

Abstract
EpsE is a cytoplasmic component of the type II secretion system in Vibrio cholerae. Through ATP hydrolysis and an interaction with the cytoplasmic membrane protein EpsL, EpsE supports secretion of cholera toxin across the outer membrane. In this study, we have determined the effect of the cytoplasmic domain of EpsL (cyto-EpsL) and purified phospholipids on the ATPase activity of EpsE. Acidic phospholipids, specifically cardiolipin, bound the copurified EpsE/cyto-EpsL complex and stimulated its ATPase activity 30–130-fold, whereas the activity of EpsE alone was unaffected. Removal of the last 11 residues (residues 243–253) from cyto-EpsL prevented cardiolipin binding as well as stimulation of the ATPase activity of EpsE. Further mutagenesis of the C-terminal region of the EpsL cytoplasmic domain adjacent to the predicted transmembrane helix suggested that this region participates in fine tuning the interaction of EpsE with the cytoplasmic membrane and influences the oligomerization state of EpsE thereby stimulating its ATPase activity and promoting extracellular secretion in V. cholerae.
Keywords: acidic phospholipids, ATPase, protein secretion
Introduction
The type II secretion (T2S) system exports toxins and degradative enzymes across the outer membrane of Gram-negative bacteria (Filloux, 2004; Cianciotto, 2005; Johnson et al, 2006). Components of this system are proposed to form a large multiprotein complex spanning the entire cell envelope. This complex is comprised of 15 gene products, 12 of which are required for translocation of specific substrates, including cholera toxin and hemagglutinin protease, across the outer membrane in Vibrio cholerae (Sandkvist et al, 1997). This system contains pseudopilins (EpsG,-H,-I, -J, and -K) that may form a pilus to extrude substrates into the extracellular space via a pore in the outer membrane (EpsD) using a mechanism analogous to a piston (Sandkvist, 2001; Filloux, 2004). Energy likely comes from EpsE, a cytoplasmic ATPase required for secretion in V. cholerae (Sandkvist et al, 1995; Camberg and Sandkvist, 2005). Based on its structural similarity with the type IV secretion ATPase HP0525 from Helicobacter pylori, EpsE has been proposed to function as a hexameric assembly at the cytoplasmic face of the inner membrane, even though it predominantly purifies as a monomeric protein (Sandkvist et al, 1995; Robien et al, 2003; Savvides et al, 2003; Camberg and Sandkvist, 2005). A recent study demonstrated that the EpsE homolog XpsE from Xanthomonas campestris also purifies as a monomer, but oligomerizes in the presence of the ATP analog AMP-PNP (Shiue et al, 2006). Communication between EpsE and the secretion complex likely occurs through the bitopic inner membrane protein EpsL (Sandkvist et al, 1995; Ball et al, 1999; Possot et al, 2000; Py et al, 2001). The cytoplasmic domain of EpsL is structurally similar to members of the actin family, such as FtsA (Abendroth et al, 2004), and has been cocrystallized with the 96 N-terminal residues of EpsE (Abendroth et al, 2005). Additionally, EpsL binds to another component in the inner membrane, EpsM, which also has a single membrane-spanning domain (Michel et al, 1998; Sandkvist et al, 1999). EpsE, -L, and M form a trimolecular complex at the cytoplasmic membrane and may serve as a base for the secretion apparatus to transduce energy across the periplasmic compartment through protein contacts or participate in assembly or functional regulation of the pseudopilus.
Previously we characterized EpsE as a weak ATPase comparable to those of related ATPases, such as XpsE from X. campestris, PilT from the type IV pilus system, and PilQ from the thin pilus biogenesis system of plasmid RP4 (Sakai et al, 2001; Herdendorf et al, 2002; Camberg and Sandkvist, 2005; Shiue et al, 2006). Surprisingly, EpsE(K270A), which contains a mutation in the conserved lysine residue of the nucleotide-binding motif, retained approximately 30% of the wild-type activity leading us to suggest that conditions may exist that result in oligomerization and/or accentuate the difference in activity between wild-type and mutant proteins (Camberg and Sandkvist, 2005).
Here, we have copurified EpsE with the cytoplasmic domain of EpsL (cyto-EpsL) to test whether the ATPase activity of EpsE can be modulated by EpsL. A similar recent investigation conducted with XpsE demonstrated that its weak ATPase activity was stimulated two-fold by a fusion protein containing the cytoplasmic domain of XpsL (Shiue et al, 2006). In addition to examining the role of cyto-EpsL on the ATP-hydrolyzing activity of EpsE, we have also assessed the role of membrane phospholipids on this activity and found that the acidic phospholipids phosphatidylglycerol and cardiolipin, together with cyto-EpsL, synergistically stimulated both the oligomerization and intrinsic ATPase activity of EpsE. Deletion and modification of residues at the C-terminus of cyto-EpsL interfered with the cardiolipin-stimulated ATPase activity of EpsE. The results suggest that the membrane proximal region of the cytoplasmic domain of EpsL may participate in fine tuning the interaction of EpsE with phospholipids and thereby regulate its oligomerization and ATPase activity.
Results
Stimulation of EpsE ATPase activity by acidic phospholipids and the cytoplasmic domain of EpsL
We previously demonstrated that monomeric EpsE displayed weak ATPase activity and proposed that conditions may exist which result in oligomerization and/or stimulate this basal rate of ATP hydrolysis, such as the addition of cofactor proteins or membrane phospholipids (Camberg and Sandkvist, 2005). We therefore examined the rate of ATP hydrolysis both in the presence of purified membrane phospholipids and the cytoplasmic domain of EpsL. Two forms of the EpsL cytoplasmic domain, EpsL(1–253) and EpsL(1–242), were histidine-tagged, coexpressed, and copurified with full-length EpsE. EpsL(1–253) was truncated immediately before the predicted transmembrane domain, which constitutes residues 254–271. Initially constructed for crystallization purposes (Abendroth et al, 2004), EpsL(1–242) was lacking an additional 11 C-terminal residues but included the relatively well-conserved residues L233 to F238. Residues up to and including K241 and S242 were also included in this EpsL construct in order to end with a hydrophilic C-terminus and to decrease the probability of protein aggregation via solvent-exposed hydrophobic patches. Both EpsE/EpsL(1–253) and EpsE/EpsL(1–242) protein complexes were monomeric and consisted of one molecule each of EpsE and truncated EpsL as indicated by size exclusion chromatography (data not shown). Copurification of EpsE with EpsL(1–253) yielded a modest two-fold increase in basal ATPase activity when compared with monomeric EpsE alone (Figure 1A). These results are in close agreement with a recent report which also described a two-fold increase in the ATPase activity of the EpsE homolog XpsE in the presence of a fusion protein containing the N-terminal 215 residues of XpsL (Shiue et al, 2006). In contrast, the shorter variant, EpsL(1–242), conferred no increase in ATPase activity. The EpsE active site mutant EpsE(K270A), which has three-fold lower ATPase activity than wild-type EpsE (Camberg and Sandkvist, 2005), displayed no enhancement in activity upon copurification with EpsL(1–253).
