

Abstract
We examined the role of cell surface clustering of β2-integrin caused by protein kinase C (PKC)-activated-cPLA2 in adhesion of eosinophilic AML14.3D10 (AML) cells. Phorbol 12-myristate 13-acetate (PMA) caused time- and concentration-dependent adhesion of AML cells to plated bovine serum albumin (BSA), which was blocked by anti-CD11b or anti-CD18 monoclonal antibodies (mAb) directed against β2-integrin. Inhibition of PKC with Ro-31-8220 or rottlerin blocked PMA-induced cell adhesion in a concentration-dependent fashion. Inhibition of cytosolic phospholipase A2 (cPLA2) with trifluoromethyl ketone or methyl arachidonyl fluorophosphonate also blocked PMA-induced cell adhesion. PMA caused time-dependent p42/44 mitogen-activated protein kinase (MAPK) (ERK) phosphorylation in these cells. U0126, a MAPK/extracellular signal-regulated protein kinase kinase (MEK) inhibitor, at the concentrations that blocked PMA-induced ERK phosphorylation, had no effect on PMA stimulated AML cell adhesion. Neither p38 MAPK nor c-Jun N-terminal kinase (JNK) was phosphorylated by PMA. PMA also caused increased cPLA2 activity, which was inhibited by Ro-31-8220, but not U0126. Confocal immunofluorescence microscopy showed that PMA caused clustering of CD11b on the cell surface, which was blocked by either PKC or cPLA2 inhibition. PMA stimulation also caused up-regulation of CD11b on the AML cell surface. However, this up-regulation was not affected by cPLA2- or PKC-inhibition. Using the mAb, CBRM1/5, we also demonstrated that PMA does not induce the active conformation of CD11b/CD18. Our data indicate that PMA causes AML cell adhesion through β2-integrin by PKC activation of cPLA2. This pathway is independent of MEK/ERK and does not require change of CD11b/CD18 to its active conformation. We find that avidity caused by integrin surface clustering – rather than conformational change or up-regulation of CD11b/CD18 – causes PMA stimulated adhesion of AML cells.
Introduction
Integrins are a diverse family of αβ heterodimetric transmembrane adhesion receptors that participate in cell–cell or cell–matrix interactions within the immune system.1,2 The β2-integrin subfamily consists of four integrins, CD11a (leucocyte function-associated antigen-1; LFA-1), CD11b (Mac-1), CD11c, and CD11d, which share a common β2 subunit (CD18) and are exclusively expressed on leucocytes.3 CD11b/CD18 is expressed primarily on cells of the myelomonocytic lineage and binds to intercellular adhesion molecule-1 (ICAM-1).4 Although the integrins on resting leucocytes bind poorly to ligand, stimulation of cells with a variety of agonists causes increased binding activity.5–7 Three mechanisms:
up-regulation of surface integrin molecules8
the avidity of existing receptors11,12 have been suggested in causing cell adhesion to counterligands. The amount of cell surface CD11b, unlike CD11a, on granulocytes and monocytes can be up-regulated rapidly by translocation from an intracellular pool to the cell surface in response to cell activation.13,14 Some prior investigations, however, suggest that quantitative changes of surface CD11b/CD18 molecules do not regulate β2 integrin adhesion.5,6 Prior investigations also have focused on conformational change in integrin as a mechanism causing cell adhesion;10 however, several recent investigations have suggested that avidity (i.e. clustering) caused by cell surface clustering of integrin may also have a substantial role in integrin-mediated adhesion. It has been reported that phorbol esters such as phorbol 12-myristate 13-acetate (PMA), an activator of protein kinase C (PKC), cause only a small increase in the binding affinity of the leucocyte integrin CD11a/CD1815 and that the major effect of PMA-induced cell adhesion is exerted through postreceptor events affecting the clustering of integrins.16–18 Like CD11a, microclustering of CD11b is induced upon stimulation of neutrophils with PMA in the absence of ligand.19 However, the signal transduction pathways causing clustering of integrins have not been elucidated.
We recently have reported that cytosolic phospholipase A2 (cPLA2) is the critical regulatory enzyme in integrin-mediated adhesion of human eosinophils.20,21 cPLA2 is phosphorylated by PKC or p42/44 mitogen-activated protein kinase (MAPK) in vitro, but only phosphorylation by MAPK has been reported to cause a significant increase in cPLA2 activity.22,23 A p42/44 MAPK (ERK)-dependent cPLA2 activation pathway also has been found in spontaneous β1-integrin-mediated adhesion of eosinophils to fibronectin.24 However, other pathways regulating cPLA2 activation have not been explored.
The objective of this study was to determine the putative role of a PKC pathway for phosphorylation of cPLA2. We hypothesized that this phosphorylation could cause cell surface clustering of β2-integrin to adhesion of eosinophilic AML14.3D10 (AML) cells.25,26 We found that PMA stimulation results in activation of cPLA2 and subsequent cell adhesion through clustering rather than the conformational change or up-regulation of CD11b. Our data also indicate cPLA2 activation by PMA was induced by PKC and was independent of ERK phosphorylation.
