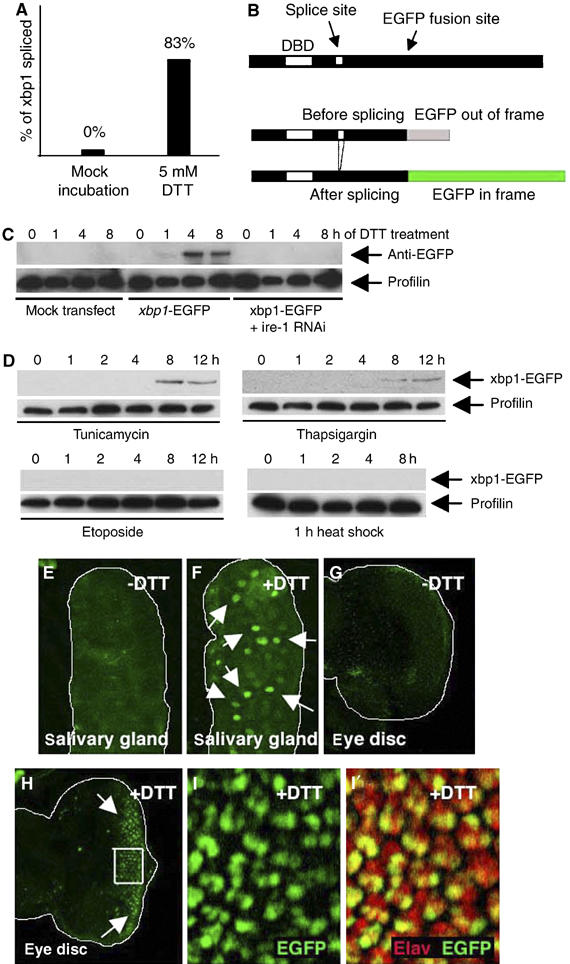
ER stress activates xbp1 splicing. (A) The percentage of clones of xbp1-RB form sequenced from tissues mock incubated in Schneider's medium (left; n=17), and after exposure to 5 mM DTT (right; n=12). (B) The design of the xbp1-EGFP reporter. EGFP was inserted 3′ to the putative Ire-1 splice site, giving rise to a fusion protein that lacks the xbp1 C-terminal region (upper). EGFP is out of frame without Ire-1-mediated splicing (middle), but comes in frame after splicing (lower). (C) xbp1-EGFP marker is activated by Ire-1 in response to DTT treatment. The top panel shows anti-GFP Westerns whereas the lower panel shows anti-Profilin as a control. S2 cells with or without xbp1-EGFP transfection were incubated with 5 mM DTT for up to 8 h. xbp1-EGFP activation is seen at 1 h and peaks at 4 h. Pretreating these cells with ire-1 dsRNA abolishes xbp1-EGFP marker activation. (D) Specificity of xbp1-EGFP activation. S2 cells transfected with xbp1-EGFP were treated with chemicals or heat-shock stress. The upper panels show anti-GFP Westerns to detect xbp1-EGFP activation, whereas the lower panels show anti-Profilin blots as a loading control. The cells were incubated with tunicamycin (10 μg/ml), thapsigargin (2 μM) or etoposide (10 μM) in S2 medium for up to 12 h. For heat-shock stress, cells were heat shocked for 1 h (37°C) and allowed to recover for up to 8 h to resume protein synthesis. (E–I) Activation of the xbp1-EGFP marker in response to DTT (genotype: arm-Gal4/uas-xbp1-EGFP). EGFP is labeled in green. Activated xbp1-EGFP markers are indicated by arrows. (E, F) Third instar salivary glands that were mock incubated without DTT (E) or with 5 mM DTT for 3 h (F). (G, H) Eye imaginal discs that were mock incubated without DTT (G) or incubated with 5 mM DTT for 2 h (H). Spliced Xbp1-EGFP accumulates in the nucleus of these cells. (I) A higher magnification image of the inset in (H). Single channel of EGFP (I) and double labeling with anti-Elav (red) in (I′) show that xbp1-EGFP is more readily activated in photoreceptor cells.