Figure 1.
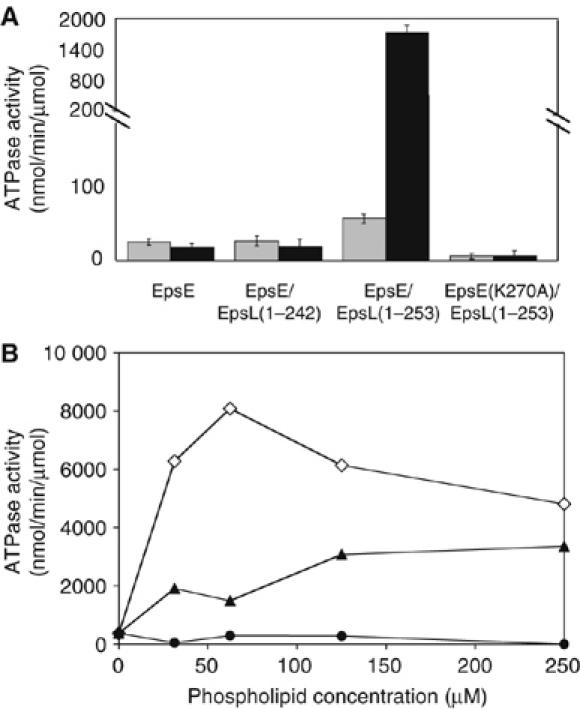
Acidic phospholipids stimulate the ATPase activity of EpsE in the presence of cyto-EpsL (residues 1–253). (A) EpsE ATP hydrolysis was examined in the absence or presence of cyto-EpsL (residues 1–242 or 1–253), phosphatidylglycerol, ATP, and MgCl2. Gray bars represent the ATPase activity for the no-phospholipid reactions, black bars correspond to the ATPase activity for the reactions containing phosphatidylglycerol. The mutant EpsE(K270A) copurified with EpsL(1–253) was also analyzed in this assay. (B) ATP hydrolysis by EpsE/EpsL(1–253) was tested in the presence of increasing concentrations (31.25, 62.5, 125, and 250 μM) of cardiolipin (open diamonds), phosphatidylglycerol (solid triangles), and phosphatidylethanolamine (solid circles) in the presence of ATP and MgCl2.
To test the effect of membrane phospholipids on the rate of ATP hydrolysis, we added purified Escherichia coli-derived phosphatidylglycerol to the ATP hydrolysis assay. Phosphatidylglycerol was chosen because it, as well as other acidic phospholipids, has been shown to activate ATPases such as SecA and FliI (Lill et al, 1990; Auvray et al, 2002). While no stimulatory effect with phosphatidylglycerol was observed for wild-type EpsE alone or for EpsE/EpsL(1–242) complex, a 30-fold increase in specific activity was observed for EpsE/EpsL(1–253) complex when compared to the no phospholipid reaction (Figure 1A) (1831.5 nmol/min/μmol and 60.5 nmol/min/μmol, respectively). As predicted, the K270A mutation in EpsE prevented stimulation of ATP hydrolysis by phosphatidylglycerol in the presence of EpsL(1–253).
Over a range of concentrations, phosphatidylglycerol stimulated the activity of EpsE/EpsL(1–253) and appeared to level off at a concentration of 125 μM (Figure 1B). Cardiolipin, another negatively charged phospholipid, displayed a clear maximal activation of the hydrolysis reaction at 62.5 μM, which reached 8075 nmol/min/μmol, a greater than 130-fold increase over the no phospholipid reaction. In contrast to phosphatidylglycerol and cardiolipin, the neutral phospholipid phosphatidylethanolamine did not stimulate the ATPase activity of EpsE/EpsL(1–253) (Figure 1B).
Activation occurs through direct binding of EpsE/EpsL(1–253) complex to acidic phospholipids
Since the activity of EpsE/EpsL(1–253) was stimulated by acidic phospholipids, we questioned whether EpsE/EpsL(1–253) was directly binding to phospholipids. To test this, we incubated purified EpsE/EpsL(1–253), EpsE/EpsL(1–242), and EpsE with individual phospholipids in the presence of ATP and MgCl2, then subjected the samples to centrifugation to pellet the phospholipids and phospholipid-bound protein and analyzed supernatants and pellets by SDS–PAGE. In the presence of either phosphatidylglycerol or cardiolipin, EpsE/EpsL(1–253) fractionated in the pellet with the phospholipids, whereas it remained free in the supernatant in either the absence of phospholipids or in the presence of the neutral phosphatidylethanolamine (Figure 2A). This suggests that acidic phospholipids are specifically binding to EpsE/EpsL(1–253). While greater than 92% of EpsE/EpsL(1–253) pelleted with the acidic phospholipid cardiolipin, EpsE/EpsL(1–242) and EpsE alone demonstrated a considerably smaller proportion of protein in the cardiolipin pellet (40 and 52%, respectively). Neither EpsE nor EpsE/EpsL(1–242) bound to phosphatidylethanolamine (data not shown).
Figure 2.
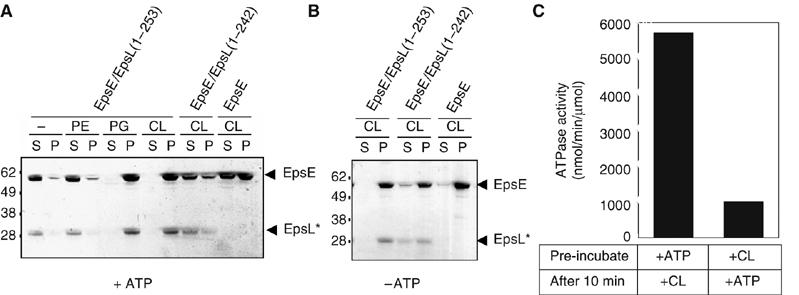
EpsE binds to acidic phospholipids. (A) Reactions containing EpsE alone or EpsE copurified with either EpsL (1–253) or EpsL(1–242) were incubated with individual phospholipids (phosphatidylethanolamine (PE), phosphatidylglycerol (PG), or cardiolipin (CL)) in the presence of ATP and MgCl2, then centrifuged at 100 000 g to pellet phospholipids and phospholipid-bound protein. Supernatants and pellets were analyzed by SDS–PAGE. (B) Reactions containing EpsE with and without cyto-EpsL (residues 1–242 or 1–253) were incubated with cardiolipin in the absence of nucleotide and centrifuged. Supernatants and pellets were analyzed as described. EpsL* marks the position of cyto-EpsL, either EpsL(1–253) or EpsL(1–242) where indicated. The numbers on the left indicate the position of molecular mass markers (in kDa). (C) ATP hydrolysis was tested following pre-incubation of EpsE/EpsL(1–253) with cardiolipin or ATP and MgCl2. After 10 min, the remaining components were added and the reactions were assayed for ATP hydrolysis.
