

Abstract
The Drosophila element Mos1 is a class II transposon, which moves by a ‘cut-and-paste' mechanism and can be experimentally mobilized in the Caenorhabditis elegans germ line. Here, we triggered the excision of identified Mos1 insertions to create chromosomal breaks at given sites and further manipulate the broken loci. Double-strand break (DSB) repair could be achieved by gene conversion using a transgene containing sequences homologous to the broken chromosomal region as a repair template. Consequently, mutations engineered in the transgene could be copied to a specific locus at high frequency. This pathway was further characterized to develop an efficient tool—called MosTIC—to manipulate the C. elegans genome. Analysis of DSB repair during MosTIC experiments demonstrated that DSBs could also be sealed by end-joining in the germ line, independently from the evolutionarily conserved Ku80 and ligase IV factors. In conjunction with a publicly available Mos1 insertion library currently being generated, MosTIC will provide a general tool to customize the C. elegans genome.
Keywords: DSB repair, homologous recombination, NHEJ, targeted mutagenesis
Introduction
Gene knockout and knock-in techniques have emerged as powerful tools to study gene function in eukaryotic model organisms such as yeast (Scherer and Davis, 1979), mouse (Doetschman et al, 1987; Thomas and Capecchi, 1987) and more recently Drosophila (Rong and Golic, 2000; Bibikova et al, 2002; Bibikova et al, 2003; Beumer et al, 2006). These techniques rely on recombination between an engineered DNA fragment and the locus to target via regions of homology provided in the DNA fragment. The engineered DNA is introduced into cells by transformation, usually as a linear fragment, or produced in vivo as for Drosophila. In the nematode Caenorhabditis elegans, spontaneous recombination between the genome and exogenous sequences is inefficient. This might be due to the ability of C. elegans germline cells to rapidly concatemerize DNA fragments introduced in the gonad and build stable extrachromosomal transgenes (Stinchcomb et al, 1985), a process that might outcompete recombination with chromosomal DNA. Very few examples of genome engineering by homologous recombination have been documented so far in C. elegans (Plasterk and Groenen, 1992; Broverman et al, 1993; Barrett et al, 2004; Berezikov et al, 2004; Jantsch et al, 2004).
One strategy to increase the recombination rate at a specific locus consists of generating a DNA double-strand break (DSB) at the locus. DSBs are very deleterious lesions, which are repaired by the cellular machinery using a number of different mechanisms conserved among eukaryotes (Haber, 2000) (Figure 1A). DSB repair mechanisms can be split in two classes: non-homologous end-joining (NHEJ) (for reviews see Dudasova et al, 2004; Daley et al, 2005; Hefferin and Tomkinson, 2005), which involves the rejoining of DNA ends by ligation, and homologous recombination (Paques and Haber, 1999). NHEJ can be achieved by several pathways (Lieber et al, 2003; Hefferin and Tomkinson, 2005). The canonical pathway depends on the evolutionarily conserved Ku and ligase IV proteins, which ensure protection from exonucleolytic degradation of broken ends and their sealing by ligation, respectively. This process will restore the initial sequence if the ends are cohesive, or introduce small footprints if ends are not complementary. In this paper, repair processes where little or no processing of the DSB is observed will be referred to as conservative. Recombination pathways are initiated by exonucleolytic processing of the DSB end by a 5′-to-3′ exonuclease, which exposes a single-stranded region of DNA that is engaged in a search for homology. If complementary strands of homologous regions flanking the DSB can anneal within the same chromosome, a process called single-strand annealing is initiated. Non-homologous 3′ end tails will be removed, new DNA synthesis and ligation occur, and the intervening sequence is lost, resulting in deletions at the DSB site. Alternatively, the single-stranded DNA can invade a homologous donor sequence and act as a primer for new DNA synthesis. One subsequent pathway, among others, can lead to non-reciprocal transfer of DNA from the donor to the recipient broken allele, a process called gene conversion. Various homologous sequences can potentially be used as repair templates, including sister chromatids and homologous chromosomes, or transgenes containing sequences homologous to the DSB flanking regions. If the transgenic fragments have been modified, these modifications will be copied in the broken locus during gene conversion, thus providing the possibility of engineering custom alleles.
Figure 1.
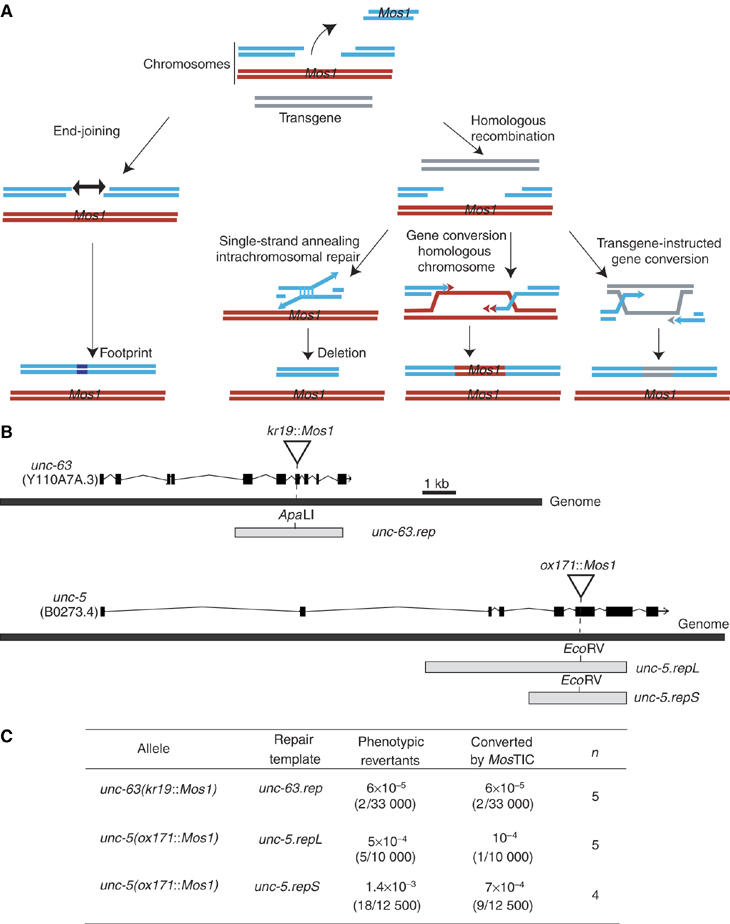
MosTIC strategy and efficiency. (A) Main pathways potentially used to repair a Mos1 excision-induced DSB during MosTIC experiments. In this example, homologous chromosomes (in red and blue) carry the same Mos1 insertion and a homologous repair template is provided on a transgene (in gray). Repair of the broken chromosome (in blue) is based either on end-joining or homologous recombination (single-strand annealing or gene conversion) (see Introduction). (B) Schematic representation of the exon/intron structure of the Mos1-containing alleles unc-63(kr19∷Mos1) and unc-5(ox171∷Mos1) with the repair templates (unc-63.rep, unc-5.repL and unc-5.repS) used in MosTIC experiments. Mos1 elements are indicated by triangles. The restriction sites ApaLI and EcoRV were introduced into the repair templates to identify MosTIC events. The repair templates did not contain enough sequences to rescue the mutant phenotypes. However, copying these sequences into the Mos1-mutated genomic loci would restore functional genes. (C) MosTIC efficiency at the unc-63 and unc-5 loci. Frequencies correspond to the number of phenotypic revertants in the progeny of transgenic animals where Mos1 excision was triggered by heat-shock. MosTIC events were identified among the phenotypic revertants by the presence of an ApaLI site (unc-63 locus) or an EcoRV site (unc-5 locus) copied into their genome. n, number of independent experiments.