Materials and Methods
Reagents
Bovine serum albumin (BSA) fraction V, butyric acid, PMA, RPMI-1640, 2-mercaptoethanol (2-ME), sodium bicarbonate, sodium pyruvate, and gentamicin were purchased from Sigma Chemical Co. (St. Louis, MO). Ro-31-8220, Gö 6976, rottlerin, and U0126, were purchased from Calbiochem (San Diego, CA). Trifluoromethyl ketone (TFMK) and methyl arachidonyl fluorophosphonate (MAFP) were purchased from Cayman (Ann Arbor, MI). Interleukin-5 (IL-5) was purchased from R&D (Minneapolis, MN). Fetal bovine serum (FBS) was purchased from HyClone Laboratories, Inc. (Logan, UT). Polystyrene 96-well microtitre plates were obtained from Costar (Cambridge, MA). 1-palmitoyl-2-[14C]arachidonyl phosphatidylcholine (PAPC) was purchased from New England Nuclear (Boston, MA). Anti-phospho-ERK1/2 antibody was purchased from Promega (Madison, WI). Anti-ERK1/2, antiphospho-p38 MAPK, anti-p38 MAPK, antiphospho-[c-Jun N-terminal kinase (JNK)], and anti-JNK antibodies were purchased from New England Biolabs (Beverly, MA). Goat anti-rabbit immunoglobulin conjugated with horseradish peroxidase (HRP) was purchased from Amersham (Arlington Heights, IL). Anti-CD11b monoclonal antibody (mAb; clone 44) was purchased from Endogen (Woburn, MA). Anti-CD11a (clone 25.3), anti-CD11c (clone BU15), anti-CD18 (clone 7E4), anti-CD29 (clone Lia1/2), and anti-CD49d mAb (clone HP2/1) were purchased from Immunotech (Westbrook, ME). The CBRM1/5 mAb against activated CD11b was a gift from Dr T. A. Springer (Harvard Medical School). Mouse immunoglobulin G (IgG) was purchased from Becton-Dickinson (Mountain View, CA).
Cell culture
The eosinophilic AML cell line was grown in RPMI-1640 containing 10% FBS, 50 µm 2-ME, 1 mm sodium pyruvate, and 50 µg/ml gentamicin. A stock of 50 mm butyric acid in phosphate-buffered saline (PBS) was prepared and stored at 4° before use. Butyric acid (0·5 mm) was added to growing cells in flasks at a starting density of 2×105 cells/ml. Two days later 10 ng/ml IL-5 was added to the medium, and cells were cultured for another 5 days, and then used.27
Cell adhesion assay
Cell adherence was assessed as residual eosinophil peroxidase (EPO) activity of adherent cells. One hundred µl of 10 µg/ml BSA dissolved in 0·05 m NaHCO3 coating buffer (15 mm NaHCO3 and 35 mm Na2CO3, pH 9·2) was added to flat-bottom 96-well microtitre plates and incubated at 4° overnight. BSA was decanted, and 200 µl/well of neat FBS was added to coated wells; after 60-min incubation at 37°, the wells were decanted and washed with Hank's balanced salt solution (HBSS) before the addition of AML cells. Cells (3×104/100 µl HBSS/0·1% gelatin) were added to each well of BSA-coated microplates with or without PMA and allowed to settle for 10 min on ice. Plates were rapidly warmed to 37° and incubated for indicated times. After a wash with HBSS, 100 µl of HBSS/0·1% gelatin was added to the reaction wells, and serial dilutions of the original cell suspension were added to the empty wells to generate a standard curve. One hundred µl of EPO substrate (1 mm H2O2, 1 mm o-phenylenediamine dihydrochloride (OPD), and 0·1% Triton-X-100 in Tris buffer, pH 8·0) then was added to the wells. After a 30-min incubation at room temperature, 50 µl of 4 m H2SO4 was added to stop the reaction. Absorbance was measured at 490 nm in a microplate reader (Thermomax, Molecular Devices, Menlo Park, CA). All assays were performed in duplicate. Data storage and analysis were facilitated by the use of computer software interfaced with the reader (Softmax, Molecular Devices). The detection of EPO by this assay was linear between concentrations of 1·0×103−1·5×104 cells/well, as determined by a standard curve.
The blocking effect of mouse monoclonal antibodies against various adhesion molecules, anti-CD11a (clone 25.3), anti-CD11b (clone 44), activated anti-CD11b (clone CBRM1/59), anti-CD11c (clone BU15), anti-CD18 (clone 7E4), anti-CD29 (clone Lia1/2) and anti-CD49d (HP2/1) was tested by preincubation of antibodies with cells for 30 min on ice before adding the cells to a 96-well microplate. The effect of pharmacological inhibitor on integrin-mediated adhesion was tested by preincubation cells with Ro-31-8220, Gö 6976, rottlerin, TFMK, MAFP, or U0126 for 30 min at 37°.
Immunoblot analysis of MAPK
AML cells (2×106/group) were stimulated with PMA for various times, and the reaction was stopped by centrifugation at 12 000 g for 30 s. The pellets then were lysed in 80 µl lysis buffer [20 mm Tris-HCl (pH 7·5), 150 mm NaCl, 1 mm disodiumethylenediaminetetra-acetic acid, 1 mm egtazic acid (EGTA), 1% Triton X-100, 2·5 mm sodium pyrophosphate, 1 mmβ-glycerophosphate, 1 mm Na3VO4, 1 µg/ml leupeptin, and 1 mm phenymethylsulphonyl fluoride (PMSF)]. After 20 min on ice, the sample was centrifuged at 12 000 g for 2 min to remove nuclear and cellular debris. The supernatants then were mixed with 14 µl of 6× sample buffer and boiled for 5 min. The samples were collected and saved at −70°.