Interestingly, when EpsE and EpsE/EpsL(1–242) were incubated with cardiolipin in the absence of ATP, binding of both EpsE forms was seen (Figure 2B). This was in contrast to the EpsE/EpsL(1–253), which fully bound to cardiolipin either with or without ATP, and suggests that EpsE undergoes a conformational change in response to ATP binding whether it is alone or in complex with EpsL(1–242). We were also able to demonstrate that EpsE in complex with EpsL(1–253) likewise appears to undergo a conformational change in response to ATP in a subsequent experiment by altering the order of addition of ATP and cardiolipin in the ATP hydrolysis assay. We found that pre-incubation of EpsE/EpsL(1–253) with cardiolipin before addition of ATP abolished the synergistic stimulation of ATP hydrolysis, whereas addition of ATP before cardiolipin retained the stimulated rate of ATP hydrolysis (Figure 2C), consistent with the activity described in Figure 1B. In addition to showing that EpsE in complex with EpsL(1–253) responds differently to cardiolipin in the absence and presence of nucleotide, these data also suggest that EpsE/EpsL(1–253) must be in a nucleotide-bound form in order to bind to phospholipids in a conformation compatible with stimulated hydrolysis.
To further detect conformational differences between EpsE/EpsL(1–253) and EpsE/EpsL(1–242) complexes in the presence of ATP and cardiolipin, we compared the protease sensitivity patterns of both protein complexes by SDS–PAGE and immunoblotting following limited proteolysis with trypsin (Figure 3). EpsE/EpsL(1–253) and EpsE/EpsL(1–242) were incubated with cardiolipin and ATP, digested with trypsin, then centrifuged to separate phospholipid-bound protein from non-bound protein. The majority of EpsE/EpsL(1–253) localized to the cardiolipin pellet supporting the result described in Figure 2A. According to SDS–PAGE, treatment with increasing amounts of trypsin degraded and released EpsE from the phospholipid vesicles, whereas EpsL(1–253) remained phospholipid bound and was considerably resistant to proteolysis. As full-length EpsE was becoming less visible in the pellet fractions, a 50 kDa band accumulated in the supernatant fractions that was reactive to anti-EpsE antibody. Although most EpsL(1–253) was pellet-associated, the small fraction in the supernatant was sensitive to proteolysis at high trypsin concentrations and cleaved into a slightly smaller form. The majority of EpsE/EpsL(1–242) was not localized to the phospholipid pellet as previously shown in Figure 2A. Immunoblotting for EpsE showed both the full-length protein and the initial cleavage product (50 kDa) in the supernatant fractions. Similarly, anti-EpsL antibodies detected EpsL(1–242) in the supernatant at all trypsin concentrations, and a slightly smaller cleavage product accumulated as the trypsin concentration increased. Although similar-sized proteolytic fragments were detected for both EpsE/cyto-EpsL complexes, the dissimilarity in accumulation of individual fragments and their phospholipid associations suggest that there are substantial differences in conformation between EpsL(1–253) and EpsL(1–242) in the presence of cardiolipin, which, in turn, affects the interaction of EpsE with phospholipids and its ability to hydrolyze ATP.
Figure 3.
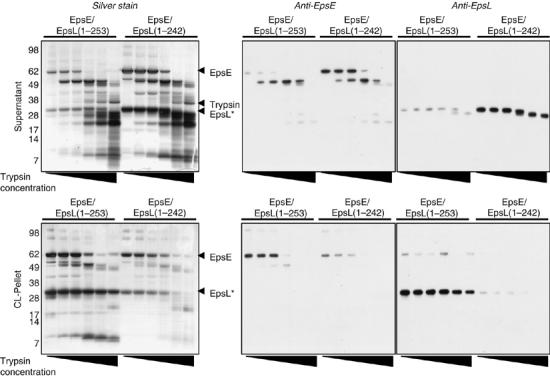
Trypsin proteolysis of EpsE/EpsL(1–253) and EpsE/EpsL(1–242) in the presence of cardiolipin and ATP. Reactions containing either EpsE/EpsL(1–253) or EpsE/EpsL(1–242), ATP, MgCl2, and cardiolipin were incubated at 37°C for 15 min, then digested with different concentrations of trypsin (0, 3, 10, 30, 100, and 300 μg/ml). After adding protease inhibitors to stop digestion, reactions were centrifuged to pellet phospholipids and phospholipid-bound protein. Supernatants and pellets were analyzed by SDS–PAGE and silver staining or immunoblotting using antibodies to EpsE and EpsL. The numbers on the left indicate the position of molecular mass markers (in kDa). EpsL* marks the position of cyto-EpsL, either EpsL(1–253) or EpsL(1–242) where indicated.
Copurification of EpsE and the cytoplasmic domain of EpsL is required for stimulation of ATPase activity
EpsL(1–253) remained associated with cardiolipin (Figure 3) under conditions in which EpsE was degraded, suggesting that the interaction between EpsL(1–253) and phospholipids can be maintained in the absence of the intact ATPase component EpsE. To test this possibility, the binding of purified EpsL(1–242) and EpsL(1–253) in the absence of EpsE was tested in a cardiolipin pelleting assay and examined by SDS–PAGE. As demonstrated in Figure 4A, both proteins remained predominantly soluble without cardiolipin, but became pellet-associated when cardiolipin was added to the reaction. This result indicates that they are both capable of binding to phospholipids, consistent with the results described for the EpsE complexed forms of EpsL(1–242) and EpsL(1–253) in the absence of ATP (Figure 2B). The binding properties of the cyto-EpsL proteins, however, differ from those of the EpsE/cyto-EpsL complexes when the latter complexes are incubated with ATP (Figure 2A). This, therefore, suggests that not only does cyto-EpsL affect the interaction of EpsE with phospholipids, but reciprocally the phospholipid binding properties of cyto-EpsL are influenced by the nucleotide-bound form of EpsE.