The feasibility of transgene-instructed DSB repair in the C. elegans germ line was demonstrated by Plasterk and Groenen (1992), who remobilized a copy of the endogenous DNA transposon Tc1 out of the unc-22 gene. When Tc1 excision was triggered in the presence of a transgene carrying a unc-22 fragment with silent polymorphisms, in rare instances repair was achieved using the transgene as a repair template and point mutations were copied in the unc-22 locus. Similar experiments were recently performed to show that, apart from introducing point mutations, a similar strategy can be used to engineer deletions and insertions at a Tc1 excision site (Barrett et al, 2004). However, a major drawback of using endogenous C. elegans transposons to introduce DSBs at given loci is inability to control transposition. One haploid genome of the wild-type C. elegans N2 strain contains at least 70 copies of active DNA transposons. Transposition can be observed in somatic cells but is completely repressed in the germ line. Derepression of germline transposition is only achieved in some mutant backgrounds, the ‘mutator' strains, causing a high rate of spontaneous mutations and resulting in the accumulation of transposon copies in the genome (Bessereau, 2006). Besides problems related to the morbidity of these mutant strains, there is a significant probability of recovering uncontrolled mutations tightly linked to the locus that is to be engineered when performing transgene-instructed gene conversion in mutator backgrounds.
To circumvent these problems, the controlled excision of a heterologous transposon could be used to generate DSBs at loci to be engineered. The transposon Mos1 was first isolated in Drosophila (Jacobson and Hartl, 1985; Jacobson et al, 1986) and experimentally introduced in C. elegans (Bessereau et al, 2001). Mos1 is a 1280 bp DNA transposon of the Tc1/mariner family and transposes via a ‘cut-and-paste' mechanism (Benjamin and Kleckner, 1992; van Luenen et al, 1994; Lampe et al, 1996). The Mos transposase is the only factor required to achieve transposition. It binds the terminal inverted repeats present at each site of the transposon and catalyzes Mos1 excision and reinsertion. Excision leaves behind a DSB, which must be repaired by the cellular machinery. Mos1 was initially used in C. elegans to perform insertional mutagenesis and rapidly identify mutated genes (Bessereau et al, 2001; Granger et al, 2004; Williams et al, 2005). Mos1 transposition could be achieved by driving the expression of the Mos transposase under the control of a heat-shock-inducible promoter. The Mos transposase proved unable to mobilize endogenous transposable elements. Therefore, Mos1 transposition in the C. elegans germ line is controlled and specific.
In the present study, we triggered Mos1 excision from specific loci to generate localized chromosomal breaks and analyzed DSB repair. We observed that repair could be achieved by multiple pathways. Specifically, we demonstrated that DSBs could be efficiently repaired by transgene-instructed gene conversion in the germ line. In addition, DSBs could also be repaired by conservative NHEJ. Although this process required the Ku80 and ligase IV in somatic cells, conservative repair could be achieved in the germ line in the absence of these proteins. Therefore, controlled excision of Mos1 gives access to DSB repair mechanisms and provides an efficient way to engineer the C. elegans genome.
Results
Transgene-instructed gene conversion can be triggered by Mos1 excision
To investigate the feasibility of engineering the C. elegans genome using transgene-instructed gene conversion triggered by Mos1 excision, we designed a strategy that would enable the detection of the gene conversion events based on the reversion of a mutant phenotype. For this purpose, we first selected Mos1 insertions causing strong visible phenotypes. Second, we constructed repair templates containing wild-type genomic sequences that flanked the Mos1 insertion point. Third, point mutations were introduced into the repair templates to generate silent restriction sites close to the Mos1 insertion point. These repair templates did not contain enough sequences to rescue the mutant phenotypes by themselves. However, copying these sequences into the Mos1-mutated genomic locus would restore a functional gene. Therefore, transgene-instructed gene conversion events could be easily identified by (i) scoring for phenotypic revertants among the progeny of mutant animals and (ii) testing the genome of these revertants for the presence of the restriction site contained in the repair template.
We first used a Mos1 insertion in the unc-63 gene, which encodes an α-subunit of nicotinic acetylcholine receptors present at neuromuscular junctions (Culetto et al, 2004) (Figure 1B). unc-63(kr19∷Mos1) mutants display severely impaired locomotion (Williams et al, 2005). We designed a repair template, unc-63.rep, containing 1.8 and 1.4 kb of genomic sequence flanking the left and right side of the Mos1 insertion point, respectively. Two point mutations were introduced to create a silent ApaLI restriction site at the Mos1 insertion point. The repair template was injected into unc-63(kr19∷Mos1) homozygous mutants together with a vector enabling the expression of the Mos transposase under the control of the heat-shock promoter hsp-16.48 (Bessereau et al, 2001). Transgenic lines were heat-shocked and screened at the next generation for animals with improved locomotion. Two phenotypic revertants were identified out of 33 000 scored progeny (n=5 experiments) (Figure 1C). PCR amplification of the genomic region initially containing Mos1 indicated that Mos1 was no longer present in unc-63. Furthermore, restriction analysis and sequencing of the PCR products demonstrated that the ApaLI restriction site that was initially contained in the repair template had been copied into the unc-63 genomic locus. Based on these results, we concluded that these revertants resulted from transgene-instructed gene conversion after excision of the Mos1 element out of the unc-63 locus.
We subsequently performed similar experiments with a Mos1 insertion in the unc-5 gene (kind gift from W Davis and E Jorgensen), which encodes a netrin receptor (Hedgecock et al, 1990; Leung-Hagesteijn et al, 1992). unc-5(ox171∷Mos1) homozygous mutants display strong axonal outgrowth defects and are severely paralyzed. As the length of homologous sequence contained in the repair template might influence the efficiency of the gene conversion process (Barrett et al, 2004), we designed two repair templates. Both contained a silent EcoRV restriction site at the Mos1 insertion site. unc-5.repL (for unc-5.repairLong template) contained 4.5 kb plus 1.4 kb genomic fragments flanking the left and right side of the Mos1 insertion point, respectively; unc-5.repS (Short) contained 1.5 kb plus 1.4 kb genomic fragments. Transgenes carrying one repair template and the Mos transposase expression vector were introduced into unc-5(ox171∷Mos1) homozygous mutants. Transgenic lines were heat-shocked and screened at the next generation for wild-type moving worms (Figure 1C). Reversion events were recovered at a frequency of 5 × 10−4 using unc-5.repL (5 in 10 000 scored progeny, n=5 experiments) and 1.4 × 10−3 using unc-5.repS (18 in 12 500, n=4). PCR analysis demonstrated that all revertants lost the Mos1 element from one unc-5 locus. However, only a fraction of these revertants contained the EcoRV site provided in the repair templates (one out of five derived from unc-5.repL and nine out of 18 from unc-5.repS). Analysis of additional revertants confirmed that transgene-instructed repair only accounted for a fraction of the reversion events (unc-5.repL: 3/15 revertants analyzed; unc-5.repS: 12/27). Further characterization of the revertants that were generated by other mechanisms is presented below.
Together, these results demonstrated that we were able to use transgene-instructed gene conversion following Mos1 excision to introduce point mutations in the C. elegans genome at two different loci, with frequencies varying from 6 × 10−5 to 7 × 10−4 events per generation. We called this technique MosTIC for Mos1 excision-induced transgene-instructed gene conversion.
Analysis of the conversion tract
During the repair of a DSB by gene conversion, sequences adjacent to the DSB site are copied from the repair template to the broken chromosome (Gloor et al, 1991; Plasterk and Groenen, 1992). To characterize the gene conversion tracts during MosTIC experiments, we introduced silent mutations in unc-5.repL to generate unc-5.repP (Polymorphic) (Figure 2A). The unc-5.repP repair template contains nine polymorphisms distributed over the unc-5 genomic fragment, in exons and introns, which can be detected by restriction analysis or DNA sequencing. An extrachromosomal array carrying unc-5.repP and the Mos transposase expression vector was generated in mutant animals homozygous for unc-5(ox171∷Mos1). MosTIC experiments were performed as described above. Phenotypic revertants were observed at a frequency of 7 × 10−4 events per generation. Twenty-one out of 48 revertants had copied the EcoRV site, which overlaps the Mos1 insertion point in unc-5.repP, into their genome and were further analyzed for the presence of additional polymorphisms (Figure 2A). The size of the conversion tracts ranged from fewer than 30 bp (two events) to up to 3 kb (one event). In one-third of the events (7/21), more than 1 kb of sequence was copied from the repair template into the genome. No discontinuous conversion tract was observed. When the frequency of conversion at a given site was plotted against the distance from the Mos1 insertion point, the curve described a bell shape centered at the Mos1 insertion point (Figure 2B), which is characteristic of a gene conversion repair mechanism (Gloor et al, 1991; Nassif and Engels, 1993; Nassif et al, 1994). These data indicate that a mutation engineered in a 1 kb region centered around the DSB site would be copied in the genome in about half of the MosTIC events, at frequencies above 10−4 events per generation.