xbp1-EGFP acts as an ER-stress marker
To detect ER stress and the activation of the UPR in vivo, we generated an xbp1-EGFP fusion construct where EGFP was subcloned after the putative Ire-1 splice site, designed to have EGFP in frame with xbp1 only after the 23-base stem–loop of xbp1 is eliminated by splicing (Figure 2B; also see Materials and methods). A similar approach was used to monitor ER stress in transgenic mice (‘ER stress-activated indicator' or ERAI) (Iwawaki et al, 2004), C. elegans (Shim et al, 2004) and human cells (Back et al, 2005). To examine the properties of xbp1-EGFP in Drosophila cells, we transfected Drosophila S2 cells with xbp1-EGFP and analyzed the emergence of the EGFP epitope in the presence or absence of ER-stress-causing agents (Figure 2C and D). While mock-transfected cells did not show immunoreactivity to GFP, adding 1 mM DTT to the media of xbp1-EGFP-transfected cells caused xbp1-EGFP activation within an hour, with the amount of this marker peaking by 4 h. xbp1-EGFP activation was ire-1 dependent, as pretreating these cells with ire-1 double-stranded RNA (dsRNA) blocked xbp1-EGFP activation (Figure 2C). xbp1-EGFP was also activated by other ER-stress-causing agents, such as tunicamycin (10 μg/ml), which inhibits glycosylation of ER proteins, or thapsigargin (2 μM), which perturbs ER-calcium homeostasis. On the other hand, etoposide (10 μM), which is a DNA-damaging agent that does not affect the ER directly, did not activate xbp1-EGFP (Figure 2D). Interestingly, subjecting the cells to heat shock for 1 h at 37°C, which elicits a strong chaperone response in the cytoplasm, did not activate xbp1-EGFP (Figure 2D). In fact, we were not able to detect xbp1-EGFP activation after up to 4 h of heat-shock treatment (data not shown). This is consistent with observations made in mammalian cells that the UPR is resilient to activation by heat-shock conditions (e.g. Harding and Ron, 1999). To further characterize the UPR in vivo, several transgenic Drosophila lines were generated harboring uas-xbp1-EGFP, and this marker was expressed through a ubiquitous promoter of the armadillo gene, using the Gal4/uas method (Brand and Perrimon, 1993) (see Materials and methods). Ubiquitous expression of xbp1-EGFP did not interfere with animal development, and the resulting adults appeared normal after hatching. Moreover, we were not able to detect significant levels of EGFP fluorescence in embryos or third instar larval imaginal discs, with only an occasional emergence of EGFP-positive cells in the cuticle, neuronal and the tracheal cells of early larval stages (Supplementary data). However, when third instar larval tissues were incubated in Schneider's medium with 5 mM DTT, green fluorescence appeared within 30 min, with the intensity increasing over time (Figure 2E–I). Owing to the nuclear localization sequence within xbp1, xbp1-EGFP accumulated in the nuclei, allowing us to easily distinguish the xbp1-EGFP signal from any nonspecific background fluorescence. Larval tissues mock incubated in Schneider's media alone did not induce green fluorescence (Figure 2E and G). Interestingly, the levels of xbp1-EGFP fluorescence, in response to DTT, varied among cell types. In eye imaginal discs, for example, DTT-induced xbp1-EGFP fluorescence first appeared in the Elav-positive neuronal population (Figure 2I), before appearing in other cell types (data not shown). Together, these observations establish xbp1-EGFP as an in vivo UPR marker.
xbp1-EGFP is activated in response to mutant Rh-1, but not by proteins that cause cytoplasmic aggregates
To examine the connection between neurodegenerative diseases and the UPR, we expressed genes that cause neurodegenerative diseases by forming protein aggregates in the cytoplasm or the ER. Among those tested were Huntingtin-Q128 (Htt-Q128) (Figure 3A), MJD-tr-Q78 (Figure 3B), tau R406W (Figure 3C) and Rh-1G69D (Figure 3D). Htt-Q128 is a human huntingtin allele, with an expanded repeat of 128 poly-glutamine (poly-Q) that causes cytoplasmic aggregates and disease in humans and Drosophila (MacDonald et al, 1993; Lee et al, 2004). MJD-tr-Q78 is another poly-Q repeat gene that causes Spinocerebellar ataxia type 3 disease in humans and neuronal toxicity during Drosophila larval development (Warrick et al, 1998). Tau 406W is a mutant allele found in human Alzheimer's disease patients that also causes neuronal degeneration when expressed in Drosophila eye discs (Wittmann et al, 2001). We also generated uas-Rh-1G69D lines enabling the expression of a mutant Rh-1 allele with a transmembrane glycine substituted to an aspartic acid residue. An endogenous allele of Rh-1 with the same mutation, ninaEG69D, causes defects in Rh-1 maturation and ER to Golgi transport (Colley et al, 1995; Kurada and O'Tousa, 1995). Although a number of studies have suggested that the expression of poly-glutamine repeat proteins and other cytoplasmic aggregates activate the UPR indirectly through overloading of the proteasome capacity (Bence et al, 2001; Kouroku et al, 2002; Nishitoh et al, 2002), Htt-Q128, MJD-tr-Q78 or tau R406W expression through the GMR-Gal4 driver in larval eye imaginal discs did not activate significant levels of the xbp1-EGFP marker (Figure 3A, B and C). By contrast, Rh-1G69D expression through identical conditions caused the UPR activation as evidenced by xbp1-EGFP fluorescence (Figure 3D). Whereas expression of MJD-tr-Q78 or tau R406W caused eye ablation, expression of Htt-Q128 and Rh-1G69D resulted in intact adult eyes, allowing us to test whether the xbp1-EGFP marker becomes activated at this later stage. Horizontal cryosections of adult eyes showed that retinas expressing Rh-1G69D continued to activate xbp1-EGFP (Figure 3F), whereas control retinas expressing ubcD1 or those expressing Htt-Q128 did not (Figure 3E–G). These observations indicate that the xbp1-EGFP reporter is not as sensitive to cytoplasmic protein aggregates as it is to ER protein misfolding.
Figure 3.
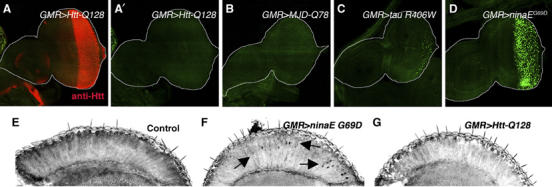
xbp1-EGFP is activated in response to mutant Rh-1, but not to proteins that cause cytoplasmic aggregates. In all panels, proteins predicted to misfold were expressed in eye imaginal discs posterior to the morphogenetic furrow and in adult retinas by using the GMR-Gal4 driver. (A) Huntingtin-Q128 expression is visualized by staining with the anti-Htt antibody (red). (A′) The anti-GFP channel only (genotype: GMR-Gal4/uas-xbp1-EGFP; uas-Htt-Q128/+). (B) MJD-tr-Q78-expressing eye disc (genotype: GMR-Gal4/uas-xbp1-EGFP; uas-MJD-tr-Q78/+). (C) Tau R406W-expressing eye disc (genotype: GMR-Gal4/uas-xbp1-EGFP; uas-tau R406W/+). (D) Rh-1G69D-expressing eye disc shows strong xbp1-EGFP activation (genotype: GMR-Gal4/uas-xbp1-EGFP; uas-Rh-1G69D/+). (E–G) Adult eye horizontal sections. (E) A control adult head expressing ubcD1 (GMR-Gal4/uas-ubcD1; uas-xbp1-EGFP). (F) Rh-1G69D expression activates xbp1-EGFP in adult retinas (arrows). (G) By contrast, xbp1-EGFP activation was not detected in Htt-Q128-expressing retinas.