Figure 4.
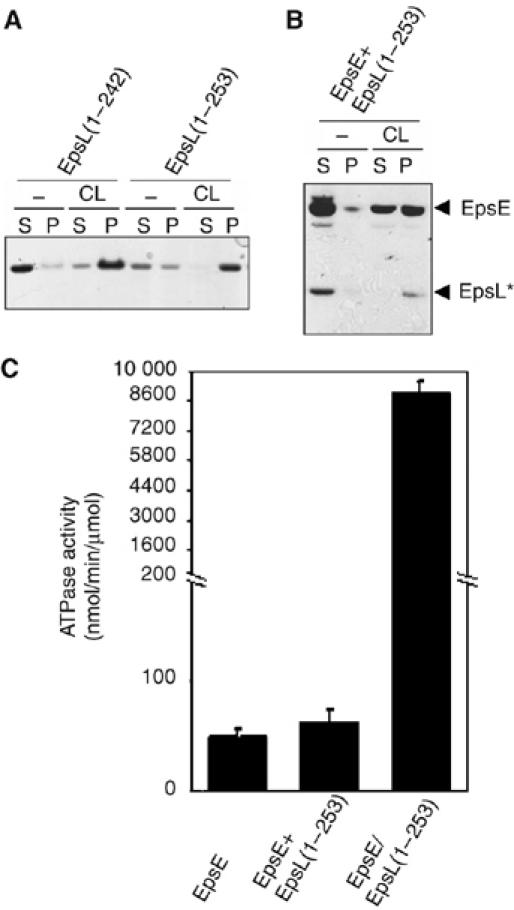
Differential phospholipid binding and activation of EpsE ATP hydrolysis for mixed and copurified EpsE and EpsL(1–253). (A) EpsL(1–242) and EpsL(1–253) were incubated with cardiolipin in the presence of ATP and MgCl2 followed by centrifugation to pellet cardiolipin and cardiolipin-bound protein and analyzed by SDS–PAGE. (B) Individually purified EpsE and EpsL(1–253) were mixed and incubated in the presence of ATP and MgCl2 with and without cardiolipin and subjected to centrifugation and analysis for cardiolipin binding as in (A). (C) Cardiolipin-stimulated ATP hydrolysis was measured for individually purified, mixed, and copurified EpsE and EpsL(1–253) in reactions containing ATP, MgCl2, and cardiolipin. EpsL* marks the position of EpsL(1–253).
We next asked whether we could add purified EpsL(1–253) to EpsE and replicate the same pelleting behavior in the presence of ATP as for the copurified EpsE/EpsL(1–253) complex. Without phospholipids, both EpsE and EpsL(1–253) were soluble, but when cardiolipin was added, EpsL(1–253) became associated with the phospholipid pellet, whereas only 55% of EpsE was localized to the pellet (Figure 4B). This proportion of pellet associated EpsE is similar to the 52% detected for EpsE alone (Figure 2A), suggesting that the two Eps proteins did not form a complex when they were purified separately.
As the addition of EpsL(1–253) did not promote further cardiolipin binding of EpsE, we hypothesized that the addition of EpsL(1–253) would likewise not stimulate the ATPase activity of EpsE in the presence of cardiolipin. The chart in Figure 4C demonstrates that only the copurified EpsE/EpsL(1–253) complex can be stimulated by cardiolipin. As expected, the addition of exogenously purified EpsL(1–253) to EpsE did not significantly change the rate of ATP hydrolysis. Taken together, these and earlier data suggest that EpsE needs to be co-produced with the intact cytoplasmic domain of EpsL in order to fold into a conformation that is responsive to structural changes in the presence of acidic phospholipids and that results in stimulation of ATPase activity.
Oligomerization of EpsE in the presence of cardiolipin and EpsL(1–253)
One possible mechanism for the increased ATPase activity of EpsE in the presence of EpsL(1–253) and acidic phospholipids may involve oligomerization as EpsE has previously been shown to form oligomers, although very inefficiently (Camberg and Sandkvist, 2005). We subjected EpsE/EpsL(1–253) to the crosslinker dithiobis succinimidyl propionate (DSP) at increasing concentrations in the presence of cardiolipin and ATP and visualized the presence of crosslinked products by SDS–PAGE and immunoblot analysis with antibodies to either EpsE or EpsL (Figure 5). The crosslinked protein profile was compared with that of the nonstimulated EpsE/EpsL(1–242) complex. While EpsE/EpsL(1–253) crosslinked into larger molecular species recognized by both anti-EpsE and anti-EpsL antibodies in a concentration-dependent manner, the amount of crosslinked products did not appreciably increase for EpsE/EpsL(1–242) as the crosslinker concentration increased.
Figure 5.
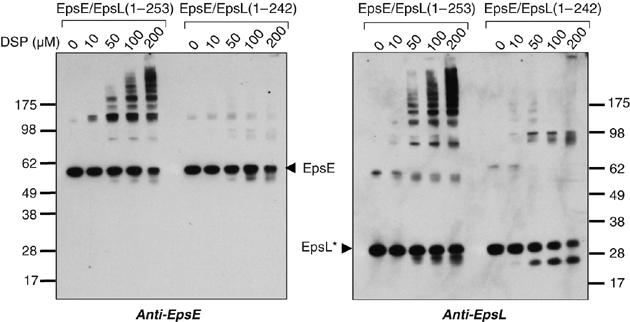
Oligomerization of EpsE/EpsL(1–253) in the presence of cardiolipin and ATP. Reactions containing EpsE/EpsL(1–253) or EpsE/EpsL(1–242) with cardiolipin, ATP and MgCl2 were crosslinked with the addition of increasing concentrations of DSP (0, 10, 50, 100, and 200 μM). Reactions were analyzed by SDS–PAGE and immunoblotting with antibodies to EpsE or EpsL. The positions of monomeric forms of EpsE and cyto-EpsL, as well as molecular mass markers are indicated. EpsL* marks the position of cyto-EpsL, either EpsL(1–253) or EpsL(1–242) where indicated.
Modification of the carboxy-terminal end of the EpsL cytoplasmic domain interferes with cardiolipin-stimulated activation of EpsE
Close examination of the residues at the C-terminus of EpsL(1–253) reveals the presence of hydrophobic and positively charged residues at regular intervals. The presence of hydrophobic and basic residues is also apparent in the region proximal to the transmembrane helix in an alignment comprised of EpsL homologs reported by Abendroth et al (2004). The enrichment of these residues and the noticeable lack of acidic amino acids may support the suggestion that this region is directly responsible for interaction with acidic phospholipids.