Figure 2.
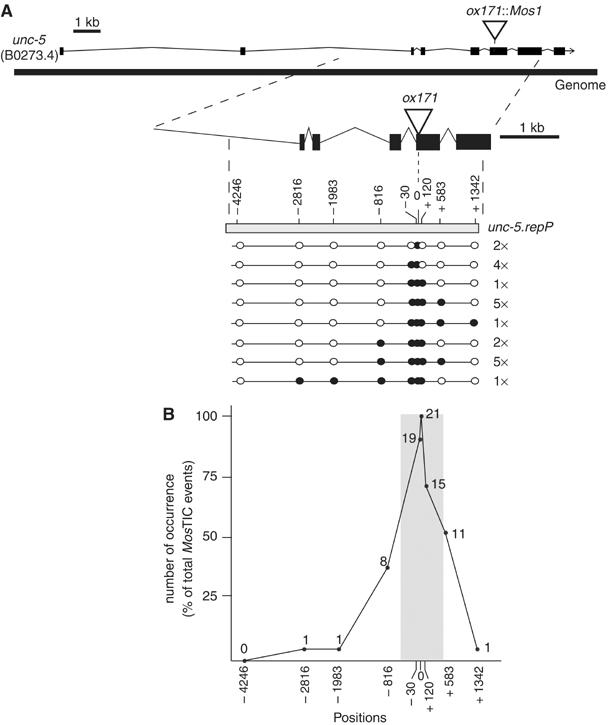
Analysis of MosTIC conversion tract. (A) Repair template engineered to monitor the MosTIC conversion tract. The restriction sites and SNPs introduced into the repair template are designated by their position relative to the Mos1 insertion point and are as follows: −4246 nt=AclI, −2816=HindIII, −1983=NheI, −816=Acc65I, −30=SNP1, 0=EcoRV, +120=SNP2, +583=SacI and +1342=SmaI. The conversion tracts analyzed are represented below the repair template scheme. Black and white dots show sites that were present and absent, respectively, in the chromosome after gene conversion. Numbers to the right of conversion tracts indicate the occurrence of each tract (out of 21). (B) The number of occurrences per site was plotted against their position in the repair template. The numbers close to the dots are the raw data. The gray zone corresponds to the 1 kb region centered at the Mos1 insertion point.
Engineering gene knockouts and gene knock-ins with MosTIC
Apart from creating point mutations in the C. elegans genome, the MosTIC technique could be used to generate deletions and introduce exogenous sequences into the chromosomes. First, we attempted to generate a deletion in the unc-5 locus. Two templates, unc-5.Ldel and unc-5.Sdel, carrying 800 nt deletions, were derived from unc-5.repL and unc-5.repS, respectively (see Materials and methods). unc-5.Ldel and unc-5.Sdel were introduced into a homozygous background for unc-5(ox171∷Mos1) together with the Mos transposase expression vector. As we could not rely on phenotypic reversion to detect MosTIC events, the progeny of heat-shocked animals were screened by PCR for the presence of the unc-5 deletion using primers on each side of the deleted region. One primer was present in both the genome and the transgene sequence, whereas the second primer was present in the genome but absent from the repair template. According to this strategy, a specific 1.6 kb PCR fragment would be amplified only if the deletion had been introduced into the genome by MosTIC. However, we could amplify such a 1.6 kb PCR fragment from every tested pool of transgenic animals, including pools of non-heat-shocked control animals (data not shown). We hypothesized that amplification of this fragment resulted from a process described as ‘PCR jumping' or ‘PCR bridging' (Paabo et al, 1990; Liu et al, 2002): during PCR, ‘bridging' occurs between single-stranded DNA molecules elongated from the genome and from the transgene because these two templates contain a region of sequence similarity. This generates a fusion fragment that is efficiently amplified at the next cycles. In our experiments, the amplification of the PCR bridging product could be minimized by decreasing the annealing time and increasing the annealing temperature. Using optimized PCR conditions, we screened pools of 100 animals. Sibs from positive pools were further analyzed to isolate strains homozygous for the deletions initially designed in the repair templates. Using three independent transgenic lines containing unc-5.Ldel, we isolated two deleted lines out of 11 000 screened animals (Figure 3B). Sequencing analysis of the unc-5 locus demonstrated the presence of a deletion identical to the one as in the repair template. No MosTIC event was detected in 10 000 screened F1 animals using unc-5.Sdel, suggesting that the regions of homology left in this repair template might be too short to drive efficient gene conversion.
Figure 3.
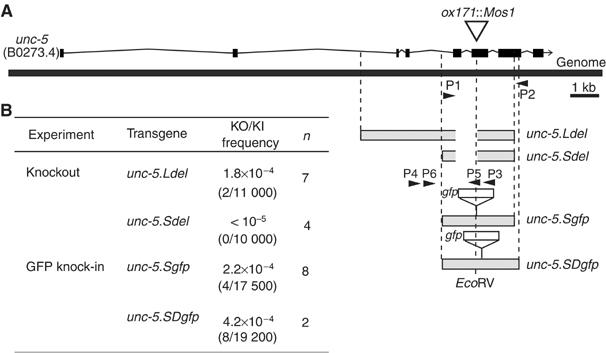
Knockout and knock-in by MosTIC at the unc-5 locus. (A) Scheme of the repair templates designed to engineer deletions and insertions in the unc-5 gene. The primers used to screen by PCR for MosTIC-engineered animals are indicated by arrows. Limits of the repair templates are indicated by dotted lines. The EcoRV site present in unc-5.SDgfp is shown. (B) Frequencies of the different events. n, number of independent experiments.
Second, we used MosTIC to introduce the GFP coding sequence into the unc-5 locus. The repair template unc-5.Sgfp, carrying the gfp sequence at the EcoRV site, was derived from unc-5.repS (Figure 3A). MosTIC experiments were performed as described above and recombinants containing chromosomal insertion of the GFP in the unc-5 locus were screened by PCR (Figure 3A). Out of 175 pools of 100 F1 animals, we isolated four lines containing genomic insertion of the GFP sequence (Figure 3B). gfp insertion was confirmed by direct sequencing of the unc-5 engineered loci. Despite the fact that GFP was fused in-frame with the UNC-5 coding sequence, insertion of the GFP in the middle of the protein likely disrupts the function of the UNC-5 receptor as demonstrated by the absence of mutant phenotype rescue and the absence of GFP detectable by fluorescence microscopy (data not shown). Although we demonstrated that MosTIC was efficient at introducing point mutations within a 1 kb region surrounding the Mos1-triggered DSB, we wondered whether the presence of a region of non-homology, such as gfp, at a distant site from the transposon in the repair template would modify recombination efficiency. To answer this question, we inserted the gfp sequence in-frame with the UNC-5 coding sequence 180 nt away from the DSB site (unc-5.SDgfp repair template; Figure 3A). Animals containing full-length gfp in the unc-5 locus were recovered with a frequency of 4.2 × 10−4 (8 in 19 200 scored progeny, n=2; Figure 3B). Such frequency is similar to what was obtained when introducing SNPs at a similar distance from the Mos1 insertion (Figure 2) and suggests that the presence of heterologous sequence in the donor template does not significantly affect the gene conversion process.
Together, these results demonstrate that MosTIC can be used to engineer gene knockouts and gene knock-ins at frequencies higher than 2 × 10−4 MosTIC events per F1 animal.
Mos1-triggered DSB can be repaired by end-joining in the germ line
Over the course of MosTIC experiments conducted to modify the unc-5 locus, it was systematically possible to identify wild-type moving animals that did not copy the polymorphisms contained in the repair templates (Table I). These events accounted for a significant fraction of the revertant animal population: for instance, they represented 80% of revertants generated using the repair template unc-5.repL (12/15), 56% of the revertants isolated while using unc-5.repS (15/27) and 54% of the revertants analyzed in the MosTIC experiments performed with unc-5.repP (27/48). PCR and sequencing analysis demonstrated that these revertants had lost the Mos1 insertion from the unc-5 locus. However, the absence of the EcoRV restriction site contained in the repair template suggested that these events did not result from repair by MosTIC. At the position that corresponded to the Mos1 insertion site, we observed footprints that introduced frame-preserving mutations, deletions or insertions (Table I), indicating that the corresponding region in the UNC-5 protein can tolerate significant changes without altering the overall functionality of UNC-5. These data demonstrated that apart from transgene-instructed repair, a DSB caused by Mos1 excision could be efficiently repaired by an additional process in the C. elegans germ line. The lack of recovery of similar events while performing MosTIC experiments at the unc-63 locus is likely explained by the insertion of Mos1 in a unc-63 exon encoding a highly conserved region among AChR subunits, which probably cannot tolerate mutations without inactivating the AChR.