Characterization of the xbp1 mutant allele
An xbp1 mutant line was isolated through the BDGP Gene Disruption Project (Spradling et al, 1999). The BDGP information on the P-element flanking sequence indicates that the P-element is inserted 71 bp upstream of the predicted transcription initiation site of the transcription start site (Figure 4A). RT–PCR of xbp1 of 4-day-old larvae revealed that the P-element insertion severely reduces transcript levels compared to similarly aged wild-type controls (Figure 4B). The mutant allele, xbp1k13803, is recessive lethal, and mutant animals show growth retardation and die before reaching the pupal stage (Figure 4C). Analysis of the mouth hook morphology of the xbp1−/− larvae indicated that the development stalled at the second instar larval stage (data not shown). The mutant larvae also lacked imaginal discs, as evidenced by the absence of anti-Eyes Absent (Eya) antibody labeling eye discs associated with larval brains (Figure 4D and E). However, the xbp1−/− phenotype in imaginal disc growth is not a direct effect on cell proliferation or survival, as clones of xbp1−/− cells can be observed within imaginal discs (see Figure 5E). Two observations indicate that these phenotypes can be attributed to the disruption of xbp1 function. First, a precise excision of the P-element reverted the phenotypes. Second, the xbp1k13803 allele failed to complement Df (2R)F36, a deletion encompassing the xbp1 locus (breakpoints 57B17;57C7). These results indicate that xbp1 is an essential gene in Drosophila.
Figure 4.
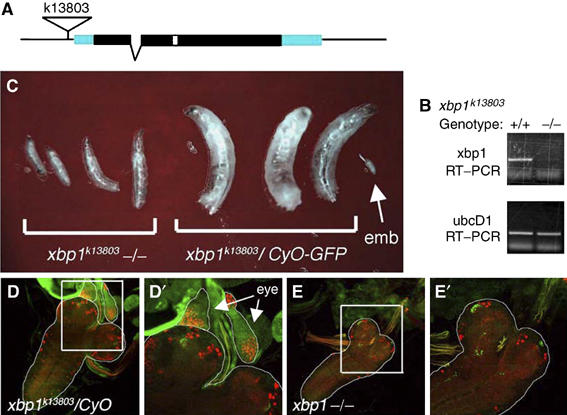
xbp1k13803−/− mutant phenotype. (A) The structure of xbp1 genomic locus. In the k13803 allele, a P-element is inserted 71 bp upstream of the putative transcription start site. The coding region is depicted in black boxes, the untranslated region in blue, one intron by a broken line and the unconventional ire-1 spliced intron in white. (B) RT–PCR reveals the loss of xbp1 transcripts in the xbp1k13803−/− larvae, compared to the wild-type controls (top). ubcD1, a ubiquitously expressed gene, is used as a loading control (bottom). (C) xbp1k13803 is a recessive lethal allele and shows larval growth retardation. Representative xbp1k13803−/− larvae are in the left and the xbp1/CyO (−/+) sibling control is in the right, at day 4 of development. Arrow points to an embryo (emb). (D, E) xbp1−/− larvae do not have imaginal discs. (D) Control larvae show eye imaginal discs on top of larval brains that are labeled with anti-Eya (red) and anti-Dronc (green; expressed ubiquitously in imaginal discs) (genotype: xbp1k13803/CyO-GFP). (D′) A magnified image of (D) in which eye discs are outlined and pointed with arrows. (E) xbp1−/− larvae do not have eye imaginal discs associated with larval brains (genotype: xbp1k13803/Df (2R)F36. (E′) A magnified image of the inset in (E).
Figure 5.
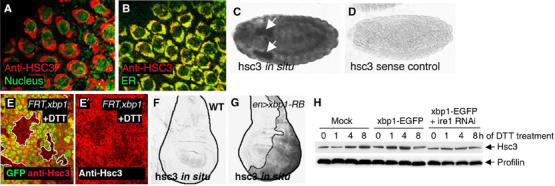
Hsc3 is an ER chaperone regulated by the Ire-1/Xbp1 pathway. (A, B) Hsc3 localizes to the ER. (A) Co-labeling with Hsc3 (red) and the nuclear membrane marker, wheat germ agglutinin (green), shows perinuclear labeling. (B) Co-labeling with the ER-YFP marker (green) shows colocalization of Hsc3 to the ER compartment (genotype: sqh-ER-YFP). (C, D) hsc3 in situ hybridization in embryos. (C) hsc3 in situ signal in embryos. Arrows point to salivary glands. (D) hsc3 sense control does not label under identical conditions. (E) Hsc3 induction by DTT requires xbp1. An eye imaginal discs with eyeless-Flipase-induced clones of xbp1−/− are labeled by the absence of GFP (green) and outlined (white boundary) (genotype: eyFLP; FRT42D xbp1k13803/FRT42DubiGFP). The discs were treated with 5 mM DTT (in S2 cells medium) for 16 h. Upregulation of Hsc3 (red) occurs in the wild-type tissue but not in xbp1 mutant tissue. (F) In situ hybridization for hsc3 in wild-type wing imaginal discs. (G) hsc3 mRNA levels are enhanced in discs expressing xbp1-RB in the posterior compartment (right half of the disc and outlined; genotype: engrailed-Gal4/uas-xbp1-RB; tub-Gal80ts: induced at 29°C for 18 h). (H) Hsc3 induction by DTT treatment requires ire-1. The upper panel shows anti-Hsc3 Western blot, whereas the lower panel shows anti-Profilin as a loading control. While Hsc3 is induced by DTT in nontransfected or xbp1-EGFP-transfected cells, pretreatment of ire-1 dsRNA blocks Hsc3 induction.