To test the requirement for hydrophobic and positively charged residues in EpsL for their functioning in vivo, we expressed different forms of full-length EpsL containing amino-acid substitutions within the last 15 residues of its cytoplasmic domain (Figure 6A). We tested for ability of the EpsL mutant proteins to restore secretion of protease in an epsL knockout mutant strain using a fluorescence-based assay (Figure 6B). While expression of plasmid-encoded wild-type epsL and several mutant epsL genes in the epsL knockout mutant restored secretion of protease, expression of epsL with K239A, K241A, W244E, and L245E substitutions did not (Figure 6B). It is possible that these latter mutations interfered with the phospholipid interaction, and as a consequence the mutant EpsL proteins were made sensitive to proteolysis as their steady-state level was reduced (Figure 6C). The material that escaped proteolysis, however, was found in the membrane pellet following subcellular fractionation (not shown), indicating that membrane insertion of these mutant proteins via their transmembrane domains was not affected.
Figure 6.
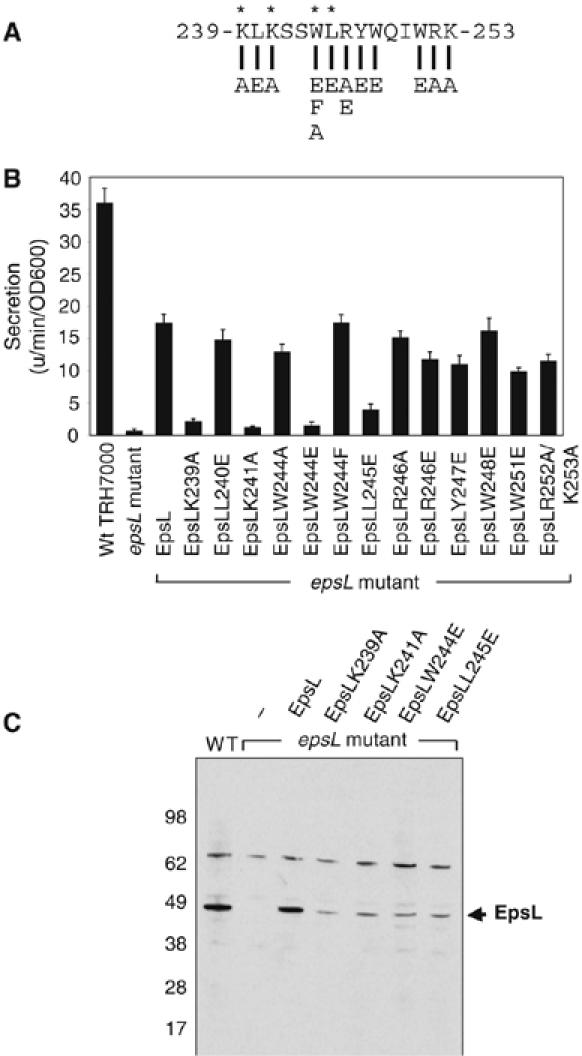
Mutations in the cytoplasmic domain of EpsL disrupt its normal function. (A) Sequence containing the C-terminal portion of the EpsL cytoplasmic domain depicts substitution mutations tested in vivo. Asterisks indicate residues that affect function of EpsL when mutated. (B) V. cholerae wild-type TRH7000 and epsL mutant strain mut 8 expressing either wild-type or mutant EpsL proteins were grown in LB at 37°C overnight. Supernatants were separated from cells and tested for the presence of extracellular protease using the proteolytic substrate N-tert-butoxy-carbonyl-Gln-Ala-Arg-7-amido-4-methyl-coumarin and compared with wild-type cells. (C) Wild-type as well as epsL mutant V. cholerae strains expressing either wild-type or mutant EpsL proteins were grown as described above. Cells were pelleted, resuspended in SDS-sample buffer, and analyzed by SDS–PAGE and immunoblotting using antibodies to EpsL. Arrow indicates the position of EpsL.
To determine the role of specific residues at the C-terminal end of the EpsL cytoplasmic domain in stimulation of EpsE ATP hydrolysis, the four mutations that prevented the function of EpsL in vivo were introduced into EpsE/EpsL(1–253). Like the EpsE/EpsL(1–253) complex, each mutant protein purified as a monomeric complex (not shown). When analyzed in the presence of cardiolipin, each mutant protein was found to display a reduced rate of ATP hydrolysis compared to the nonmutated EpsE/EpsL(1–253) (Figure 7A). The ATPase activity for the most drastically affected mutant, W244E, was reduced by 80%, whereas the other mutations lowered the activity by 20–29%. When the EpsE/EpsLW244E(1–253) complex was incubated with cardiolipin in the presence of ATP, it pelleted slightly less efficiently than EpsE/EpsL(1–253) (Figure 7B). The amount of EpsE/EpsL W244E(1–253) in the cardiolipin pellet was reduced by 12%.
Figure 7.
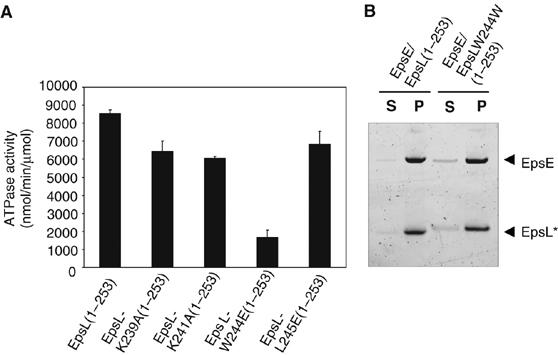
EpsE, copurified with mutant EpsL(1–253) proteins, displays reduced rates of cardiolipin-stimulated ATP hydrolysis. (A) EpsE, copurified with either wild-type or mutant forms of EpsL(1–253), were assayed for ATP hydrolysis in the presence of cardiolipin, ATP, and MgCl2. (B) Cardiolipin association of EpsE/EpsLW244E(1–253) was compared with EpsE/EpsL(1–253) in a phospholipid pelleting assay containing ATP, MgCl2, and cardiolipin. Following centrifugation, supernatants and pellets were analyzed by SDS–PAGE. EpsL* marks the position of cyto-EpsL, either EpsL(1–253) or EpsL-W244E(1–253).