Table 1.
Footprints generated in the germ line during MosTIC experiments at the unc-5 locusa
 |
Mos1 excision leaves non-complementary 3′ protruding single strands (3′-PSS) that are 3 nt long at each side of the break. To gain further insight into possible DSB repair mechanisms, we analyzed the DNA footprints left at the Mos1 excision site. They could be classified into three categories. Class I contained small footprints ranging from a few base-pair insertions contained in Mos1-derived 3′-PSS to small deletions (less than 10 nt). Among these events, we could further distinguish between footprints where both TA dinucleotides that flank the 3′-PSS were intact (class Ia) and footprints where at least one nucleotide in the 3′-PSS flanking sequence had been deleted (class Ib). These footprints represented 72% (55/76) of the analyzed events. Such footprints can be considered conservative and are usually found after repair by a canonical NHEJ mechanism. Class II footprints (3/76) consisted of larger deletions. Remarkably, a 633 bp deletion was able to reconstitute an in-frame sequence encoding a functional UNC-5 protein. Class III footprints (18/76) contained insertions that could be up to 36 nt long. The inserted sequences always corresponded to small direct duplications of sequences adjacent to the DSB point. In every case, it was possible to find microhomologies between one broken end, usually in the 3′-PSS, and the sequence immediately 5′ to the duplicated region, suggesting that this end was used to prime DNA synthesis. In most cases, nucleotides immediately 3′ to the duplication could pair with the other broken end, suggesting that sealing the two ends might use a microhomology-based repair mechanism involving the de novo-synthesized strand and one broken end (see Discussion and Supplementary Figure S1).
These results demonstrate that during MosTIC experiments, DSB caused by Mos1 excision can be repaired by various mechanisms including end-joining, in addition to transgene-instructed gene conversion.
Ku–ligase IV-dependent NHEJ mechanisms are preferentially used in C. elegans somatic cells but are dispensable in the germ line for DSB repair after Mos1 excision
The data presented above demonstrated that both end-joining repair and transgene-instructed gene conversion operate in the germ line to heal Mos1-triggered DSBs, hence raising the possibility that they might compete during MosTIC experiments. Therefore, we tested whether MosTIC efficiency would be increased in genetic backgrounds defective for end-joining repair. Specifically, we used mutants expected to be defective for the Ku80 and ligase IV, two factors highly conserved from yeast to humans that define a canonical NHEJ pathway (Hefferin and Tomkinson, 2005). These factors were recently shown to affect the sensitivity to ionizing radiations in somatic cells but not in the C. elegans germ line (Clejan et al, 2006). Yet, DSB repair was not analyzed at the sequence level.
To characterize the involvement of CKU-80 and LIG-4 in the repair of Mos1-triggered DSBs, we first used a PCR-based assay that could identify most end-joining repair events, including those generating sequences encoding non-functional proteins and those that occur in somatic cells. A transgene carrying the Mos transposase-encoding gene under the control of a heat-shock promoter was introduced into animals homozygous for the unc-5(ox171∷Mos1) allele. As opposed to the previous experiments in which DSB repair events were analyzed in the progeny of heat-shocked animals, transgenic animals were harvested 18 h after heat-shock and directly analyzed by PCR using primers in the unc-5 sequence flanking the Mos1 insertion (Figure 4). In every experiment, we could amplify two PCR products: a predicted 2.6 kb fragment, which contained Mos1, and a 1.3 kb fragment corresponding to end-joining products (Figure 4A). Subcloning the total PCR products and sequencing 1.3 kb inserts confirmed that Mos1 was absent from these fragments. The small amount of end-joining products in non-heat-shocked animals most likely reflects leaky expression of the Mos transposase in somatic tissues from the heat-shock promoter, as observed with an hsp∷gfp transgene (data not shown). We analyzed footprints generated at the excision site (Figure 4B). They were mainly class Ia (12/14), suggesting that the broken ends were efficiently protected against exonucleolytic degradation as observed during repair by a conservative NHEJ mechanism. In addition, six class II deletions were identified as well as one deletion containing a remaining Mos1 fragment at the undeleted break point. Such events are frequently observed after transposon excision repair and might result from a mechanism called synthesis-dependent strand annealing (SDSA) (Nassif et al, 1994; Adams et al, 2003; Izsvak et al, 2004). Class III duplications were under-represented as compared to what we observed previously in the germ line during MosTIC experiments. This suggests that DSB repair products detected by the PCR-based assay might occur mostly in somatic cells where canonical NHEJ might predominate. Accordingly, similar results were obtained using early L2 larvae in which the germ line was not yet developed (data not shown).
Figure 4.

PCR analysis of DSB repair in lig-4/cku-80-defective backgrounds. (A) Details of the experimental procedure used to study DSB repair by PCR. unc-5(ox171∷Mos1) adults were heat-shocked (t=0) to induce Mos1 excision and DSB repair was analyzed by PCR with primers flanking the Mos1 insertion point (t=18 h). PCR products were analyzed on an agarose gel: (−) non-heat-shocked, (+) heat-shocked samples, M=size marker (1 kb Plus DNA Ladder, Invitrogen). (B) Sequence analysis of footprints generated in wild-type and cku-80 and lig-4 mutants. See Table I footnotes for legends. Numbers in parentheses represent the numbers of footprints analyzed per genotype.
To test the involvement of Ku80 and ligase IV in the conservative repair process detected by PCR, we performed similar experiments in lig-4(tm750) and cku-80(ok861) mutant backgrounds. The abundance of the shorter PCR fragment was strongly decreased in both mutants with respect to the Mos1-containing product (Figure 4A). As NHEJ factors were previously reported to regulate Sleeping Beauty transposition in mammalian cells (Izsvak et al, 2004), we cannot rule out that this decrease might reflect a change of Mos1 excision frequency in this assay. To further characterize a potential impairment of NHEJ in cku-80/lig-4 mutants, we analyzed the sequence of the footprints generated in those backgrounds (Figure 4B). In cku-80(ok861) mutants, conservative repair was still detected. However, no class Ia event could be isolated (0/8), in contrast to the class I events recovered in the wild type. This suggests that in the absence of CKU-80, the broken ends are no longer efficiently protected against exonucleolytic degradation. In lig-4 mutants, class I events were almost absent (only one event was recovered, which might have been generated in the germ line; see below). In contrast, class II footprints were not significantly affected by the absence of either CKU-80 or LIG-4. These results indicated that in C. elegans somatic cells, at least two end-joining mechanisms coexist. One prominent conservative mechanism depends on LIG-4 and partially on CKU-80 and generated small footprints in which the genomic sequences flanking the DSB are intact, whereas a second mechanism, which was mostly CKU-80- and LIG-4-independent, generated deletions of the flanking genomic sequences and could also be used to generate small direct duplications.
As the results described above demonstrated that cku-80 and lig-4 mutations impaired NHEJ, we tested whether the relative frequency of end-joining repair events versus transgene-instructed gene conversion would be affected in cku-80 and lig-4 mutant backgrounds during MosTIC experiments. MosTIC experiments were performed as described previously at the unc-5 locus in cku-80 and lig-4 mutants. Phenotypic reversion and MosTIC events were recovered at frequencies similar to those obtained in a wild-type background (Table II). Similarly, we were not able to distinguish any qualitative difference between the repair events generated in wild-type or mutant backgrounds by analyzing the sequences of the repair events. Specifically, the class I and III footprints that we observed in cku-80 and lig-4 mutants were similar to that observed in the wild-type background (Table I). Therefore, these results indicated that repair of DSBs induced by Mos1 excision in the germ line, including conservative end-joining, can be achieved in the absence of CKU-80 and LIG-4.
Table 2.