Samples were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE), using 10% acrylamide gels under reducing condition (15 mA/gel). Electrotransfer of proteins from the gels to polyvinylidene fluoride membrane was achieved using a semidry system (400 mA, 60 min). The membrane was blocked with 1% BSA for 60 min, then incubated with 1/5000 antiphosphorylation-specific ERK1/2 antibody, 1/1000 anti-ERK1/2 antibody, 1/1000 antiphosphorylation-specific p38 MAPK, 1/1000 antip38 antibody, 1/1000 antiphosphorylation-specific JNK, or 1/1000 anti-JNK antibody diluted in Tris buffered saline with tween 20 (TBST) overnight. The membranes then were washed three times for 20 min with TBST. Goat anti-rabbit IgG conjugated with HRP was diluted 1/3000 in TBST and incubated with polyvinylidene fluoride membrane for 60 min. The membrane was again washed three times with TBST and assayed by an ECL chemiluminescence system (Amersham).
Analysis of surface integrin expression by immunofluorescence flow cytometry and confocal microscopy
AML cells were preincubated with each inhibitor and then stimulated by 10−7 m PMA for 30 min. Thereafter, cells were centrifuged at 400 g for 10 min, and the pellets were resuspended in PBS/0·1% BSA. Aliquots of 5×105 cells were incubated with 10 µg/ml of mAb directed against CD11b, CBRM1/5, or isotype-matched control antibody for 30 min at 4°. After two washes, the cells were incubated with an excess of fluoroscein isothiocyanate (FITC)-conjugated goat anti-mouse immunoglobulin for 20 min at 4°. The cells were washed twice, resuspended in 1% paraformaldehyde, and kept at 4° until analysed. Flow cytometry was performed by FACScan (Becton Dickinson, Mountain View, CA). Fluorescence intensity was determined on at least 5000 cells from each sample. The results were expressed as the specific mean fluorescence intensity (sMFI) (control antibody fluorescence subtracted). As a positive control, further experiments were performed using 5 mm Mn2+ to stimulate CBRM1/5 up-regulation. To confirm the distribution of CD11b, CD11b staining also was performed on cytospin preparations (Cytospin 2; Shandon, Pittsburgh, PA), and fluorescence was analysed using a Zeiss Axiovert confocal microscope equipped with an external argon-krypton laser (488 nm).
Determination of cPLA2 enzyme activity
The cPLA2 activity assay was modified from the method of Kim et al.28 Briefly, 2×106 AML cells were preincubated with Ro-31-8220 or U0126 for 30 min and then stimulated with or without 100 nm PMA for various times. The reaction was stopped by centrifugation, and the pellets were resuspended in 70 µl sonication buffer (20 mm Tris, pH 8·0, 2·5 mm EDTA, 10 µg/ml leupeptin, 5 µg/ml aprotinin, 1 mm PMSF, 2 mm Na3VO4, 50 mm NaF, and 5 µg/ml pepstatin) and sonicated briefly (4×10 s, at a power setting of 3). Lysates were pretreated with 5 mm dithiothreitol (DTT) on ice for 5 min to inactivate secretory PLA2, and 10 µl of 50 mm CaCl2 was then added to each sample. A total of 10 µl substrate ([14C]PAPC) was dried under a stream of N2 and resuspended in 200 µl 10% ethanol in H2O with vigorous vortex mixing. The reaction was initiated by adding 10 µl portion of the substrate (final concentration 9 µm) to cell lysate. The reaction was conducted for 30 min at 37° and was stopped by adding 560 µl Dole's reagent (heptane-isopropyl alcohol−1 N H2SO4, 400 : 390 : 10 by volume), followed by 110 µl H2O, vortexed for 20 s and then centrifuged at 12 000 g. The upper layer (180 µl) was transferred to 800 µl hexane containing 25 mg silica gel. A total of 750 µl of sample then was mixed with 2 ml scintillation fluids, and the radioactivity was counted in a liquid scintillation counter.
Statistical analysis
All measurements were expressed as mean±SEM. Variation between two groups was tested using Student's t-test. Variation among more than two groups was tested using anova followed by Fisher's protected least significant difference. A value of P < 0·05 was accepted as statistically significant.
Results
Kinetics and concentration-dependent effect of PMA on AML cell adhesion to BSA
Experiments were conducted to determine the kinetics of AML cell adhesion to BSA-coated plates. Cells were incubated with or without 100 nm PMA on BSA-coated plates for various times ≤60 min at 37°. Cell adhesion in the presence of PMA was detected within 5 min of incubation on BSA coated plates; maximal adhesion occurred within 15–30 min (Fig. 1a). Accordingly, a 30-min incubation time was used for BSA adhesion in subsequent experiments. PMA-induced adhesion to BSA was evident at 10 nm, and was maximal at 100 nm (Fig. 1b).
Figure 1.

(a) Kinetics of PMA (100 nm)-induced AML cell adhesion to BSA-coated wells. (b) Concentration-dependent effect of PMA on adhesion to BSA following 30 min incubation in AML cells. Each point represents the mean±SEM of four separate experiments. **P < 0·01 compared with each control.
Demonstration of β2-integrin causing PMA-stimulated adhesion of AML cells to BSA
The contribution of the β2-integrin to the adhesion of AML cells to BSA was validated by inhibition assays using specific blocking mAb. Adhesion of these cells to BSA was demonstrated to be β2-integrin mediated. Adhesion of stimulated cells was significantly inhibited by anti-CD11b mAb (clone 44) to 54·9±6·5% of isotype control (P < 0·01; Fig. 2). Similarly, blockade of the common β2-chain by anti-CD18 mAb (clone 7E4) caused a decrease in adhesion to 14·0±4·3% of isotype control (P < 0·01). Neither anti-CD11a nor anti-CD11c had any measurable inhibitory effects on AML cell adhesion. Adhesion to BSA was specific for β2-integrin. Neither anti-CD49d nor anti-CD29 mAb caused blockade of AML cell adhesion to BSA. Both mAbs were bioactive, as they block spontaneous eosinophil adhesion to either vascular cell adhesion molecule-1 (VCAM-1) or fibronectin.20,24
Figure 2.