Discussion
In this study we report the synergistic stimulation of EpsE ATP hydrolysis by acidic phospholipids and the cytoplasmic domain of EpsL (residues 1–253). While phosphatidylglycerol and cardiolipin had no stimulatory effect on the enzymatic activity of EpsE alone, they increased the specific activity of a protein complex composed of EpsE and EpsL(1–253) by 30- and 130-fold, respectively. In previous studies of EpsE, we speculated that the active form of EpsE is hexameric (Robien et al, 2003; Camberg and Sandkvist, 2005). Here we show by limited trypsin digestion and chemical crosslinking that EpsE in complex with EpsL(1–253) undergoes conformational changes and oligomerizes in the presence of acidic phospholipids; however, it is unknown whether the acidic phospholipids specifically induce hexamerization. These findings, along with the earlier discovery that EpsE is detected in both the cytoplasm and inner membrane following subcellular fractionation of V. cholerae cells (Sandkvist et al, 1995), suggest that EpsE may transition between two conformational states, possibly a monomer with minimal catalytic activity and an oligomer with increased ability to hydrolyze ATP. The conversion between these two states may be sensitive to the occupancy of the nucleotide-binding site, and may be directed via the interaction between the cytoplasmic domain of EpsL and the N-terminal domain of EpsE (Sandkvist et al, 1995; Abendroth et al, 2005).
Acidic phospholipids such as cardiolipin are essential for function of many membrane-spanning proteins that participate in ATP synthesis, energy transduction, and protein transport in both mitochondrial and bacterial membranes (Mileykovskaya and Dowhan, 2005), and have been suggested to promote assembly of protein complexes into larger supercomplexes. Via their negatively charged surface, they may adsorb and concentrate soluble proteins such as SecA and other ATPases onto membrane surfaces (Lill et al, 1990). This may, in fact, be a driving mechanism to localize ATP-hydrolyzing enzymes to sites where ATP is generated. There are also suggestions that acidic phospholipids may not be uniformly distributed but are rather concentrated at distinct sites within the cytoplasmic membrane (Mileykovskaya and Dowhan, 2000; Kawai et al, 2004). As a consequence, proteins with affinity for these phospholipids may also exhibit similar localization patterns. Recently, it was suggested that a cluster of positively charged residues on the surface of the mechanosensitive channel protein MscL that is close to one of the transmembrane helices produces a ‘hot spot' for anionic phospholipid binding (Powl et al, 2005). Inspection of the sequence that precedes the transmembrane helix of EpsL and the corresponding regions in EpsL homologs (see alignment in Abendroth et al, 2004) reveals a conserved enrichment of positively charged and hydrophobic residues, which may similarly form a hot spot for binding to acidic phospholipids. Replacement of two of these charged residues, K239 and K241, to alanine in EpsL supports this suggestion as it resulted in nonfunctional mutant proteins. When expressed in the V. cholerae epsL mutant strain, neither EpsL-K239A nor EpsL-K241A could restore extracellular secretion, and degradation products accumulated, which were consistent with the size of the cytoplasmic domain (Figure 6). Disruption of normal positioning at the membrane due to these substitutions may have exposed protease-sensitive sites. Two lines of evidence suggest that these lysine residues may be required, but are not sufficient for interaction with acidic phospholipids and stimulation of EpsE ATP hydrolysis. First, both K239 and K241 are present in the EpsE/EpsL(1–242) complex that could not be activated by phospholipids in the ATPase assay. Second, when the K239A and K241A mutations were individually introduced into the EpsE/EpsL(1–253) complex, they only reduced the phospholipid-stimulated ATPase activity of the purified proteins by 25 and 29%, respectively (Figure 7). It is likely, therefore, that the lysine residues act in concert with other residues in the C-terminal region of the cytoplasmic domain of EpsL to bind to phospholipids and stimulate the ATPase activity of EpsE. This is supported by two additional mutant proteins, EpsL-W244E and EpsL-L245E, which were also unable to complement the secretion defect in the epsL mutant strain and which were likewise sensitive to proteolysis, suggesting that hydrophobic residues are also important for the correct localization of the cytoplasmic domain with respect to the membrane. When the W244E and L245A mutations were introduced into EpsL(1–253) and the resulting mutant EpsL(1–253) proteins were copurified with EpsE, they were also found to exhibit reduced ATPase activity in the presence of acidic phospholipids. The W244E mutant was most severely affected displaying an 80% reduced activity compared with the nonmutated form, whereas the activity of the L245E mutant was reduced by 20% (Figure 7). Although a negatively charged residue was not tolerated at residue position 244, we found that there is not an absolute requirement for tryptophan in the full-length EpsL. Replacement of W244 with either phenylalanine or alanine produces a functional protein and suggests that a hydrophobic residue may be sufficient at this position in EpsL to support protein secretion in V. cholerae.
As EpsE and XpsE display similar ATPase activities (60 versus 20 nmol/min/μmol) and are equally two-fold stimulated by the cytoplasmic domains of EpsL and XpsL, respectively, it is likely that the activity of XpsE can be further stimulated by phospholipids via a mechanism that may be common to all T2S ATPases. The requirement for both positively charged and hydrophobic residues suggests that electrostatic as well as hydrophobic interactions between the C-terminal end of the cytoplasmic domain of EpsL and the phospholipid bilayer are a prerequisite for stimulation of EpsE's ATPase activity. We speculate that under normal circumstances in vivo, once ATP binds to the EpsE–EpsL complex, hydrophobic residues in the C-terminal end of the cytoplasmic domain of EpsL may embed into the lipid bilayer, while positively charged residues interact with the negatively charged phospholipids on the surface of the membrane. This may, in turn, bring ATP-bound EpsE within close proximity of the membrane where it may oligomerize and become active. Alternatively, the positively charged residues may interact electrostatically with the negatively charged phospholipid surface, while residues at positions 244 and 245 are in hydrophobic contact with a hydrophobic patch on EpsL or ATP-bound EpsE, thus inducing a conformational change in EpsE that mediates oligomerization and stimulation of ATP hydrolysis. A recent model for T2S in X. campestris suggests that nucleotide binding induces oligomerization of XpsE and precedes binding to membrane-bound XpsL (Shiue et al, 2006). In our system, occupancy of the nucleotide-binding domain in EpsE is not a requirement for complex formation with EpsL, however stimulation of ATP hydrolysis by cardiolipin requires pre-loading of the EpsE/EpsL(1–253) complex with ATP. A key requirement for cardiolipin stimulation is the membrane-proximal region of EpsL that responds to the nucleotide occupancy state of EpsE. This region, which maintains a peripheral interaction between the EpsE/cytoEpsL complex and acidic phospholipids, may illustrate a novel mechanism to control the activity of a protein or protein complex that is already inserted in a membrane. While the transmembrane helix is required for insertion and maintenance of EpsL in the cytoplasmic membrane, the hydrophobic and positively charged residues in the cytoplasmic domain may fine tune its interaction with the cytoplasmic membrane, thus activating EpsE, the energy-providing component of the T2S apparatus.