Footprints generated in the germ line of lig-4/cku-80-defective animals during MosTIC experimentsa
 |
Discussion
A by-product of type II transposon mobilization is the generation of a DSB at the transposon excision site. In this study, we took advantage of the ability to control Mos1 transposition in the C. elegans germ line to introduce chromosomal breaks at given loci. Analysis of the repair process indicated that Mos1-triggered DSB can be repaired by end-joining and homologous-recombination mechanisms. First, we demonstrated that the well-conserved canonical Ku80–ligase IV-dependent end-joining pathway is also present in C. elegans where it is mostly used in somatic cells, whereas a second mechanism is Ku80- and ligase IV-independent and functions in germline cells to achieve conservative end-joining. Second, we showed that a homologous recombination repair pathway can be used to achieve transgene-instructed gene conversion. Based on this observation, we developed a novel technique, which we named MosTIC. It allows the engineering of custom alleles and provides an efficient tool to manipulate the C. elegans genome.
Mos1 excision repair generates diverse footprints, including direct duplications
During DSB repair, the structure of the broken ends affects the end-joining process. Complementary and blunt termini can be rapidly repaired by a single ligation event, whereas complex substrates require multiple processing steps to achieve break repair (Pfeiffer and Vielmetter, 1988). The Mos1 transposase catalyzes DSBs at each end of the Mos1 element. The 5′ cut of the non-transferred strand is made 3 bases within the transposon, whereas the 3′ cut is made exactly at the end of the transposon. As a result, Mos1 excision leaves non-complementary 3′-PSS that are 3 nt long at each side of the break (Dawson and Finnegan, 2003). In our experiments, end-joining repair generated several categories of footprints.
The first class of events generated small footprints ranging from the insertion of few bases contained in the 3′-PSS to the deletion of fewer than 10 bases immediately flanking the break point. Such footprints are very similar to those usually observed after DNA transposon excision (see, for instance, examples reported for Mos1 in Drosophila (Bryan et al, 1990), Tc1 and Tc3 in C. elegans (Ruan and Emmons, 1987; Plasterk, 1991; Plasterk and Groenen, 1992; van Luenen et al, 1994) or Sleeping Beauty in mammalian cells (Fischer et al, 2001; Izsvak et al, 2004)), and are typical of repair by canonical NHEJ. These events were prominent in somatic cells, where they required Ku80 and ligase IV activities.
A second class of repair events was Ku–ligase IV-independent and generated deletions from one hundred to one thousand base pairs long, which were mostly asymmetrical. This class of footprints was Ku80- and ligase IV-independent. The generation of similar deletions after Tc1 excision has been widely used to inactivate genes in C. elegans. As short direct repeats were sometimes found at the end points of these deletions, it was proposed that the two broken DNA ends scan each other's flank until a match is found, after which the break is sealed and the intervening sequence is lost (Zwaal et al, 1993). In our experiments, careful analysis of the sequences flanking deletion points could only identify microhomologies limited to one or a few nucleotides. An alternative pathway called SDSA has been proposed for transposition-induced DSB repair based on the observation that repair often generates a deleted version of the transposon (Nassif et al, 1994; Adams et al, 2003). In this scenario, DSB repair uses a homologous template, which in our case would be the homologous chromosome or the sister chromatid, which contains a Mos1 copy, and possibly the repair template during MosTIC experiments. If this mechanism is processive enough, it regenerates a transposon copy at the excision site and is therefore not detected. If the process aborts before the whole template is copied, a Ku–ligase IV-independent, microhomology-mediated end-joining pathway is used to seal the broken ends and often generates deleted versions of transposable elements (Verkaik et al, 2002; Adams et al, 2003). SDSA was shown to be used in different organisms during transposon excision repair (Nassif et al, 1994; Adams et al, 2003; Yant and Kay, 2003; Izsvak et al, 2004; McVey et al, 2004). In our experiments, footprints containing fragments of the transposon were found in the PCR approach. Similarly, a unc-5 revertant was found over the course of MosTIC experiments to contain a fragment of the GFP tag copied from the repair template together with deletion of the adjacent genomic sequences (Supplementary Figure S2). These footprints suggest that SDSA is active in C. elegans. Finally, besides SDSA, direct end-joining might occur independently of Ku–ligase IV activities via microhomology-dependent processes after one or both broken ends have been subjected to exonucleolytic processing (Ma et al, 2003).
The third class of footprints consists of small direct duplications. Sparse examples of such repair events after transposon excision can be found in the literature. Repaired DSB sites containing duplications have been described in human cells (Roth et al, 1985) and Drosophila (Adams et al, 2003; McVey et al, 2004), and were also found at the site of chromosomal translocations in follicular lymphomas (Jager et al, 2000) and promyelocytic leukemias (Welzel et al, 2001). Several mechanistic hypotheses have been put forward to account for these duplications. In human cells, slipped mispairing was proposed (Roth et al, 1985). In Drosophila, footprints containing templated nucleotides were thought to be ‘characteristic of aberrant end-joining after aborted homologous recombination' (Adams et al, 2003). The existence of an error-prone polymerase involved in the generation of those events was also postulated (McVey et al, 2004). In our experimental situation, the duplication events that we recovered might result from DNA synthesis primed at the 3′-PSS. In most cases, we could identify microhomology between one of the 3′-PSS and the region immediately 5′ to the duplicated sequence. After pairing with the opposite chromosomal broken end, eight bases were copied on average (from 3 to 32), then followed by microhomology-directed end-joining between the de novo-synthesized sequence and the broken end (Supplementary Figure S1). It would be interesting to test whether such a mechanism is only observed in the repair of DSB with 3′-PSS ends or if it is a more general mechanism able to generate small direct duplications.
A conservative NHEJ mechanism might function independently of Ku–ligase IV in the C. elegans germ line
Analysis of DSB repair after Mos1 excision was performed in adult C. elegans hermaphrodites, which contain 959 somatic cells and more than 1000 germline nuclei. Therefore, detection of repair products using PCR on whole animal lysates potentially identifies events that might occur in the soma and in the germ line. Using this strategy, we confirmed the expected function of cku-80 and lig-4 genes in NHEJ in C. elegans. The Ku80 and Ku70 proteins are widely conserved among phyla, from yeast to human (for review see Hefferin and Tomkinson, 2005). They form heterodimers that bind broken ends, protect them from degradation and might serve as alignment factors. In cku-80 mutants, the detected amount of NHEJ products was dramatically reduced in our PCR assay. Some end-joining products could be identified but in every case we observed limited exonucleolytic degradation of at least one of the broken ends, including the 3′-PSS and a few adjacent nucleotides, and repair occurred at sites of microhomologies. In vertebrate cells, the loss of Ku80 causes the disappearance of the Ku70 protein (Errami et al, 1996; Gu et al, 1997; Singleton et al, 1997). Repair of DSBs in the absence of Ku80 mostly generates large deletions or SDSA repair products containing partial sequences of the transposon (Feldmann et al, 2000; Guirouilh-Barbat et al, 2004; Izsvak et al, 2004), as also observed in this study. The occurrence of imperfect NHEJ events suggests that, even in the absence of Ku activity, the ligase IV can achieve end-joining before the broken ends have been subjected to extensive degradation. However, we cannot totally exclude that a residual Ku70 activity functions in C. elegans in the absence of Ku80 and still protects broken ends, but less efficiently than a Ku70–Ku80 dimer.
During conservative end-joining, the ligase IV provides the ligase activity required to seal broken ends. In contrast to mouse Lig4 mutants that die during development due to early neurodegeneration (Barnes et al, 1998; Frank et al, 2000), C. elegans lig-4 mutants are viable and fertile and do not exhibit gross phenotypes, as in Drosophila (Gorski et al, 2003; McVey et al, 2004; Romeijn et al, 2005). However, we demonstrated that NHEJ repair was heavily impaired in the absence of ligase IV. PCR amplification of the unc-5 locus after Mos1 excision hardly detected any amplification products corresponding to precise repair of the broken ends. Out of 19 events that were sequenced, only one corresponded to precise end-joining (and might actually have been generated in the germ line; see below). Most events were deletions, either symmetrical or asymmetrical, with occasional insertions at the repair site. These data are very similar to what was observed after repair of transposon excision in other systems such as Drosophila in the absence of ligase IV (Romeijn et al, 2005) or of its cofactor XRCC4 in mammalian cells (Izsvak et al, 2004). Altogether, our results demonstrate that a canonical Ku–ligase IV-dependent NHEJ pathway is at work in C. elegans to perform conservative end-joining of DSBs.