Effect of mAb directed against β1- or β2-integrin on PMA-stimulated adhesion of AML cells to BSA. AML cells were preincubated with an optimal concentration of mAb against β1 or β2 integrins or isotype control and incubated with 100 nm PMA for 30 min in BSA-coated wells. Each point represents the mean±SEM of four separate experiments. **P < 0·01 compared with control.
Effects of PMA or cPLA2 inhibition on PMA-induced adhesion of AML cells to plated BSA
Three PKC inhibitors (Ro-31-8220, Gö 6976, and rottlerin) were used to determine whether PKC is involved in AML cell adhesion. As shown in Fig. 3, Ro-31-8220, Gö 6976, and rottlerin inhibited PMA-induced adhesion of AML cells (IC50s=1·94×10−7±0·11×10−7, 5·82×10−7±0·91×10−7, and 1·12×10−6±0·17×10−6 m, respectively).
Figure 3.

Effect of the PKC inhibitors, Ro-31-8220 (closed squares), Gö 6976 (closed triangles), and rottlerin (closed circles) on PMA-induced AML cell adherence to BSA. AML cells were preincubated with the specified concentrations of inhibitors for 30 min at 37°, followed by addition of PMA and incubation for 30 min at 37°. The adherence of AML cells was assessed as detailed in Materials and Methods in the presence of inhibitors. Each point represents the mean±SEM of four separate experiments. **P < 0·01 compared with each control.
The role of cPLA2 in PMA stimulated adhesion of AML cells was examined using competitive cPLA2 active-site inhibitor, TFMK29 and the non-competitive active-site-directed inhibitor, MAFP.30 TFMK caused concentration-dependent suppression of PMA-stimulated AML cell adhesion to BSA-coated plates (IC50=4·38×10−6±0·34×10−6 m), and MAFP caused comparable inhibition (IC50=6·68×10−6±0·73×10−6 M) (Fig. 4). Complete inhibition of this adhesion was achieved at 10 µm TFMK (P < 0·01) or at 30 µm MAFP (P < 0·01).
Figure 4.

Effect of the cPLA2 inhibitors, TFMK (closed circles) and MAFP (closed triangles), on AML cell adhesion to BSA. Each point represents the mean±SEM of four separate experiments. *P < 0·05 and **P < 0·01 compared with each control.
Effect of MAPK inhibition on PMA-induced AML cell adhesion
As PMA has been shown to induce cPLA2 activation through MAPK31 we examined whether MAPK was involved in PMA-induced adhesion of AML cells. Figure 5 indicates that MAPK phosphorylation is not involved in PMA-stimulated adhesion of AML cells to BSA. Figure 5(a) demonstrates that PMA caused time-dependent ERK1/2 phosphorylation (top panel), which was observed within 30 s and peaked at 5–20 min; equivalent loading of ERK1/2 was demonstrated at all times (bottom panel). ERK1/2 phosphorylation caused by PMA was only partially inhibited by 10 µm Ro-31-8220 (Fig. 5b), suggesting that PMA may cause ERK phosphorylation at least in part through an alternative (e.g. non-PKC-dependent) pathway. PMA-induced ERK1/2 phosphorylation was completely inhibited by U0126, an inhibitor of ERK1/2 activation secondary to its inhibition of upstream MAP/ERK kinase (MEK) (Fig. 5c).32 However, 10 µm U0126, which completely blocked PMA-induced ERK phosphorylation (Fig. 5c), did not suppress AML cell adhesion to plated BSA at 30 min (Fig. 6). Neither p38 nor JNK phosphorylation was observed in PMA-stimulated AML cells (data not shown).
Figure 5.

ERK phosphorylation in PMA-stimulated AML cells. (a) Time-dependent effects of PMA on ERK1/2 phosphorylation. AML cells were incubated with 100 nm PMA for indicated times. AML cells were lysed, and the lysates were mixed with sample buffer and loaded on 10% SDS-PAGE, followed by immunoblotting with antiphosphorylation-specific ERK1/2 (a, top panel), anti-ERK1/2 (phosphorylated+non-phosphorylated) (a, bottom panel) as described in Materials and Methods. Effects of Ro-31-8220 (b) or U0126 (c) on PMA-stimulated ERK1/2 phosphorylation. AML cells were preincubated with different concentrations of Ro-31-8220 or U0126 for 30 min, and stimulated with 100 nm PMA for 10 min ERK1/2 phosphorylation (each top panel) and total ERK1/2 (each bottom panel) were measured as in (a). The result shown is representative of three different experiments.
Figure 6.

Effect of U0126, a MEK inhibitor, on PMA-induced AML cell adherence to BSA. AML cells were preincubated with the specified concentrations of U0126 for 30 min at 37°, followed by addition of PMA and incubation for 30 min at 37°. The adherence of AML cells was assessed as detailed in Materials and Methods in the presence of inhibitors. Each point represents the mean±SEM of four separate experiments.
