

Abstract
C-reactive protein (CRP) is the prototypic acute-phase protein in man which performs innate immune functions. CRP-mediated phagocytosis may be indirect, through activation of complement and complement receptors, or direct, through receptors for the Fc portion of immunoglobulin G (IgG; FcγRs) or even a putative CRP-specific receptor. No strong evidence has been shown to indicate which receptors may be responsible for phagocytosis or signalling responses. Using BIAcore technology, we confirm that CRP binds directly to the extracellular portion of FcγRI with a threefold higher affinity than IgG (KD = 0·81 × 10−9 m). Binding is Ca2+ dependent and is inhibited by IgG1 but not by phosphorylcholine (PC). CRP opsonization (using CRP concentrations within the normal human serum range) of PC-conjugated sheep erythrocytes increased phagocytosis of these particles by COS-7 cells transfected with FcγRI-II chimaera or FcγRI/γ-chain. Interferon-γ-treated U937 cells, which signal through FcγRI to activate phospholipase D (PLD) in response to cross-linked IgG, were also activated by CRP without any requirement for further cross-linking. These studies indicate that CRP is capable of binding to and cross-linking FcγRI thereby resulting in PLD activation and increased phagocytosis. Uptake by FcγRI has been reported to promote various acquired immune responses suggesting that CRP could act in a similar way.
Introduction
C-reactive protein (CRP) is the prototypic acute-phase serum protein in humans with serum levels increasing by up to 1000-fold within 24 hr of severe inflammation.1 Its main role appears to be in innate immune responses where it can activate complement, cause opsonization and protect against infection. CRP is synthesized not only by hepatocytes, which account for the vast majority of CRP in plasma, but also by macrophages and subsets of lymphocytes.2 CRP is a pentamer of non-covalently linked subunits, each of which has a calcium-dependent binding site for phosphorylcholine (PC). The multivalent structure of CRP with ligand-binding sites on the same face of the pentamer3 leads to the possibility of high-avidity interactions with ligands and receptors.
CRP binds to PC-substituted carbohydrate on the surface of Streptococcus pneumoniae and also binds to less well-defined structures on a variety of other micro-organisms including, Diplococcus pneumoniae, Escherichia coli, Aspergillus fumigans and Candida albicans.4 PC is not the only ligand for CRP, a repeating phosphorylated disaccharide ligand has been defined on Leishmania donovani.5 CRP-mediated phagocytosis has been demonstrated for several of these organisms.5–7 Investigations of phagocytic mechanisms are complicated by the fact that both direct binding to cells and indirect binding occur since in vivo CRP can activate complement and thus the complement receptors CR1 and CR3 may be involved in uptake. The protective effect of CRP on S. pneumoniae infection, however, is only partially dependent on complement.8,9
CRP binding to FcγRI was first suggested when it was shown that CRP binding to monocytic cells could be partially inhibited with immunoglobulin G (IgG)10 and was further demonstrated following transfection of COS-7 cells with FcγRI which increased CRP binding.11 The site on CRP involved in FcγRI binding was suggested to be the sequence YLGGP, since this was homologous to a region in IgG (LLGGP) known to be involved in receptor interaction.12 When mutated to YEGGP this led to a protein that failed to bind transfected cells.11 FcγRI alone, however, cannot account for the range of cells that bind CRP since only a small fraction (20%) of the total CRP binding to U937 cells could be inhibited by monomeric IgG.13 FcγRIIa R131, but not FcγRIIa H131, homozygous cells generated a calcium response to CRP, which suggested that this might be the second CRP receptor.14 Various binding studies with CRP are difficult to interpret, however, and whether the FcγRIIa R131 allele is the other major receptor for CRP remains unclear.15,16 Evidence for a specific CRP receptor has arisen from cross-linking studies with U937 cells and inhibition of binding with a monoclonal antibody.17,18
The three families of Fc receptor for IgG are defined by the differences in their affinity for IgG and recognition by monoclonal antibodies. Fc receptors play a co-ordinating role in immune responses. They are important in phagocytosis, antibody-dependent cellular cytotoxicity,19 antigen presentation20–22 and inflammation, for example in the context of immune complex peritonitis.23 FcγRI is the high-affinity receptor for IgG Fc expressed on macrophages, monocytes and dendritic cells which can bind monomeric IgG and plays a pivotal role linking the cellular and humoral arms of the immune system.24 FcγRI is predicted to possess three extracellular, immunoglobulin binding domains and a short cytoplasmic domain.25 Aggregation of FcγRI after IgG cross-linking triggers a variety of different effector functions, including endocytosis and phagocytosis,26 mostly through association with γ-chain (as for FcεRI and FcγRIII) to recruit tyrosine kinases for signal transduction. Aggregation of FcγRI leads to phosphorylation of the associated γ-chains at the immunoreceptor tyrosine-based activation motif (ITAM) and the consequent recruitment and activation of non-receptor tyrosine kinases including Hck and Lyn. This generates a binding site for SH2-containing proteins such as Syk, Ras GTPase activating protein, Shc, Grb2, or the p85 subunit of phosphatidylinositol 3-kinase, phospholipase Cγ1, or phospholipase Cγ2.27
In this study, we have specifically looked at the interaction between CRP and FcγRI, with the aim of further definition of CRP and Fc receptor interaction using surface plasmon resonance to confirm directly the interaction between the two molecules and to demonstrate the potential for high avidity.