To test whether impairing NHEJ would favor transgene-instructed repair in the germ line after Mos1 excision, we performed MosTIC experiments in cku-80 and lig-4 mutants. DSB repair was detected based on reversion of the uncoordinated phenotype of unc-5(ox171∷Mos1) mutants. By contrast with the results obtained in PCR-based experiments, the frequency and the nature of the repair events were unchanged in cku-80 and lig-4 mutants as compared to wild type. Specifically, class I events that derive from conservative end-joining of broken chromosome ends were recovered at the same frequency in mutant and wild-type backgrounds. Several hypotheses can be proposed to explain how these footprints are generated. First, they might result from an SDSA process that would abort immediately. Testing this hypothesis would require the analysis of Mos1-induced DSB repair in homologous recombination-defective mutants, such as rad-51, which mediates strand invasion required for SDSA. Unfortunately, rad-51 is an essential gene in C. elegans, which precludes analysis of DSB repair in the germ line. Second, conservative end-joining observed in the absence of Ku80–ligase IV could be reminiscent of microhomology-dependent end-joining processes that have been described in yeast and Drosophila. In yeast, microhomology-mediated end-joining was shown to be independent of Ku and partially independent of ligase IV (Ma et al, 2003), leading to small deletions at the repair site. In Drosophila, DSB repair could still be performed in the germ line in the absence of the ligase IV, even after the homologous recombination machinery was genetically disrupted (McVey et al, 2004). However, it is important to note that microhomologies were not often found at the site of repair after Mos1-induced DSB repair, as opposed to what was observed in cases described above. Altogether, our results revealed that two processes coexist in C. elegans to generate conservative end-joining. One process, which resembles well-characterized canonical NHEJ repair pathways, depends on Ku and ligase IV and seems to be mostly active in somatic cells. A second process is capable of achieving conservative repair of broken chromosomes in the absence of Ku80 and ligase IV, in agreement with recent results indicating that Ku80 and ligase IV are dispensable in the germ line for DNA repair after exposure to ionizing radiations (Clejan et al, 2006).
MosTIC, a novel technique to engineer the C. elegans genome
Apart from end-joining, mechanisms based on homologous recombination are able to repair DSBs in the C. elegans germ line. Because this study was performed in animals homozygous for Mos1 insertions, repair events based on homologous recombination using the intact chromosome as a repair template could not be identified. Repair by homologous recombination-based mechanisms was only observed when providing homologous transgenes that could be used as repair templates. During this process, sequences contained in the transgene were copied into the genome, resulting in gene conversion. The length of the gene conversion tract was investigated using an asymmetric repair template carrying 1.4 and 4.5 kb of homologous sequences with silent mutations distributed along both arms of the repair template. Mutations were copied in the chromosome at frequencies distributed along a bell-shaped curve centered at the Mos1 excision site. The median length of the conversion tract was roughly 500 bp on each side of the DSB and in one example conversion extended up to 2.8 kb from the Mos1 excision site. This distribution is highly reminiscent of the data obtained in the Drosophila germ line while performing targeted gene replacement via P element-induced gap repair (Gloor et al, 1991). Similar conversion tracts are also observed in yeast (Borts and Haber, 1989; Palmer et al, 2003) and mammalian cells (Taghian and Nickoloff, 1997; Elliott et al, 1998), although they tend to be shorter. Interestingly, the presence of non-homologous sequence in the donor template, such as gfp, does not cause abrupt termination of the conversion tract. Therefore, the MosTIC technique provides a means to modify a sequence at distance from a given Mos1 insertion.
Transgene-instructed gene conversion triggered by Mos1 excision, a technique that we called MosTIC, provides an efficient way to engineer custom alleles in the C. elegans genome. MosTIC events were recovered at frequencies ranging from 10−5 to 10−4 per generation. This efficiency is much higher than the initial gene-conversion experiments performed after Tc1 mobilization in a mut-6 mutator background (Plasterk and Groenen, 1992) and comparable to what was reported more recently after repeating those experiments in mut-2 and mut-7 backgrounds (Barrett et al, 2004). Several hypotheses were raised to account for the improved efficiency in the most recent study. First, the use of specific mutator backgrounds, which were needed to derepress Tc1 transposition, was proposed to improve transposition excision rate. However, apart from global derepression of all endogenous transposable elements, mut-2 and mut-7 also participate in other regulatory pathways such as RNA interference and germline co-suppression (Ketting et al, 1999; Tabara et al, 1999; Dernburg et al, 2000; Ketting and Plasterk, 2000; Vastenhouw et al, 2003; Robert et al, 2005) that could have modified gene-conversion efficiency. In our system, Mos1 transposition was achieved in a wild-type background. To test if a mutator background would modify MosTIC efficiency, some MosTIC experiments were repeated in a mut-7(pk204) background. Gene conversion events were recovered at similar frequencies as in the wild type (data not shown). This indicates that MosTIC experiments can be performed in a wild-type background without losing efficiency. Second, the use of long asymmetric templates containing about 7–9 kb of homologous sequence was proposed to be more efficient than the symmetrical 3 kb template used by Plasterk and Groenen (1992). In MosTIC experiments performed to target the unc-5 locus, we obtained the same efficiency with 6 kb asymmetrical templates and 2.9 kb symmetrical templates. Interestingly, similar MosTIC frequencies were obtained when the region of homology was interrupted by a deletion or by the presence of a GFP-coding fragment. These results suggest that the search for homology to engage recombination is restricted to the regions adjacent to the DSB. As a practical correlate, it is possible to mutate a C. elegans gene by MosTIC starting with a fragment of less than 3 kb that contains the modification to be introduced in the genome.
In conclusion, MosTIC provides an efficient way to manipulate the C. elegans genome that relies on the mobilization of Mos1 at a specific locus. When Mos1 transposes in the C. elegans germ line, an average of 2.6 insertions occur per haploid genome (Williams et al, 2005). These insertions are unlinked and can easily be separated by out-crossing. Therefore, MosTIC experiments can be performed in animals where a single Mos1 insertion is present, hence minimizing the risk of introducing uncontrolled mutations at other loci. In addition, the only requirement to achieve efficient germline Mos1 excision is a transgenic Mos transposase source, which can be easily eliminated once the conversion experiment is performed without further genetic manipulation of the MosTIC-engineered strains. The prerequisite of this technique is to have a Mos1 element inserted into the genomic region to be engineered. Such insertions can be recovered in genetic screens using Mos1-mediated mutagenesis (Williams et al, 2005). In addition, a European effort (http://elegans.imbb.forth.gr/nemagenetag/) is in progress to generate a comprehensive Mos1 insertion library. To date, around 5000 Mos1 insertions have been isolated. They are annotated in Wormbase (http://www.wormbase.org/) and can be freely obtained on simple request. As the distribution of Mos1 insertions is relatively uniform in the genome, MosTIC potentially provides an efficient means to manipulate a large proportion of the C. elegans genes.
Materials and methods
C. elegans strains and alleles
MosTIC lines: Microinjections (Stinchcomb et al, 1985) were performed in EN19 (unc-63(kr19∷Mos1) I) or EN59 (unc-5(ox171∷Mos1) IV) strains with DNA mix containing pJL44[hsp-16.48∷transposase] (50 ng/μl), repair template (50 ng/μl) and pPB118.33[myo-2:gfp] (5 ng/μl). Transgenic F1 animals were selected based on GFP expression in the pharynx and transgenic lines were cultivated at 25°C.
NHEJ-defective strains: FX750 (lig-4(tm750) III) was generated by the National Bioresource Project for the Experimental Animal ‘Nematode C. elegans' (Japan) and RB873 (lig-4(ok716) III) and RB964 (cku-80(ok861) III) by the OMRF Knockout Group (Canada).