The role of CD11b clustering in PMA-stimulated adhesion of AML cells
Pretreatment with butyric acid and IL-5 caused CD11b (25·8±0·46–39·9±3·1 sMFI) and CBRM1/5 up-regulation (6·1±1·4–10·6±2·2 sMFI) in AML cells. PMA caused up-regulation of CD11b expression on AML cells (P < 0·01; Fig. 7a), but none of the three PKC inhibitors nor TFMK, a cPLA2 inhibitor, blocked the up-regulation of CD11b induced by PMA (Fig. 7a). In control studies we demonstrated the ability of Mn2+ to cause up-regulation of CBRM1/5. However, PMA did not induce expression of a CD11b activation epitope recognized by the mAb, CBRM1/59 (Fig. 7b), suggesting that a conformational change of CD11b does not occur in PMA-induced AML cell adhesion to BSA. Confocal immunofluorescence microscopy showed that PMA caused clustering of CD11b on the AML cell surface, which was inhibited by pretreatment with Ro-31-8220 or TFMK (Fig. 8). Thus, both PKC and cPLA2 are involved in the regulation of CD11b clustering in PMA-induced AML cell adhesion.
Figure 7.

(a) Induction of surface CD11b expression by PMA and effects of various inhibitors. AML cells (0·5×106) were preincubated with Ro-31-8220 (1·0 µm), Gö 6976 (10 µm), rottlerin (10 µm), or TFMK (10 µm) for 30 min at 37°, followed by addition of PMA and incubation for 30 min at 37°. (b) CBRM1/5 epitope expression before and after PMA. Mn2+ at a concentration of 5 mm was used as a positive control stimulus for up-regulation of CBRM1/5. Surface CD11b and CBRM1/5 expression was measured by flow cytometry as described in Materials and Methods. Data are the mean±SEM of four separate experiments. **P < 0·01 compared with the positive (PMA +) control.
Figure 8.
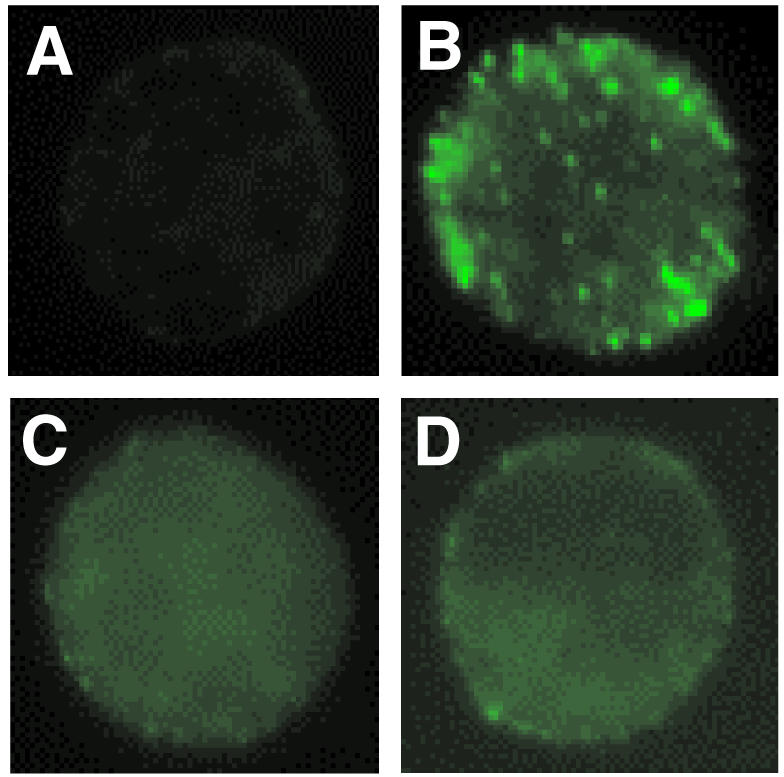
Distribution of CD11b as determined by immunocytochemistry and confocal microscopy. CD11b was detected by anti-CD11b followed by FITC-conjugated goat anti-mouse immunoglobulin as described in Materials and Methods. CD11b is localized in clusters on PMA-treated cells (b), whereas it is dispersed on either cells without PMA-stimulation (a) or cells pretreated with 1·0 µm Ro-31-8220 (c), and 10 µm TFMK (d).
PMA-induced cPLA2 activation and effects of PKC inhibitors
To establish the biological significance of PKC in causing PMA-induced cPLA2 activation, we examined PMA-induced cPLA2 activation in the presence of the PKC inhibitor, Ro-31-8220, or the MEK inhibitor, U0126, in AML cells. cPLA2 activity was increased from 1·77±0·33 pm/2×106 cells/30 min for non-stimulated AML cells to 4·81±0·47 pm/2×106 cells/30 min after 15-min PMA stimulation (P < 0·01) (Fig. 9a). Although 10 µm U0126, which completely blocked PMA-induced ERK phosphorylation (Fig. 5c), did not prevent PMA-induced cPLA2 activation (Fig. 9b), pretreatment with 1·0 µm Ro-31-8220 suppressed completely this enhanced activity (P < 0·01) (Fig. 9b), suggesting that PKC is the upstream kinase for cPLA2.
Figure 9.

cPLA2 activity in lysates of PMA-stimulated AML cells. (a) Time-dependent effect of PMA on cPLA2 activity. AML cells stimulated with 100 nm PMA for indicated times. cPLA2 activity in the AML cell lysates was measured, as described in Materials and Methods, using substrate [14C]PAPC. (b) Effect of Ro-31-8220 and U0126 on cPLA2 activity. AML cells were pretreated with Ro-31-8220 or U0126 for 30 min and then stimulated with 100 nm PMA for 30 min at 37° and assayed for cPLA2 activity. Each point represents the mean±SEM of three separate experiments.