Ire-1/xbp1 pathway regulates the ER-chaperone Hsc3
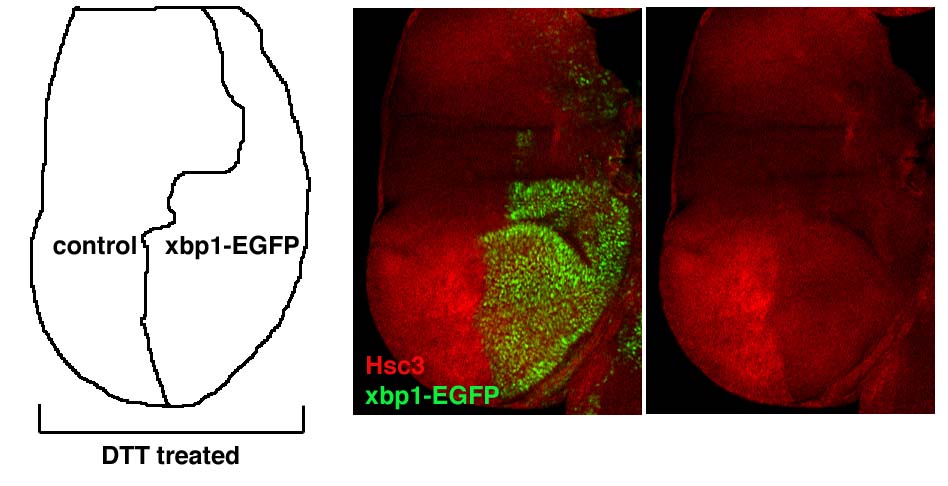
A hallmark of the UPR is the transcriptional activation of ER chaperones. In mammals, xbp1 contributes to such transcriptional regulation. The Drosophila homolog of the ER chaperone GRP78/BiP (Hendershot et al, 1988; Kozutsumi et al, 1989) is encoded by hsc3 (Rubin et al, 1993). A Western blot on embryo protein extracts with anti-Hsc3 antibody detected a 75 kDa band (data not shown), consistent with the predicted molecular weight of Hsc3. The immunohistochemistry of the embryonic amnioserosa cells revealed that Hsc3 staining colocalizes with an ER-YFP marker (LaJeunesse et al, 2004), which is consistent with the idea that Hsc3 is an ER chaperone (Figure 5A and B). The transcript of hsc3 is broadly expressed in embryos, with higher expression levels found in developing secretory organs, such as the salivary glands (Figure 5C and D). To test if xbp1 is required for hsc3 induction in response to ER stress, we generated mosaic clones of xbp1−/− cells in imaginal discs and treated them with DTT. After 16 h of DTT treatment, anti-Hsc3 immunoreactivity was enhanced in the xbp1+ population, whereas the xbp1−/− clones showed lower amounts of Hsc3 (Figure 5E). This indicated that xbp1 is required for enhanced Hsc3 expression in response to DTT treatment. We also tested whether the spliced xbp1 (xbp1-RB) can activate hsc3 transcription; we generated transgenic lines harboring the RB form of Drosophila xbp1. When xbp1-RB was conditionally expressed in the wing imaginal disc posterior (P) compartment, hsc3 was transcriptionally induced in response (Figure 5F and G). These data show that the Drosophila hsc3 expression is regulated by xbp1. To further confirm that the observed regulatory mechanism occurs through the ire-1/xbp1 pathway, we analyzed Hsc3 protein levels in S2 cells preteated with or without ire-1 dsRNA. Whereas Hsc3 levels increased after DTT treatment of cells in nontransfected or xbp1-EGFP-transfected cells, such increase was blocked by ire-1 dsRNA treatment (Figure 5H). In this experiment, we also noticed a weak dominant-negative effect of xbp1-EGFP on the induction of Hsc3 after 8 h of DTT treatment. The dominant-negative activity of xbp1-EGFP was further confirmed in vivo, where xbp1-EGFP-expressing imaginal discs cells showed lower amount of Hsc3 induction, compared to control cells when treated with DTT (Supplementary data). A similar dominant-negative effect was reported for the mammalian xbp1-GFP reporter (Iwawaki et al, 2004) and for unspliced xbp1, which is thought to form nonfunctional dimers with the spliced xbp1, thereby blocking its transcriptional activity (Lee et al, 2003; Yoshida et al, 2006). Collectively, these results show that blocking the formation or function of xbp1-RB compromises a cell's ability to induce Hsc3 during the UPR.
The UPR is activated in the ninaEG69D−/+ retina
With the tools to detect and examine the functional significance of the UPR in Drosophila, we tested if the UPR is activated in the background of ninaEG69D, a class III Drosophila Rh-1 mutant allele that serves as a model for ADRP in humans. Previous studies demonstrated that ninaEG69D protein has defective transport from ER to Golgi and accumulates in the ER (Colley et al, 1995; Kurada and O'Tousa, 1995). These observations prompted us to test if splicing of the xbp1-EGFP marker is activated in this mutant. We assayed this by driving the expression of xbp1-EGFP in either the ninaEG69D−/+ or the wild-type background. Splicing of the marker did not occur in the wild-type background, even with anti-GFP antibody to further enhance the sensitivity levels of the assay. In contrast, xbp1-EGFP splicing was readily detected in the ninaEG69D−/+ background, where EGFP specifically accumulated in the nuclei of Rh-1-expressing photoreceptor cells (Figure 6A and B). We also assayed for hsc3 transcript levels by in situ hybridization. ninaEG69D−/+ eyes had marked increase in hsc3 expression compared to wild-type controls (Figure 6C and D). These observations establish that the UPR is activated in ninaEG69D−/+ animals.