Plasmid construction
unc-63.rep: Genomic sequences between positions 5 145 834 and 5 149 076 of chromosome I were cloned into pBS KSII+ (Stratagene) digested by XhoI and EcoRV. Positions 5 147 665 and 5 147 668 were respectively changed into a ‘g' and a ‘c' to create an ApaLI site.
unc-5.repS and unc-5.repL: Genomic sequences between positions 5 497 225 and 5 500 133 and positions 5 497 225 and 5 503 280 of chromosome IV were respectively cloned into pBS KSII+. Positions 5 498 637–5 498 643 were replaced by ‘gatatc' to create the EcoRV site.
unc-5.repP: unc-5.repL was mutagenized using the QuickChange® II XL site-directed mutagenesis and QuickChange® multi-site-directed mutagenesis kits (Stratagene). Introduced polymorphisms are described Figure 2.
unc-5.Ldel and unc-5.Sdel: unc-5.Ldel and unc-5.Sdel were constructed starting respectively from unc-5.repL and unc-5.repS, which were both digested with EcoRV and SalI. After filling the SalI site with the klenow enzyme, the plasmids were religated. The EcoRV–SalI junctions were sequenced in unc-5.Ldel (…tcagattcgacg…) and unc-5.Sdel (…tcagatcgacg…).
unc-5.Sgfp: unc-5.Sgfp was obtained by cloning a gfp PCR-amplified (Phusion, Finnzyme) fragment in-frame into the EcoRV site of unc-5.repS.
unc-5.SDgfp: unc-5.SDgfp was obtained in two steps: first, a gfp PCR-amplified (Phusion, Finnzyme) fragment was cloned in-frame into the BstBI site of unc-5.repS. Second, a 275-bp-long unc-5 genomic fragment (primers: gcgaggttaatgctagctgg and ggggtacccatcgcatgagaatccagg) was added between NheI and KpnI sites of this construct.
Transgene instructed gene repair experiments
Heat-shock treatments were performed on mixed populations of transgenic and non-transgenic animals from MosTIC lines for 1 h at 33°C, followed by 1 h at 15°C and 1 h at 33°C. Heat-shocked animals were allowed to recover overnight at 20°C and transgenic individuals were transferred as pools on fresh plates. When working with repair templates unc-63.rep, unc-5.repL, unc-5.repS or unc-5.repP, pools of heat-shocked animals were grown for 5 days at 20°C and their progeny were scored for wild-type moving animals. During scoring, counting was performed to estimate the size of the scored populations. When working with repair template unc-5.Ldel, unc-5.Sdel or unc-5.Sgfp, pools of heat-shocked animals were grown at 20°C for 1 week and PCR screens were performed on populations collected from starved plates. We estimated that approximately 100 F1 animals were generated from five heat-shocked P0 animals. Lysis was performed for 3 h at 65°C followed by 20 min at 95°C in 50 μl of buffer (50 mM KCl, 10 mM Tris pH 8.2, 2.5 mM MgCl2, 0.45% NP-40, 0.45% Tween 20, 0.01% gelatin) complemented with proteinase K (1 mg/ml). Lysates were diluted 10 or 100 times before performing PCR. MosTIC events possibly generated with repair templates unc-5.Ldel and unc-5.Sdel were scored using primers CGAATGGTCCCCGTGGATCG (P1 on Figure 3) and CCATACACTTTCCATTGCTG (P2) (2.5 mM MgCl2; annealing temperature: 55°C). MosTIC events possibly generated using unc-5.Sgfp were screened using primers AAAGGGCAGATTGTGTGGAC (P3) and TCAGATCCGAAAGCAGAGGT (P4) for the first PCR (2.5 mM MgCl2; annealing time: 15 s; annealing temperature: 68°C) and primers TCACCTTCACCCTCTCCACT (P5) and GCGGCACATTTCAAAAGAAT (P6) for the nested PCR (2.5 mM MgCl2; annealing time: 30 s; annealing temperature: 65°C).
Detection of end-joining events by PCR
Heat-shock treatments were performed for 1 h at 33°C, followed by 1 h at 15°C and 1 h at 33°C on wild-type or mutant young hermaphrodites containing a Mos transposase-expressing vector. Eighteen hours after heat-shock, lysis was performed on single animals for 1 h. Alternatively, heat-shock treatments were performed on L2 stage larvae which were lysed 9 h after heat shock. Primers AGTCATGTACCGTTCCACCTC and ATCGCAATGAAGTCCGCTATT were used to detect end-joining repair events. Subsequent TA cloning of the total PCR product was performed with the pGEM-T® Vector System I (Promega, Madison, WI) before sequencing.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
We are thankful to E Jorgensen, W Davis, R Weimer and T Boulin for providing reagents as well as to H Gendrot and S Vidal for technical support. Some of the strains used in this study were provided by the Caenorhabditis Genetic Center (which is funded by the NIH National Center for Research Resources (NCRR)), the C. elegans gene knockout consortium and the Japanese National BioResources Project. We are grateful to P Kuwabara, S Malinsky, S Marcand, B Lopez and E Meyer for critical reading of the manuscript. VR was supported by a European Union Grant (6th Framework program, PL-503334, code NemageneTAG). This work was funded by Nemagenetag and an AVENIR grant from the Institut National de la Santé et de la Recherche Médicale.
References
- Adams MD, McVey M, Sekelsky JJ (2003) Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science 299: 265–267 [DOI] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T (1998) Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol 8: 1395–1398 [DOI] [PubMed] [Google Scholar]
- Barrett PL, Fleming JT, Gobel V (2004) Targeted gene alteration in Caenorhabditis elegans by gene conversion. Nat Genet 36: 1231–1237 [DOI] [PubMed] [Google Scholar]
- Benjamin HW, Kleckner N (1992) Excision of Tn10 from the donor site during transposition occurs by flush double-strand cleavages at the transposon termini. Proc Natl Acad Sci USA 89: 4648–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Bargmann CI, Plasterk RH (2004) Homologous gene targeting in Caenorhabditis elegans by biolistic transformation. Nucleic Acids Res 32: e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessereau JL (2006) Transposon in C. elegans. In Wormbook, WormBook e (ed.) Vol. WormBook, doi:10.1895/wormbook.1.70.1, http://www.wormbook.org [Google Scholar]
- Bessereau JL, Wright A, Williams DC, Schuske K, Davis MW, Jorgensen EM (2001) Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature 413: 70–74 [DOI] [PubMed] [Google Scholar]
- Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D (2006) Efficient gene targeting in Drosophila with zinc finger nucleases. Genetics 12: 2391–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D (2003) Enhancing gene targeting with designed zinc finger nucleases. Science 300: 764. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Golic M, Golic KG, Carroll D (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161: 1169–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borts RH, Haber JE (1989) Length and distribution of meiotic gene conversion tracts and crossovers in Saccharomyces cerevisiae. Genetics 123: 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broverman S, MacMorris M, Blumenthal T (1993) Alteration of Caenorhabditis elegans gene expression by targeted transformation. Proc Natl Acad Sci USA 90: 4359–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan G, Garza D, Hartl D (1990) Insertion and excision of the transposable element mariner in Drosophila. Genetics 125: 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clejan I, Boerckel J, Ahmed S (2006) Developmental modulation of nonhomologous end joining in Caenorhabditis elegans. Genetics 173: 1301–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culetto E, Baylis HA, Richmond JE, Jones AK, Fleming JT, Squire MD, Lewis JA, Sattelle DB (2004) The Caenorhabditis elegans unc-63 gene encodes a levamisole-sensitive nicotinic acetylcholine receptor alpha subunit. J Biol Chem 279: 42476–42483 [DOI] [PubMed] [Google Scholar]
- Daley JM, Palmbos PL, Wu D, Wilson TE (2005) Nonhomologous end joining in yeast. Annu Rev Genet 39: 431–451 [DOI] [PubMed] [Google Scholar]
- Dawson A, Finnegan DJ (2003) Excision of the Drosophila mariner transposon Mos1. Comparison with bacterial transposition and V(D)J recombination. Mol Cell 11: 225–235 [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Zalevsky J, Colaiacovo MP, Villeneuve AM (2000) Transgene-mediated cosuppression in the C. elegans germ line. Genes Dev 14: 1578–1583 [PMC free article] [PubMed] [Google Scholar]
- Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, Thompson S, Smithies O (1987) Targeted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330: 576–578 [DOI] [PubMed] [Google Scholar]
- Dudasova Z, Dudas A, Chovanec M (2004) Non-homologous end-joining factors of Saccharomyces cerevisiae. FEMS Microbiol Rev 28: 581–601 [DOI] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M (1998) Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol 18: 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errami A, Smider V, Rathmell WK, He DM, Hendrickson EA, Zdzienicka MZ, Chu G (1996) Ku86 defines the genetic defect and restores X-ray resistance and V(D)J recombination to complementation group 5 hamster cell mutants. Mol Cell Biol 16: 1519–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann E, Schmiemann V, Goedecke W, Reichenberger S, Pfeiffer P (2000) DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res 28: 2585–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Wienholds E, Plasterk RH (2001) Regulated transposition of a fish transposon in the mouse germ line. Proc Natl Acad Sci USA 98: 6759–6764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5: 993–1002 [DOI] [PubMed] [Google Scholar]