Discussion
In this study, we demonstrated that PMA caused AML cell adhesion to plated BSA that was β2-integrin (CD11b/CD18) dependent and that this adhesion results from clustering of CD11b/CD18. PMA-induced CD11b clustering and subsequent adhesion to BSA was regulated by PKC-dependent activation of cPLA2. However, activation of cPLA2 was independent of MAPK. Adhesion also did not depend upon conformational changes in β2-integrin after activation with PMA.
Prior investigations have emphasized the role of conformational changes of integrins in regulation their affinity to their counterligands.9,10 Recently, it has been suggested that clustering may be an alternative mechanism for regulation of integrin adhesion.12 Although IL-5 caused a CD11b conformational change in human eosinophils, we have shown previously that adhesion is suppressed by cPLA2 inhibition without affecting this conformational change.20 In this study, PMA did not increase expression of the activation-associated epitopes of CD11b recognized by CBRM1/5. Accordingly, adhesion in this system was not mediated by a conformational change in β2-integrin. Our data suggest that avidity (i.e. clustering of integrin) – but not conformational up-regulation of affinity – is responsible for adhesion of AML cells. Although PMA caused CD11b up-regulation on the AML cell surface, no inhibitor that blocked cell adhesion affected CD11b up-regulation. Thus, the quantitative change of CD11b alone is not sufficient to cause β2-integrin adhesion. Furthermore, our data did not establish why inhibition of PKC activation caused by PMA had no effect on PMA-induced CD11b expression in this study. We did establish in control studies that AML cells are capable of expressing the active conformation of CD11b/CD18 (Fig. 6b). One possible explanation is that PMA also activates a non-PKC pathway resulting in CD11b up-regulation. PMA has been shown to act on non-PKC proteins, such as Unc-13 and the chimaerins.33,34
We previously have demonstrated that adhesion to BSA decreases after approximately 30 min after stimulation of IL-5 in human peripheral blood eosinophils.35 Davey et al.36 also demonstrated that PMA-induced polymorphonuclear leucocyte (PMN) adhesion to fibrinogen or ICAM-1 decreases after 30 min after PMA stimulation. These authors have suggested that the decrease in adhesion might result from CD11b proteolysis. Thus, it is possible that the mechanism of decrease in adhesion of AML14.3D10 cells in our study is due to CD11b proteolysis. Alternatively, we have shown that eosinophil adhesion depends upon stable phosphorylation of cPLA2.24 The mechanisms regulating this phosphorylation are not yet fully defined.
CPLA2 is known to be activated by MAPK37 a proline-directed serine/threonine kinase.38 The activation of PKC also has been shown to play a role in cPLA2 activation.23,31,39,40 Some studies suggest PKC activates cPLA2 through ERK activation31,39 while other studies suggest PKC can activate cPLA2 directly.23,40 In the present study, we found that neither ERK, p38 nor JNK is involved in adhesion of AML cells. This suggests that adhesion caused by PMA is activated by PKC-induced activation of cPLA2 through a pathway that does not utilize MAPK.
In the present study, three PKC inhibitors (Ro-31-8220, Gö 6796, and rottlerin) were used in an attempt to dissect which isoenzymes might play a role in AML cell adhesion. Ro-31-8220 is a broad-spectrum PKC inhibitor41 while rottlerin is originally reported to be a PKCδ-specific inhibitor (IC50=3–6 µm)42 and Gö 6796 blocks selectively the activity of conventional PKC family members with an IC50 of 2·3–7·9 nm.43 As shown in Fig. 3, Ro-31-8220, Gö 6976, and rottlerin inhibited PMA-induced AML cell adhesion with IC50s of 0·25, 1·7, and 1·6 µm, respectively. One hundred-fold more Gö 6976 was required for inhibition of adhesion than catalytic activity. Accordingly, our data suggest specific isoforms of PKC could be involved in PMA-induced adhesion in AML cells. However, the PKC inhibitors used in this study have been shown not to be specific for PKC isoforms, and recently rottlerin has been shown not to inhibit PKCδ.44 Further studies using cells stably transfected with PKC isoenzyme are required to affirm this conclusion.
It is important to recognize some limitations of our findings. In these investigations, we modeled mechanisms of cellular adhesion using a cultured cell line of AML14.3D10 eosinophilic cells. This insured uniformly of cell surface expression and biochemical regulation, which is difficult to achieve from human peripheral blood cells obtained from multiple donors.45 Our data suggest that: (1) PKC can regulate β2-integrin adhesion even after increased cell surface expression of β2-integrin; (2) change to active conformation may not be essential for β2-integrin adhesion; and (3) induction of cell surface clustering of β2-integrin may play a critical role in integrin adhesion. We also find that cPLA2 activation caused by PKC is independent of MAPK in AML cells. Because these studies are performed in a cultured cell line, confirmation of the applicability of this putative mechanism of adhesive regulation in normal human cells remains to be established.
Acknowledgments
We thank Drs Cassandra Paul and Michael Baumann (Wright State University, Dayton, OH) for providing AML cells and Dr Timothy Springer (Harvard Medical School, Boston, MA) for providing mAb CBRM1/5. This work was supported by National Heart, Lung, and Blood Institute Grants HL-46368, by NHLBI SCOR Grant HL-56399 (A.R.L), American Lung Association Research Grant RG-003-N (X.Z), and by the GlaxoSmithkline Center of Excellence Award (A.R.L).