Second, since previous studies have examined uptake of zymosan into mouse bone-marrow macrophages which can enter cells through many potential receptors and the systems used in those studies were mouse : human,28 we sought a more defined system to investigate receptor usage. We have devised a human transfected system to examine the specificity of receptors involved in CRP-mediated phagocytosis of PC-labelled erythrocytes.
We demonstrate that binding to this receptor in the presence of γ-chain (in the absence of FcγRIIa) is sufficient to cause CRP-mediated phagocytosis of PC-conjugated sheep erythrocytes by COS-7 cells expressing Fc receptors. Consistent with this we also demonstrate that, when added to interferon-γ (IFN-γ) -treated U937 cells [in which phospholipase D (PLD) activation can be mediated through FcγRI but not FcγRIIa cross-linking] CRP initiates the PLD pathway of calcium mobilization.
Materials and methods
Sources of CRP and serum amyloid P
CRP was extracted from acute-phase serum by affinity chromatography using PC–Sepharose beads and purified by ion exchange chromatography and gel filtration as previously described.29 Serum amyloid P (SAP) was purified from human serum by affinity chromatography, anion exchange and zinc chelate chromatography.30
Cells and cell culture
COS-7 cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies, Paisley, UK) supplemented with 2 mm glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 0·015 mg/ml gentamicin and 10% fetal bovine serum. Cells were plated at 4 × 105/ml in 60-mm Petri dishes to reach 50% confluence overnight.
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats over Ficoll gradients.31 PBMCs were adhered for 2 hr without serum, washed and incubated for 6 days in RPMI-1640 containing 100 U/ml penicillin G, 100 μg/ml streptomycin sulphate, 2 mm glutamine, 1 mm sodium pyruvate and 10 mm HEPES in the presence of 10% fetal calf serum prior to use.
Transient expression
The simian virus 40-based expression vector CDM32 was used for the transient expression of the cDNAs of all clones in COS-7 cells using the diethylaminoethyl–dextran method.25 The cDNAs (5 μg/3 ml Petri dish) used for transfection were FcγRI,25γ-chain,33 FcγRI/II chimaera,34 FcγRI-glycosylphosphatidylinositol (GPI)26 and Syk. All experiments were performed 2 or 3 days post-transfection, when surface expression is maximal.
Sheep erythrocyte coupling to PC and opsonization with CRP and IgG1
Sheep erythrocytes were coupled to PC using a modification of a previously described method for erythrocyte labelling.35 Briefly, 5 ml of 20 mg/ml bovine serum albumin (BSA) in 0·02 m sodium phosphate buffer (pH 4·5) was mixed with an equal volume of 2% (w/v) ethyl(dimethylaminopropylcarbodiimide) (EDC; Pierce and Warriner, Chester, UK) for 5 min. Forty milligrammes ρ-amino-phenyl-phosphorylcholine (PC, Sigma, Poole, Dorset, UK) was added and mixed overnight at room temperature. The PC-coupled BSA was subsequently purified on a Sephacryl 300 column and the major peak was stored at − 20°. PC-BSA and unlabelled aliquots of BSA were dialysed overnight against phosphate-buffered saline (PBS) containing 1 mm ethylenediaminetetraacetic acid. 2-Iminothiolane (Sigma) was added to a final concentration of 0·3 mm and incubated for 45 min at room temperature. The resultant mixture was filtered through a PD10 column pre-equilibrated with PBS and aliquots were stored at − 20°.
Sheep erythrocytes were washed four times in PBS. Sulphosuccinimidyl −4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (sulfo-SMCC; Pierce and Warriner, Chester, UK) was added to the cells (109/ml) at a final concentration of 0·5 mm and mixed for 1 hr at room temperature in the dark. After washing four times with PBS, the erythrocytes were finally mixed with 0·8 mg of either PC-BSA-sulphydryl (SH) or BSA-SH for 1 hr at room temperature, washed with PBS and resuspended at ∼5 × 108/ml.