- Gloor GB, Nassif NA, Johnson-Schlitz DM, Preston CR, Engels WR (1991) Targeted gene replacement in Drosophila via P element-induced gap repair. Science 253: 1110–1117 [DOI] [PubMed] [Google Scholar]
- Gorski MM, Eeken JC, de Jong AW, Klink I, Loos M, Romeijn RJ, van Veen BL, Mullenders LH, Ferro W, Pastink A (2003) The Drosophila melanogaster DNA Ligase IV gene plays a crucial role in the repair of radiation-induced DNA double-strand breaks and acts synergistically with Rad54. Genetics 165: 1929–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger L, Martin E, Segalat L (2004) Mos as a tool for genome-wide insertional mutagenesis in Caenorhabditis elegans: results of a pilot study. Nucleic Acids Res 32: e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Jin S, Gao Y, Weaver DT, Alt FW (1997) Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc Natl Acad Sci USA 94: 8076–8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS (2004) Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell 14: 611–623 [DOI] [PubMed] [Google Scholar]
- Haber JE (2000) Partners and pathways repairing a double-strand break. Trends Genet 16: 259–264 [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH (1990) The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron 4: 61–85 [DOI] [PubMed] [Google Scholar]
- Hefferin ML, Tomkinson AE (2005) Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair (Amst) 4: 639–648 [DOI] [PubMed] [Google Scholar]
- Izsvak Z, Stuwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z (2004) Healing the wounds inflicted by sleeping beauty transposition by double-strand break repair in mammalian somatic cells. Mol Cell 13: 279–290 [DOI] [PubMed] [Google Scholar]
- Jacobson JW, Hartl DL (1985) Coupled instability of two X-linked genes in Drosophila mauritiana: germinal and somatic mutability. Genetics 111: 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson JW, Medhora MM, Hartl DL (1986) Molecular structure of a somatically unstable transposable element in Drosophila. Proc Natl Acad Sci USA 83: 8684–8688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager U, Bocskor S, Le T, Mitterbauer G, Bolz I, Chott A, Kneba M, Mannhalter C, Nadel B (2000) Follicular lymphomas' BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood 95: 3520–3529 [PubMed] [Google Scholar]
- Jantsch V, Pasierbek P, Mueller MM, Schweizer D, Jantsch M, Loidl J (2004) Targeted gene knockout reveals a role in meiotic recombination for ZHP-3, a Zip3-related protein in Caenorhabditis elegans. Mol Cell Biol 24: 7998–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp TH, van Luenen HG, Plasterk RH (1999) Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell 99: 133–141 [DOI] [PubMed] [Google Scholar]
- Ketting RF, Plasterk RH (2000) A genetic link between co-suppression and RNA interference in C. elegans. Nature 404: 296–298 [DOI] [PubMed] [Google Scholar]
- Lampe DJ, Churchill ME, Robertson HM (1996) A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J 15: 5470–5479 [PMC free article] [PubMed] [Google Scholar]
- Leung-Hagesteijn C, Spence AM, Stern BD, Zhou Y, Su MW, Hedgecock EM, Culotti JG (1992) UNC-5, a transmembrane protein with immunoglobulin and thrombospondin type 1 domains, guides cell and pioneer axon migrations in C. elegans. Cell 71: 289–299 [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4: 712–720 [DOI] [PubMed] [Google Scholar]
- Liu S, Thaler DS, Libchaber A (2002) Signal and noise in bridging PCR. BMC Biotechnol 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JL, Kim EM, Haber JE, Lee SE (2003) Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol 23: 8820–8828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Radut D, Sekelsky JJ (2004) End-joining repair of double-strand breaks in Drosophila melanogaster is largely DNA ligase IV independent. Genetics 168: 2067–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassif N, Engels W (1993) DNA homology requirements for mitotic gap repair in Drosophila. Proc Natl Acad Sci USA 90: 1262–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassif N, Penney J, Pal S, Engels WR, Gloor GB (1994) Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol 14: 1613–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paabo S, Irwin DM, Wilson AC (1990) DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem 265: 4718–4721 [PubMed] [Google Scholar]
- Palmer S, Schildkraut E, Lazarin R, Nguyen J, Nickoloff JA (2003) Gene conversion tracts in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res 31: 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer P, Vielmetter W (1988) Joining of nonhomologous DNA double strand breaks in vitro. Nucleic Acids Res 16: 907–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RH (1991) The origin of footprints of the Tc1 transposon of Caenorhabditis elegans. EMBO J 10: 1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RH, Groenen JT (1992) Targeted alterations of the Caenorhabditis elegans genome by transgene instructed DNA double strand break repair following Tc1 excision. EMBO J 11: 287–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert VJ, Sijen T, van Wolfswinkel J, Plasterk RH (2005) Chromatin and RNAi factors protect the C. elegans germline against repetitive sequences. Genes Dev 19: 782–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeijn RJ, Gorski MM, van Schie MA, Noordermeer JN, Mullenders LH, Ferro W, Pastink A (2005) Lig4 and rad54 are required for repair of DNA double-strand breaks induced by P-element excision in Drosophila. Genetics 169: 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YS, Golic KG (2000) Gene targeting by homologous recombination in Drosophila. Science 288: 2013–2018 [DOI] [PubMed] [Google Scholar]
- Roth DB, Porter TN, Wilson JH (1985) Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol 5: 2599–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan KS, Emmons SW (1987) Precise and imprecise somatic excision of the transposon Tc1 in the nematode C. elegans. Nucleic Acids Res 15: 6875–6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer S, Davis RW (1979) Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci USA 76: 4951–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton BK, Priestley A, Steingrimsdottir H, Gell D, Blunt T, Jackson SP, Lehmann AR, Jeggo PA (1997) Molecular and biochemical characterization of xrs mutants defective in Ku80. Mol Cell Biol 17: 1264–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcomb DT, Shaw JE, Carr SH, Hirsh D (1985) Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol Cell Biol 5: 3484–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC (1999) The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 99: 123–132 [DOI] [PubMed] [Google Scholar]
- Taghian DG, Nickoloff JA (1997) Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol 17: 6386–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR (1987) Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51: 503–512 [DOI] [PubMed] [Google Scholar]
- van Luenen HG, Colloms SD, Plasterk RH (1994) The mechanism of transposition of Tc3 in C. elegans. Cell 79: 293–301 [DOI] [PubMed] [Google Scholar]
- Vastenhouw NL, Fischer SE, Robert VJ, Thijssen KL, Fraser AG, Kamath RS, Ahringer J, Plasterk RH (2003) A genome-wide screen identifies 27 genes involved in transposon silencing in C. elegans. Curr Biol 13: 1311–1316 [DOI] [PubMed] [Google Scholar]
- Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC (2002) Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur J Immunol 32: 701–709 [DOI] [PubMed] [Google Scholar]
- Welzel N, Le T, Marculescu R, Mitterbauer G, Chott A, Pott C, Kneba M, Du MQ, Kusec R, Drach J, Raderer M, Mannhalter C, Lechner K, Nadel B, Jaeger U (2001) Templated nucleotide addition and immunoglobulin JH-gene utilization in t(11;14) junctions: implications for the mechanism of translocation and the origin of mantle cell lymphoma. Cancer Res 61: 1629–1636 [PubMed] [Google Scholar]
- Williams DC, Boulin T, Ruaud AF, Jorgensen EM, Bessereau JL (2005) Characterization of Mos1-mediated mutagenesis in Caenorhabditis elegans: a method for the rapid identification of mutated genes. Genetics 169: 1779–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, Kay MA (2003) Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells. Mol Cell Biol 23: 8505–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaal RR, Broeks A, van Meurs J, Groenen JT, Plasterk RH (1993) Target-selected gene inactivation in Caenorhabditis elegans by using a frozen transposon insertion mutant bank. Proc Natl Acad Sci USA 90: 7431–7435 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2