Abbreviations
- PKC
protein kinase C
- cPLA2
cytosolic phospholipase A2
- PAF
platelet activating factor
- MAPK
mitogen-activated protein kinase
- ERK
p42/44 MAPK
- MEK
MAPK/extracellular signal-regulated protein kinase kinase
- JNK
c-Jun N-terminal kinase
- TFMK
trifluoromethyl ketone
- MAFP
methyl arachidonyl fluorophosphonate
- EPO
eosinophil peroxidase
- sMFI
specific mean fluorescence intensity
- PAPC
1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine
References
- 1.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–34. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 2.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. The leukocyte integrins. J Biol Chem. 2000;275:23409–12. doi: 10.1074/jbc.R000004200. [DOI] [PubMed] [Google Scholar]
- 4.Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, Springer TA. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b:/CD18) J Cell Biol. 1990;111:3129–39. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright SD, Meyer BC. Phorbol esters cause sequential activation and deactivation of complement receptors on polymorphonuclear leukocytes. J Immunol. 1986;136:1759–64. [PubMed] [Google Scholar]
- 6.Lo SK, Detmers PA, Levin SM, Wright SD. Transient adhesion of neutrophils to endothelium. J Exp Med. 1989;169:1779–93. doi: 10.1084/jem.169.5.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Detmers PA, Lo SK, Olsen-Egbert E, Walz A, Baggiolini M, Cohn ZA. Neutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J Exp Med. 1990;171:1155–62. doi: 10.1084/jem.171.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neeley SP, Hamann KJ, White SR, Baranowski SL, Burch RA, Leff AR. Selective regulation of expression of surface adhesion molecules Mac-1 1–selectin, and VLA–4 on human eosinophils and neutrophils. Am J Respir Cell Mol Biol. 1993;8:633–9. doi: 10.1165/ajrcmb/8.6.633. [DOI] [PubMed] [Google Scholar]
- 9.Weber C, Katayama J, Springer TA. Differential regulation of beta 1 and beta 2 integrin avidity by chemoattractants in eosinophils. Proc Natl Acad Sci USA. 1996;93:10939–44. doi: 10.1073/pnas.93.20.10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hato T, Pampori N, Shattil SJ. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin alphaIIb beta3. J Cell Biol. 1998;141:1685–95. doi: 10.1083/jcb.141.7.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Kooyk Y, Weder P, Heije K, Figdor CG. Extracellular Ca2+ modulates leukocyte function-associated antigen-1 cell surface distribution on T lymphocytes and consequently affects cell adhesion. J Cell Biol. 1994;124:1061–70. doi: 10.1083/jcb.124.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krauss K, Altevogt P. Integrin leukocyte function-associated antigen-1-mediated cell binding can be activated by clustering of membrane rafts. J Biol Chem. 1999;274:36921–7. doi: 10.1074/jbc.274.52.36921. [DOI] [PubMed] [Google Scholar]
- 13.Miller LJ, Bainton DF, Borregaard N, Springer TA. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. J Clin Invest. 1987;80:535–44. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bainton DF, Miller LJ, Kishimoto TK, Springer TA. Leukocyte adhesion receptors are stored in peroxidase-negative granules of human neutrophils. J Exp Med. 1987;166:1641–53. doi: 10.1084/jem.166.6.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lollo BA, Chan KW, Hanson EM, Moy VT, Brian AA. Direct evidence for two affinity states for lymphocyte function-associated antigen 1 on activated T cells. J Biol Chem. 1993;268:21693–700. [PubMed] [Google Scholar]
- 16.Faull RJ, Kovach NL, Harlan JM, Ginsberg MH. Stimulation of integrin-mediated adhesion of T lymphocytes and monocytes: two mechanisms with divergent biological consequences. J Exp Med. 1994;179:1307–16. doi: 10.1084/jem.179.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danilov YN, Juliano RL. Phorbol ester modulation of integrin-mediated cell adhesion: a postreceptor event. J Cell Biol. 1989;108:1925–33. doi: 10.1083/jcb.108.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haverstick DM, Sakai H, Gray LS. Lymphocyte adhesion can be regulated by cytoskeleton-associated, PMA-induced capping of surface receptors. Am J Physiol. 1992;262:C916–26. doi: 10.1152/ajpcell.1992.262.4.C916. [DOI] [PubMed] [Google Scholar]
- 19.Detmers PA, Wright SD, Olsen E, Kimball B, Cohn ZA. Aggregation of complement receptors on human neutrophils in the absence of ligand. J Cell Biol. 1987;105:1137–45. doi: 10.1083/jcb.105.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, Munoz NM, Kim KP, Sano H, Cho W, Leff AR. Cytosolic phospholipase A2 activation is essential for beta 1 and beta 2 integrin-dependent adhesion of human eosinophils. J Immunol. 1999;163:3423–9. [PubMed] [Google Scholar]
- 21.Myou S, Sano H, Fujimura M, et al. Blockade of eosinophil migration and airway hyperresponsiveness by cPLA2-inhibition. Nat Immunol. 2001;2:145–9. doi: 10.1038/84244. [DOI] [PubMed] [Google Scholar]
- 22.Lin LL, Lin AY, DeWitt DL. Interleukin-1 alpha induces the accumulation of cytosolic phospholipase A2 and the release of prostaglandin E2 in human fibroblasts. J Biol Chem. 1992;267:23451–4. [PubMed] [Google Scholar]