Figure 6.
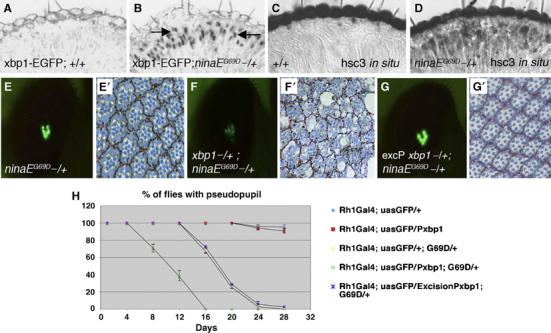
ninaEG69D mutants activate the UPR, which suppresses the course of retinal degeneration. (A, B) Horizontal sections of 5- to 10–day-old adult eyes expressing xbp1-EGFP in a wild-type background (A) or in a ninaEG69D−/+ background (B). EGFP epitope appears in the nuclei of only the ninaEG69D−/+ outer photoreceptors (R1–R6, arrows). (C, D) The transcripts of hsc3 are at low basal level in wild-type (C), but are significantly enhanced in ninaEG69D−/+ retina (D). Genotypes are Rh-1-Gal4; uas-xbp1-EGFP/+ (in A, C) and Rh-1-Gal4; ninaEG69D/uas-xbp1-EGFP (in B, D). (E, F) Representative examples of the pseudopupil image visualized with Rh1Gal4; uasGFP, at experimental day 12, for ninaEG69D−/+ (E), xbp1k13803−/+; ninaEG69D−/+ (F) and a control in the background of a precisely excised chromosome of xbp1k13803, excision Pxbp1−/+; ninaEG69D−/+ (G). (E′–G′) Representative examples of the respective tangential sections of (E–G). (H) Quantification of the degeneration process by the pseudopupil assay. For each genotype, the percentage indicates the number of flies with pseudopupil/total number of flies. Each data point is an average of at least three independent assays. xbp1k13803 has a dominant effect in accelerating the retinal degeneration of ninaEG69D.
An xbp1 mutant dominantly enhances retinal degeneration of ninaEG69D−/+
In order to find if xbp1 has any role in the degeneration of ninaEG69D−/+ retina, we tested if the xbp1k13803 allele exerts any dominant effect on the time course of retinal degeneration. The pseudopupil, an image formed deep in the retina by the projection of light from several ommatidial clusters (Franceschini and Kirschfeld, 1971), has been extensively used as an assay for photoreceptor integrity or degeneration. We used a variation of the pseudopupil assay, in which the transgenic expression of EGFP in the photoreceptors (in our case, Rh1-EGFP) allows the visualization of the pseudopupil under fluorescent light in a more sensitive manner (Pichaud and Desplan, 2001). Using this assay, we found no evidence of retinal degeneration in Rh1-EGFP flies, in the wild-type or xbp1k13803−/+ background (Figure 6). In ninaEG69D−/+ flies, however, the pseudopupil started to disappear after 16 days, in a progressive manner, and no flies with a pseudopupil image could be observed after day 24. The xbp1k13803−/+ background significantly accelerated the retinal degeneration of ninaEG69D−/+ flies; in some (around 30%) of the flies, the pseudopupil started to disappear by day 8 (i.e. 8 days earlier than ninaEG69D−/+ flies in a wild-type background) and no pseudopupils were observed after day 16. In contrast, the chromosome derived from a precise excision of the P element in xbp1k13803, thereby having a wild-type xbp1 allele, had no effect in the degeneration process, as in these flies the pseudopupil was lost at a rate similar to that of ninaEG69D−/+ flies. These observations indicated that disruption of xbp1, and not an unrelated background mutation, accounts for the enhanced degeneration rate of ninaEG69D−/+ retina. The pseudopupil results were confirmed by analyzing semi-thin sections of retinas from the different genotypes at day 12 (Figure 6E–G). Similar results were also obtained with another class III Rh-1 allele, ninaERH27, with a cysteine to tyrosine substitution in the amino acid 200 (data not shown). Collectively, these results indicate that xbp1 has a protective role against the retinal degeneration in class III ninaE alleles.
We also expressed xbp1-RB in the eye, to test if the gain of function of xbp1 can reverse the course of retinal degeneration of ninaE mutants. However, expression of xbp1-RB in the eye through the GMR promoter generated small, rough eyes with enhanced cell death, and was not suppressed by coexpression of the baculoviral caspase inhibitor p35 (data not shown). Late expression of xbp1-RB in the outer photoreceptor cells (Rh1-Gal4) also led to the degeneration of these cells 6–8 days after emergence of the flies from the pupal case, as evidenced by the lack of Rh1-GFP pseudopupils (data not shown). We cannot conclude from these experiments whether the toxic effect of xbp1-RB reflects its normal physiological role or an experimental artifact due to a high level of the protein, which is not present under physiological conditions.