Materials and methods
Cloning and expression
Plasmids pEpsE/EpsL(1–253)His6, pEpsE/EpsL(1–242)His6, pEpsL(1–253)His6,and pEpsL(1–242)His6 containing the region of epsL that encodes the cytoplasmic domain (residues 1–242 or residues 1–253) were constructed with and without epsE using the expression vector pET21d(+) (Abendroth et al, 2004, 2005) (Supplementary Figure 1). A six-histidine tag was attached to the carboxy-terminus of the EpsL fragments. A fragment of epsE containing a lysine to alanine mutation in the Walker A motif (Camberg and Sandkvist, 2005) was cloned into pEpsE/EpsL(1–253)His6 by exchange of an NotI/BamHI fragment to create pEpsE(K270A)/EpsL(1–253)His6. pEpsE/EpsL(1–253)His6 constructs with mutations (K239A, K241A, W244E, and L245E) in EpsL(1–253) were also made. The epsL fragments with and without epsE were coexpressed in E. coli BL21(DE3) cells and induced by the addition of IPTG as described (Abendroth et al, 2005). EpsL proteins and EpsE/EpsL complexes were purified using metal affinity chromatography and gel filtration (Abendroth et al, 2004, 2005). Monomeric EpsE was obtained following thrombin cleavage of a GST-EpsE fusion protein expressed from pGST-EpsE, leaving a glycine–serine overhang at the amino-terminus (Camberg and Sandkvist, 2005).
For in vivo studies, substitution mutations in full-length epsL, K239A, L240E, K241A, W244E, W244F, W244A, L245E, R246A, R246E, Y247E, W248E, W251E, and R252A/K253A were created by site-directed mutagenesis for expression in the V. cholerae epsL mutant strain mut 8 (Sandkvist et al, 1997). For in vitro studies, EpsE/EpsL(1–253) with either K239A, K241A, W244E, or L245E substitutions were constructed, expressed in E. coli and purified as described (Abendroth et al, 2005).
Phospholipids
E. coli-derived membrane phospholipids phosphatidylethanolamine, phosphatidylglycerol, and cardiolipin (Avanti, Alabaster, AL) were resuspended in 20 mM Tris pH 8.0, 150 mM NaCl, and 1 mM DTT. Large unilammellar vesicles (LUVs) were created by 10 cycles of vortexing for 2 min followed with five successive freeze–thaw cycles by immersion in dry-ice/ethanol for 1 min and a 37°C water bath for 3 min.
Activity assays
ATPase activities of EpsE-containing complexes were measured using a modified malachite green assay (Camberg and Sandkvist, 2005). End point assays containing 1 μM protein, 5 mM ATP, and 5 mM MgCl2 in Buffer A (100 mM HEPES pH 8.5, 65 mM NaCl, 5% glycerol) were incubated at 37°C for 360 min. Where indicated, phosphatidylethanolamine, phosphatidylglycerol, and cardiolipin (31.25, 62.5, 125, and 250 μM) were added to ATPase reactions containing 0.5 μM protein and incubated for 60 min. Samples were taken at the start and finish of every reaction and assayed for inorganic phosphate in duplicate. Error is reported as standard error, representative of three sample sets.
For studies involving pre-incubation of EpsE/EpsL(1–253) with either cardiolipin or ATP, reactions containing 0.5 μM EpsE/EpsL(1–253) and 125 μM cardiolipin, or 5 mM ATP with 5 mM MgCl2 were mixed in Buffer A and incubated at 37°C. After 10 min, the remaining hydrolysis reaction components were added and the rate of ATP hydrolysis was assayed.
Phospholipid-pelleting assays
EpsE, EpsE/EpsL(1–253), and EpsE/EpsL(1–242) (1 μM) were incubated with purified phospholipids (250 μM) in the absence and presence of ATP (5 mM) and MgCl2 (5 mM) at 37°C for 15 min in 100 mM HEPES pH 8.5 and 65 mM NaCl. Before setting up the reactions, proteins were pre-spun at 100 000 g for 20 min. After incubation, reactions were centrifuged at 100 000 g for 1 h at 4°C to pellet phospholipids and phospholipid-bound protein. Supernatants were aspirated and pellets were resuspended in an equal volume. Supernatants and pellets were examined by SDS–PAGE using the NuPAGE system (Invitrogen, Carlsbad, CA) and stained with GelCode Blue (Pierce, Rockford, IL). ImageQuant software was used to quantify the percentage of protein in the pellet. For reactions containing EpsL(1–242) and EpsL(1–253), 5 μM protein was used in the pelleting assay and fractions were analyzed under reducing conditions.
Protease-sensitivity assay
Reactions of EpsE/EpsL(1–253) and EpsE/EpsL(1–242) (1 μM) that contained cardiolipin (250 μM), ATP (5 mM), and MgCl2 (5 mM) were incubated at 37°C for 15 min in 100 mM HEPES buffer pH 8.5 with 65 mM NaCl and then digested with trypsin (0, 3, 10, 30, 100, 300 μg/ml) at room temperature for 5 min. Following addition of EDTA-free protease inhibitor cocktail (Roche, Indianapolis, IN), the reactions were centrifuged for 20 min at 4°C and 15 000 g, which was sufficient to pellet cardiolipin LUVs. Supernatants and pellets were collected and analyzed by SDS–PAGE and silver staining (Invitrogen). Immunoblots were performed using antibodies to EpsE and EpsL.
Chemical crosslinking
Reactions containing 1 μM EpsE/EpsL(1–253) or EpsE/EpsL(1–242) with 125 μM cardiolipin in Buffer A with 5 mM ATP and 5 mM MgCl2 were incubated for 15 min at 37°C, then disthiobis succinimidyl propionate (DSP) (Pierce) was added to a final concentration of 0, 10, 50, 100, or 200 μM. After 30 min at 23°C, 1 μl of 1 M Tris, pH 8.5, was added to the 20 μl reaction to quench residual DSP. Reactions were analyzed by SDS–PAGE and immunoblotting with anti-EpsE and anti-EpsL antibodies.