- 23.Nemenoff RA, Winitz S, Qian NX, Van Putten V, Johnson GL, Heasley LE. Phosphorylation and activation of a high molecular weight form of phospholipase A2 by p42 microtubule-associated protein 2 kinase and protein kinase C. J Biol Chem. 1993;268:1960–4. [PubMed] [Google Scholar]
- 24.Sano H, Zhu X, Sano A, Boetticher EE, Shioya T, Jacobs B, Munoz NM, Leff AR. Extracellular signal-regulated kinase 1/2-mediated phosphorylation of cytosolic phospholipase A2 is essential for human eosinophil adhesion to fibronectin. J Immunol. 2001;166:3515–21. doi: 10.4049/jimmunol.166.5.3515. [DOI] [PubMed] [Google Scholar]
- 25.Baumann MA, Paul CC. The AML14 and AML14.3D10 cell lines: a long-overdue model for the study of eosinophils and more. Stem Cells. 1998;16:16–24. doi: 10.1002/stem.160016. [DOI] [PubMed] [Google Scholar]
- 26.Paul CC, Mahrer S, Tolbert M, Elbert BL, Wong I, Ackerman SJ, Baumann MA. Changing the differentiation program of hematopoietic cells. Retinoic acid-induced shift of eosinophil-committed cells to neutrophils. Blood. 1995;86:3737–44. [PubMed] [Google Scholar]
- 27.Zimmermann N, Daugherty BL, Stark JM, Rothenberg ME. Molecular analysis of CCR-3 events in eosinophilic cells. J Immunol. 2000;164:1055–64. doi: 10.4049/jimmunol.164.2.1055. [DOI] [PubMed] [Google Scholar]
- 28.Kim K, Jung SY, Lee DK, Jung JK, Park JK, Kim DK, Lee CH. Suppression of inflammatory responses by surfactin, a selective inhibitor of platelet cytosolic phospholipase A2. Biochem Pharmacol. 1998;55:975–85. doi: 10.1016/s0006-2952(97)00613-8. [DOI] [PubMed] [Google Scholar]
- 29.Street IP, Lin HK, Laliberte F, et al. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–40. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 30.Huang Z, Payette P, Abdullah K, Cromlish WA, Kennedy BP. Functional identification of the active-site nucleophile of the human 85-kDa cytosolic phospholipase A2. Biochemistry. 1996;35:3712–21. doi: 10.1021/bi952541k. [DOI] [PubMed] [Google Scholar]
- 31.Pyne NJ, Tolan D, Pyne S. Bradykinin stimulates cAMP synthesis via mitogen-activated protein kinase-dependent regulation of cytosolic phospholipase A2 and prostaglandin E2 release in airway smooth muscle. Biochem J. 1997;328:689–94. doi: 10.1042/bj3280689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 33.Maruyama IN, Brenner S. A phorbol ester/diacylglycerol-binding protein encoded by the unc-13 gene ofCaenorhabditis elegans. Proc Natl Acad Sci USA. 1991;88:5729–33. doi: 10.1073/pnas.88.13.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Areces LB, Kazanietz MG, Blumberg PM. Close similarity of baculovirus-expressed n-chimaerin and protein kinase C alpha as phorbol ester receptors. J Biol Chem. 1994;269:19553–8. [PubMed] [Google Scholar]
- 35.Zhu X, Subbaraman R, Sano H, Jacobs B, Sano A, Boetticher E, Munoz NM, Leff AR. A surrogate method for assessment of beta (2)-integrin-dependent adhesion of human eosinophils to ICAM-1. J Immunol Meth. 2000;240:157–64. doi: 10.1016/s0022-1759(00)00192-7. [DOI] [PubMed] [Google Scholar]
- 36.Davey PC, Zuzel M, Kamiguti AS, Hunt JA, Aziz KA. Activation-dependent proteolytic degradation of polymorphonuclear CD11b. Br J Haematol. 2000;111:934–42. [PubMed] [Google Scholar]
- 37.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–78. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 38.Nishida E, Gotoh Y. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci. 1993;18:128–31. doi: 10.1016/0968-0004(93)90019-j. [DOI] [PubMed] [Google Scholar]
- 39.Hazan I, Dana R, Granot Y, Levy R. Cytosolic phospholipase A2 and its mode of activation in human neutrophils by opsonized zymosan. Correlation between 42/44 kDa mitogen-activated protein kinase, cytosolic phospholipase A2 and NADPH oxidase. Biochem J. 1997;326:867–76. doi: 10.1042/bj3260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wijkander J, Sundler R. An 100-kDa arachidonate-mobilizing phospholipase A2 in mouse spleen and the macrophage cell line J774. Purification, substrate interaction and phosphorylation by protein kinase C. Eur J Biochem. 1991;202:873–80. doi: 10.1111/j.1432-1033.1991.tb16445.x. [DOI] [PubMed] [Google Scholar]
- 41.Davis PD, Hill CH, Keech E, et al. Potent selective inhibitors of protein kinase C. FEBS Lett. 1989;259:61–3. doi: 10.1016/0014-5793(89)81494-2. [DOI] [PubMed] [Google Scholar]
- 42.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 43.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–7. [PubMed] [Google Scholar]
- 44.Soltoff SP. Rottlerin is a mitochondrial uncoupler that decreases cellular ATP levels and indirectly blocks protein kinase Cdelta tyrosine phosphorylation. J Biol Chem. 2001;276:37986–92. doi: 10.1074/jbc.M105073200. [DOI] [PubMed] [Google Scholar]
- 45.Leff AR, Herrnreiter A, Naclerio RM, Baroody FM, Handley DA, Munoz NM. Effect of enantiomeric forms of albuterol on stimulated secretion of granular protein from human eosinophils. Pulm Pharmacol Ther. 1997;10:97–104. doi: 10.1006/pupt.1997.0082. [DOI] [PubMed] [Google Scholar]