Discussion
ER stress has been implicated in a wide variety of human diseases, including many neurodegenerative disorders (Forman et al, 2003), diabetes (Ozcan et al, 2004) and cancer (Bi et al, 2005), but relatively few have been validated in animal models. Here, we showed that the basic UPR pathway is conserved from yeast, C. elegans, Drosophila and mammals. Moreover, we demonstrate that the UPR is activated in the ADRP model of Drosophila, and this plays a protective role against the progression of retinal degeneration.
xbp1 splicing occurs specifically in response to ER stress
We have shown that xbp1 splicing is a specific indicator of the UPR signaling in Drosophila. Using the xbp1-EGFP fusion construct, we were able to detect xbp1 splicing in response to ER-stress-causing agents (DTT, thapsigargin and tunicamycin), ectopic expression mutant Rh-1, and in the Drosophila model of ADRP. However, our in vivo experiments did not support previous observations that cytoplasmic aggregates can also cause ER stress indirectly, through compromising the proteasome capacity. xbp1 activation in the ADRP model can account for many phenotypes previously reported. One such phenotype is the dramatic enlargement of the ER network (Colley et al, 1995; Kurada and O'Tousa, 1995). As studies in other organisms have shown that the ire-1/xbp1 branch of UPR promotes ER membrane biogenesis (Cox et al, 1997; Reimold et al, 2001), it is most likely that the enlarged ER-network biogenesis of ninaEG69D−/+ retina is due to Drosophila xbp1 activation. In contrast to the stressed cells, we were not able to detect significant levels of xbp1 (or xbp1-EGFP) splicing in unstressed developing tissues of embryos or late third instar larvae. This is consistent with previous studies conducted in transgenic mice harboring an xbp1-GFP(venus) construct similar to ours. In these mice, xbp1 splicing was not detected in embryonic or postnatal stages, but only after late postnatal stage (16 days or older) and only in a few tissues (Iwawaki et al, 2004). We did observe, however, a small number of cells showing xbp1-EGFP splicing in earlier stage larvae, indicating that occasional xbp1 splicing occurs during normal development. As xbp1 mutation is recessive lethal, the low level of natural xbp1 splicing may account for the requirement of this gene in Drosophila development. We cannot exclude, however, that the unspliced RA form of xbp1 also plays a role during development, accounting for the lethality of xbp1-deficient animals. xbp1 is required during embryonic development in mammals (Reimold et al, 2000), but not in C. elegans, where the animals can complete their developmental program without xbp1 (Shen et al, 2001). In addition to xbp1, hsc3 and ire-1, other genes homologous to those implicated in the UPR are found in the Drosophila genome. These include the ER transmembrane kinase perk (Pomar et al, 2003) and the ER tethered transcription factor ATF6 (annotated as CG3136). Although their in vivo function has not been analyzed in Drosophila, their presence and high sequence homology suggest a conserved function in the UPR.
Implications on the mechanism of neurodegeneration
Previous studies demonstrated that the class III ninaE alleles show caspase-dependent cell death (Davidson and Steller, 1998; Galy et al, 2005). The underlying mechanism by which caspases become active in these cells remains unclear, but our finding of the UPR activation in these cells provides at least three models. First model is based on the observation that JNK signaling is activated as part of the UPR (Urano et al, 2000). In this model, activation of the UPR stimulates Ire-1/TRAF interaction, independent of xbp1 mRNA splicing, leading to JNK activation and apoptosis. In fact, Ire-1/TRAF/JNK signaling is required for apoptosis in response to poly-glutamine repeat accumulation in cultured cells (Nishitoh et al, 2002). In addition, enhanced JNK signaling is detected in the retinas of a class III ninaE allele in Drosophila (Galy et al, 2005). Finally, activation of JNK signaling in Drosophila results in the induction of the proapoptotic gene hid (Moreno et al, 2002; Ryoo et al, 2004). The second model is based on the observations that Ca2+ release from the ER can activate caspase activation and cell death (Nakagawa et al, 2000; Orrenius et al, 2003; Rizzuto et al, 2003). It is possible that the disruption of ER homeostasis by the accumulation of misfolded proteins in the ER results in the release of Ca2+ into the cytoplasm, activating a proteolytic cascade leading to caspase activation. Third, as CHOP has been implicated to mediate ER-stress-triggered apoptosis in mammals, a similar mechanism may exist in Drosophila. However, there are no obvious homologs of CHOP in the Drosophila genome. Whether these models account for the death of class III ninaE mutant photoreceptors awaits further studies.
Drosophila as a model organism for investigating the UPR
Drosophila provides a unique advantage as a model for studying human diseases associated with ER-stress-triggered cell death, as the mechanisms of stress-provoked caspase activation are largely conserved between the two species. In addition, the short lifespan of Drosophila, combined with a similarly accelerated rate of chronic disease manifestation, may help validate the in vivo significance of the UPR in a growing list of disorders, ranging from neurodegenerative disease involving cytoplasmic protein aggregates (Nakanishi et al, 2005), hypoxia during cancer progression (Bi et al, 2005) and p53-induced cell death (Qu et al, 2004). In this regard, our present work on the role of the UPR in Drosophila retinal degeneration may provide a framework for further investigations into the UPR in human disease.
Materials and methods
Fly stocks
The following genotypes were used in this study: to characterize the xbp1 mutant phenotype, xbp1k13803/CyO-GFP and Df (2R)F36/CyO-GFP were used (from the Bloomington Stock Center). The precise excision of the xbp1k13803 allele was carried out by crossing this line to the D2-3 transposase line. ninaEG69D and ninaERH27 (Colley et al, 1995; Kurada and O'Tousa, 1995) were used as models for the class III ADRP. Gal4/uas system was used for ectopic gene expression in Drosophila (Brand and Perrimon, 1993), in which the following Gal4 lines were used: GMR-Gal4 (for eye imaginal disc expression), armadillo-Gal4 (for ubiquitous expression), Rh1-Gal4 (for expression in the outer photoreceptor cells (R1–R6; Pichaud and Desplan, 2001) during late pupal life and in the adult), engrailed-Gal4 (for expression in the posterior compartment of the wing imaginal disc). eyeless-Flipase was used to generate Xbp1 mutant clones in the eye and tub-Gal80ts (McGuire et al, 2004) was used for conditional expression of xbp1- RB. uas-xbp1-EGFP was made by subcloning EGFP after the EcoRV site of xbp1, which generated a chimeric construct with EGFP fused after the 252nd amino-acid residue of the xbp1-RA isoform. uas-Rh1G69D and uas-xbp1-RB were generated through RT–PCR and subcloned into the pUAST plasmid for generation of transgenic lines. uas-Htt Q128 (Lee et al, 2004), uas-MJD-tr-Q78 (Warrick et al, 1998) and uas-tau-R406W (Wittmann et al, 2001) were obtained to express proteins that form cytoplasmic aggregates. ER-YFP driven by the squash gene promoter (sqh-ER-YFP) was used as an ER marker, as in LaJeunesse et al (2004).