Complementation of EpsL mutants
Mutant genes epsL(K239A), epsL(L240E), epsL(K241A), epsL(W244E), epsL(W244F), epsL(W244A), epsL(L245E), epsL(R246A), epsL(R246E), epsL(Y247E), epsL(W248E), epsL(W251E), and epsL(R252A/K253A) were expressed in V. cholerae epsL mutant strain mut 8 and tested for protease secretion using a modified fluorescence-based assay (Sandkvist et al, 1997). Filtered supernatants from overnight cultures grown in LB were assayed in 5 mM HEPES pH 7.5 and 0.05 mM N-tert-butoxy-carbonyl-Gln-Ala-Arg-7-amido-4-methyl-coumarin (Sigma) for 10 min at 37°C using the excitation and emission wavelengths 385 and 440 nm, respectively. Error is reported as standard error, representative of three samples.
To detect the presence of EpsL, wild-type V. cholerae TRH7000 and epsL mutant strain mut 8 expressing epsL, epsL(K239A), epsL(K241A), epsL(W244E), and epsL(L245E) were grown in LB and cells were pelleted, resuspended in SDS sample buffer, and subjected to SDS–PAGE. Proteins were immunoblotted using antibodies to EpsL.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank Michael Bagdasarian for reading the manuscript. This work was supported by grant AI49294 from the National Institutes of Health (to MS). JLC and TLJ were supported, in part, by National Institutes of Health training Grants HL007698 and AI007258, respectively.
References
- Abendroth J, Bagdasarian M, Sandkvist M, Hol WG (2004) The structure of the cytoplasmic domain of EpsL, an inner membrane component of the type II secretion system of Vibrio cholerae: an unusual member of the actin-like ATPase superfamily. J Mol Biol 344: 619–633 [DOI] [PubMed] [Google Scholar]
- Abendroth J, Murphy P, Sandkvist M, Bagdasarian M, Hol WG (2005) The X-ray structure of the type II secretion system complex formed by the N-terminal domain of EpsE and the cytoplasmic domain of EpsL of Vibrio cholerae. J Mol Biol 348: 845–855 [DOI] [PubMed] [Google Scholar]
- Auvray F, Ozin AJ, Claret L, Hughes C (2002) Intrinsic membrane targeting of the flagellar export ATPase FliI: interaction with acidic phospholipids and FliH. J Mol Biol 318: 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball G, Chapon-Herve V, Bleves S, Michel G, Bally M (1999) Assembly of XcpR in the cytoplasmic membrane is required for extracellular protein secretion in Pseudomonas aeruginosa. J Bacteriol 181: 382–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camberg JL, Sandkvist M (2005) Molecular analysis of the Vibrio cholerae type II secretion ATPase EpsE. J Bacteriol 187: 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianciotto NP (2005) Type II secretion: a protein secretion system for all seasons. Trends Microbiol 13: 581–588 [DOI] [PubMed] [Google Scholar]
- Filloux A (2004) The underlying mechanisms of type II protein secretion. Biochim Biophys Acta 1694: 163–179 [DOI] [PubMed] [Google Scholar]
- Herdendorf TJ, McCaslin DR, Forest KT (2002) Aquifex aeolicus PilT, homologue of a surface motility protein, is a thermostable oligomeric NTPase. J Bacteriol 184: 6465–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TL, Abendroth J, Hol WG, Sandkvist M (2006) Type II secretion: from structure to function. FEMS Microbiol Lett 255: 175–186 [DOI] [PubMed] [Google Scholar]
- Kawai F, Shoda M, Harashima R, Sadaie Y, Hara H, Matsumoto K (2004) Cardiolipin domains in Bacillus subtilis marburg membranes. J Bacteriol 186: 1475–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Dowhan W, Wickner W (1990) The ATPase activity of SecA is regulated by acidic phospholipids, SecY, and the leader and mature domains of precursor proteins. Cell 60: 271–280 [DOI] [PubMed] [Google Scholar]
- Michel G, Bleves S, Ball G, Lazdunski A, Filloux A (1998) Mutual stabilization of the XcpZ and XcpY components of the secretory apparatus in Pseudomonas aeruginosa. Microbiology 144 (Part 12): 3379–3386 [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W (2000) Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J Bacteriol 182: 1172–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W (2005) Role of membrane lipids in bacterial division-site selection. Curr Opin Microbiol 8: 135–142 [DOI] [PubMed] [Google Scholar]
- Possot OM, Vignon G, Bomchil N, Ebel F, Pugsley AP (2000) Multiple interactions between pullulanase secreton components involved in stabilization and cytoplasmic membrane association of PulE. J Bacteriol 182: 2142–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powl AM, East JM, Lee AG (2005) Heterogeneity in the binding of lipid molecules to the surface of a membrane protein: hot spots for anionic lipids on the mechanosensitive channel of large conductance MscL and effects on conformation. Biochemistry 44: 5873–5883 [DOI] [PubMed] [Google Scholar]
- Py B, Loiseau L, Barras F (2001) An inner membrane platform in the type II secretion machinery of Gram-negative bacteria. EMBO Rep 2: 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robien MA, Krumm BE, Sandkvist M, Hol WG (2003) Crystal structure of the extracellular protein secretion NTPase EpsE of Vibrio cholerae. J Mol Biol 333: 657–674 [DOI] [PubMed] [Google Scholar]
- Sakai D, Horiuchi T, Komano T (2001) ATPase activity and multimer formation of Pilq protein are required for thin pilus biogenesis in plasmid R64. J Biol Chem 276: 17968–17975 [DOI] [PubMed] [Google Scholar]
- Sandkvist M (2001) Biology of type II secretion. Mol Microbiol 40: 271–283 [DOI] [PubMed] [Google Scholar]
- Sandkvist M, Bagdasarian M, Howard SP, DiRita VJ (1995) Interaction between the autokinase EpsE and EpsL in the cytoplasmic membrane is required for extracellular secretion in Vibrio cholerae. EMBO J 14: 1664–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkvist M, Hough LP, Bagdasarian MM, Bagdasarian M (1999) Direct interaction of the EpsL and EpsM proteins of the general secretion apparatus in Vibrio cholerae. J Bacteriol 181: 3129–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkvist M, Michel LO, Hough LP, Morales VM, Bagdasarian M, Koomey M, DiRita VJ, Bagdasarian M (1997) General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in Vibrio cholerae. J Bacteriol 179: 6994–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, Buhrdorf R, Fischer W, Haas R, Waksman G (2003) VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J 22: 1969–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiue SJ, Kao KM, Leu WM, Chen LY, Chan NL, Hu NT (2006) XpsE oligomerization triggered by ATP binding, not hydrolysis, leads to its association with XpsL. EMBO J 25: 1426–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1