Molecular biology
RT–PCR was carried out with the xbp1 coding sequence based on the sequence information from flybase and BDGP, and subsequently subcloned into pBluescript SK+. To compare the xbp1 transcript levels between wild-type and xbp1k13803−/− larvae, total RNA was prepared from 4-day-old larvae, followed by RT–PCR for xbp1 or ubcD1. The mutant larvae were distinguished based on the loss of the CyO-GFP balancer. For the analysis of xbp1 isoforms, brains, imaginal discs and salivary glands of 40 larvae were collected and incubated in Schneider's cell medium containing 5 mM DTT, or mock incubated without DTT, for 4 h before conducting RT–PCR of xbp1. Clones were sequenced to examine the ratio of xbp1-RA and RB isoforms with and without DTT treatment. At least two independent mRNA preparations were made for the analysis. hsc3 cDNA was a gift from Karen Palter (Rubin et al, 1993), and the full-length cDNA (the 2.2 kb EcoRI fragment) was subsequently subcloned into pBluescript SK+. The T3 and T7 promoters of the pBluescript were used to generate DIG-labeled riboprobes for in situ hybridization using standard protocols. Alkaline phosphatase secondary antibody followed by NBT/BCIP reaction was used for further analysis.
Cell culture and RNAi treatment
Drosophila Schneider S2 cells were cultured under standard conditions (ref: invitrogen manual). A standard protocol was followed for dsRNA treatment of cultured cells (Armknecht et al, 2005). In brief, 20 μg of ire-1 dsRNA was added to each well containing 106 cells, which was followed by another boost of 20 μg dsRNA at day 3. At day 4, xbp1-EGFP was transfected to cells using Effectene™ (Qiagen). We split the cells at day 5 and treated them with DTT at day 6 to trigger the UPR. The ire-1 dsRNA consisted of a 509-nt region (Amplicon ID: DRSC 15606) described by the Drosophila RNAi Screening Center (http://www.flyrnai.org). The following primers were used to amplify this sequence from an embryo cDNA library: ‘R' primer: CAAAAGCAGAGCGAGAATG; ‘S' primer: TTAATGTCGCGATGCACAA. This amplicon has no predicted off-targets.
Western blots and immunohistochemistry
To generate the guinea-pig anti-Hsc3 antibody, the C-terminal half of the hsc3 coding sequence, beginning from the XhoI site to the 3′ end, was subcloned into pET14b (Novagen). The resulting 35 kDa His-tagged recombinant protein was purified to generate a polyclonal antibody. The antisera were subsequently affinity purified against the same epitope. The xbp1-EGFP marker signal was enhanced through anti-GFP antibody labeling (rabbit anti-GFP A6455 from Molecular Probes), followed by either FITC-linked (Figure 2) or alkaline phosphatase-linked (Figure 6) secondary antibodies. Other antibodies used were mouse anti-Rh1 (4C5), mouse anti-Elav (9F8a9) from University of Iowa DSHB and mouse anti-Huntingtin (Mab2166) from Chemicon. All secondary antibodies were from Jackson ImmunoResearch.
Retinal degeneration assay
Flies with the relevant genotypes were selected and cultured in vials (30–50 flies in each vial), at 25°C, in permanent light (around 2000 lux). The vials were changed frequently to avoid mixing of the flies with eventual progeny. The quantification of pseudopupils was performed on a pad under blue fluorescent light after anesthetizing the flies with CO2. Tangential plastic sections were performed as described previously (Tomlinson, 1985) and toluidine blue was used as a dye to increase the contrast.
Supplementary Material
Supplementary data 1
{kind=link}
Supplementary data 2
{kind=link}
Acknowledgments
We thank Charles Zuker, Troy Littleton, Bertrand Mollereau, Mel Feany, Nanci Bonini and the Bloomington Stock center for fly strains, Karen Palter for the hsc3 cDNA, Bertrand Mollereau, Cesar Mendes, Genevieve S Joseph, Jessica Treisman and Ramuji Dasgupta for technical advice, Rokhaya Cisse for embryo injections, the Steller lab members, Jonathan Lin and David Ron for comments and suggestions on the manuscript. We also thank Julie Hollien and Jonathan Weisman for communicating results before publication. HDR is a special fellow of the Leukemia-Lymphoma Society and HS is an investigator of the Howard Hughes Medical Institute.
References
- Armknecht S, Boutros M, Kiger A, Nybakken K, Mathey-Prevot B, Perrimon N (2005) High-throughput RNA interference screens in Drosophila tissue culture cells. Methods Enzymol 392: 55–73 [DOI] [PubMed] [Google Scholar]
- Back SH, Schroder M, Lee K, Zhang K, Kaufman RJ (2005) ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35: 395–416 [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin–proteasome system by protein aggregation. Science 292: 1552–1555 [DOI] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C (2005) ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24: 3470–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415 [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplsmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96 [DOI] [PubMed] [Google Scholar]
- Colley NJ, Cassill JA, Baker EK, Zuker CS (1995) Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc Natl Acad Sci USA 92: 3070–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Chapman RE, Walter P (1997) The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell 8: 1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson FF, Steller H (1998) Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature 391: 587–591 [DOI] [PubMed] [Google Scholar]
- Forman MS, Lee VM-Y, Trojanowski JQ (2003) Unfolding' pathways in neurodegenerative disease. Trends Neurosci 26: 407–410 [DOI] [PubMed] [Google Scholar]
- Franceschini N, Kirschfeld K (1971) In vivo optical study of photoreceptor elements in the compound eye of Drosophila. Kybernetik 8: 1–13 [DOI] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol 2: 379–384 [DOI] [PubMed] [Google Scholar]
- Galy A, Roux MJ, Sahel JA, Leveillard T, Giangrande A (2005) Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for rhodopsinPro23His human retinitis pigmentosa. Hum Mol Genet 14: 2547–2557 [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D (2002) Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol 18: 575–599 [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Harding HP, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271–274 [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10: 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot LM, Ting J, Lee AS (1988) Identity of the immunoglobulin heavy-chain-binding protein with the 78 000-dalton glucose-regulated protein and the role of posttranslational modifications in its binding function. Mol Cell Biol 8: 4250–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313: 52–53 [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K, Miura M (2004) A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med 10: 98–102 [DOI] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Jimbo A, Kikuchi T, Yamagata T, Momoi MY, Kominami E, Kuida K, Sakamaki K, Yonehara S, Momoi T (2002) Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum Mol Genet 15: 1505–1515 [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y, Normington K, Press E, Slaughter C, Sambrook J, Gething MJ (1989) Identification of immunoglobulin heavy chain binding protein as glucose-regulated protein 78 on the basis of amino acid sequence, immunological cross-reactivity, and functional activity. J Cell Sci Suppl 11: 115–137 [DOI] [PubMed] [Google Scholar]
- Kurada P, O'Tousa JE (1995) Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron 14: 571–579 [DOI] [PubMed] [Google Scholar]
- LaJeunesse DR, Buckner SM, Lake J, Na C, Pirt A, Fromson K (2004) Three new Drosophila markers of intracellular membranes. Biotechniques 36: 784–790 [DOI] [PubMed] [Google Scholar]
- Lee A-H, Iwakoshi NN, Anderson KC, Glimcher LH (2003) Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA 100: 9946–9951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT (2004) Cytoplasmic aggregates trap polyglutmamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci USA 101: 3224–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard DS, Bowman VD, Ready DF, Pak WL (1992) Degeneration of photoreceptors in rhodopsin mutants of Drosophila. J Neurobiol 23: 605–626 [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose C, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins MAA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sediacek Z, Poustka A, Frischauf A-M, Lehrach H, Buckler AJ, Church D, Doucette-Stamm L, O'Donovan MC, Riba-Ramirez L, Shah M, Stanton V, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983 [DOI] [PubMed] [Google Scholar]
- McGuire SE, Mao Z, Davis RL (2004) Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci STKE 220: p16. [DOI] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12: 1263–1268 [DOI] [PubMed] [Google Scholar]
- Mori K, Ogawa N, Kawahara T, Yanagi H, Yura T (1998) Palindorme with spacer of one nucleotide is characteristic of the cis-acting unfolded protein response element in Saccharomyces cerevisiae. J Biol Chem 273: 9912–9920 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103 [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Sudo T, Morishima N (2005) Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol 169: 555–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4: 552–565 [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461 [DOI] [PubMed] [Google Scholar]
- Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13: 349–355 [DOI] [PubMed] [Google Scholar]
- Pichaud F, Desplan C (2001) A new visualization approach for identifying mutations that affect differentiation and organization of the Drosophila ommatidia. Development 128: 815–826 [DOI] [PubMed] [Google Scholar]
- Pomar N, Berlanga JJ, Campuzano S, Hernandez G, Elias M, de Haro C (2003) Functional characterization of Drosophila melanogaster PERK eukaryotic initiation factor 2alpha (eIF2alpha) kinase. Eur J Biochem 270: 293–306 [DOI] [PubMed] [Google Scholar]
- Qu L, Huang S, Baltzis D, Rivas-Estilla A-M, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Toshimura A, Koromilas AE (2004) Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3b. Genes Dev 18: 261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH (2000) An essential role in liver development for transcription factor XBP-1. Genes Dev 14: 152–157 [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412: 300–307 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhaes PJ, Di Virgillio F, Pozzan T (2003) Calcium and apoptosis: facts and hypotheses. Oncogene 22: 8619–8627 [DOI] [PubMed] [Google Scholar]
- Rubin DM, Mehta AD, Zhu J, Shoham S, Chen X, Wells QR, Palter KB (1993) Genomic structure and sequence analysis of Drosophila melanogaster HSC70 genes. Gene 128: 155–163 [DOI] [PubMed] [Google Scholar]
- Ruegsegger U, Leber JH, Walter P (2001) Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 107: 103–114 [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the wingless signaling pathways. Dev Cell 5: 853–855 [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci 22: 10690–10698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789 [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107: 893–903 [DOI] [PubMed] [Google Scholar]
- Shim J, Umemura T, Nothstein E, Rongo C (2004) The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol Biol Cell 15: 4818–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A (2002) The unfolded protein response modulates disease severity in Pelizaeus–Merzbacher disease. Neuron 36: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM (1999) The Berkeley Drosophila Genome Project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics 153: 135–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R, Imai Y, Hattori N, Mizuno Y (2003) Parkin and endoplasmic reticulum stress. Ann NY Acad Sci 991: 101–106 [DOI] [PubMed] [Google Scholar]
- Tomlinson A (1985) The cellular dynamics of pattern formation in the eye of Drosophila. J Embryol Exp Morphol 89: 313–331 [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101: 249–258 [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinase by transmembrane protein kinase IRE1. Science 287: 664–666 [DOI] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93: 939–949 [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711–714 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Oku M, Suzuki M, Mori K (2006) pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 172: 565–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data 1
Supplementary data 2