Phagocytosis assay
To opsonize, PC-coupled erythrocytes (PCE) were washed twice in PBS and incubated at 108/ml with CRP (2 μg/ml in PBS containing 0·5 mm CaCl2; PBSC) or a subagglutinating dilution of rabbit anti-sheep erythrocyte stroma (Sigma) for 60 min at 4° and washed three times in PBSC. Opsonization/uptake of PCE was compared to uncoupled and BSA-coupled erythrocytes. Erythrocytes were added to transfected COS-7 cells in six-well plates containing sterile cover slips or PBMCs in eight-chamber slides and left for 2 hr at 37°. Unbound erythrocytes were washed away three times with PBS and hypotonic shock buffer (1 mm PBS pH 2·5) was added to half of the wells for 2 min. Cells were fixed with 2·5% glutaraldehyde in 0·2 m PBS pH 7·4. Internalized erythrocytes were visualized by staining for myeloperoxidase using hydrogen peroxide and σ-dianisidine as previously described.26 A total of 200 cells per condition were counted for phagocytic index (number of internalized erythrocytes per 100 cells) as well as the percentage of cells containing erythrocytes.
BIAcore
To confirm FcγRI as a CRP receptor, the extracellular portion of FcγRI was expressed as a GPI-anchored mutant which was cleaved from transfected COS cells by PI-PLC.36 The supernatant FcγRI-GPI was captured on the appropriately prepared BIAcore 2000 CM5 sensorchip (BIACORE, Stevenage, UK) using a specific monoclonal antibody against FcγRI as previously described.36
Ten microlitres of various concentrations of ligands for FcγRI were injected at 5–50 μl/min for 1 min and the kinetics of the interactions were analysed using BIA evaluation 2·1 software. All calculations were performed after subtraction from a reference flow cell (control) where an irrelevant primary antibody was coupled to the chip but was otherwise treated in an identical way to the experimental cell. This blank was used to account for bulk refractive index changes and non-specific binding.
In additional experiments the chimaeric FcγRI; (I.II.I37), was used and replaced FcγRI in this protocol.
Measurement of PLD activation in IFN-γ-treated U937 cells
U937 cells (106/ml) were treated with 200 ng/ml human IFN-γ and 0·185 MBq/ml [3H]palmitic acid in RPMI-1640 containing 10% (v/v) fetal calf serum for 18 hr. Cells were washed in ice-cold medium, resuspended at 2 × 106/ml, and held on ice or incubated with 10 μg/ml human monomeric IgG1 (Sigma) for 45 min at 4° to occupy surface FcγRI. The cells were diluted and centrifuged to remove unbound IgG and resuspended in ice-cold medium. Cross-linking antibody (goat anti-human IgG; Sigma) at 40 μg/ml or 20 μg/ml CRP was added in medium and butan-1-ol (0·3% final) was added as required, and incubated for 30 min at 37°.
PLD activity was measured by the trans-phosphatidylation assay as previously described.38 Briefly, cells were extracted by Bligh–Dyer phase separation. An aliquot of the lower organic phase was removed and dried down under vacuum. The samples were redissolved in 25 μl of chloroform/methanol (19 : 1, v/v), containing 40 μg of unlabelled phosphatidylbutanol (Lipid Products, Redhill, UK) as standard, and applied to pre-run, heat-activated thin-layer chromatography plates (20 × 20 cm, Silica gel 150A grooved plates, Whatman International Ltd, Maidstone, UK). The plates were developed in the organic phase of the solvent, ethyl acetate/2,2,4-trimethylpentane/acetic acid/water (11 : 5 : 2 : 10) for approximately 90 min, and the position of the phosphatidylbutanol product was detected using iodine vapour. [3H]Phosphatidylbutanol-containing silica indicated by the phosphatidylbutanol standard was then scraped into scintillation fluid and counted.
Results were calculated as a percentage of the total radioactivity incorporated in the lipids.
Statistical analyses
The Mann–Whitney U-test was performed using the spss package for Windows, release 6·1.
Results
CRP binds captured FcγRI in a calcium-dependent manner
The binding of CRP to the extracellular domain of FcγRI was analysed by BIAcore. For this, FcγRI-GPI was transiently expressed on COS cells. The extracellular domain of FcγRI was released by digestion with PIPLC36 and captured using specific monoclonal antibodies bound to the BIAcore sensor chip surface.36 Purified CRP (1 mg/ml) was injected over this surface and binding was compared to a sensor chip treated in an identical manner but lacking the extracellular domain of FcγRI. In the presence of 0·5 mm Ca2+, specific binding of CRP to FcγRI was demonstrated (Fig. 1). Direct measurements of the kinetics of association and dissociation of human CRP to FcγRI were consistent over four different concentrations of the ligand (Table 1) and over three flow rates (5, 20 and 50 μl/min, data not shown).
Figure 1.
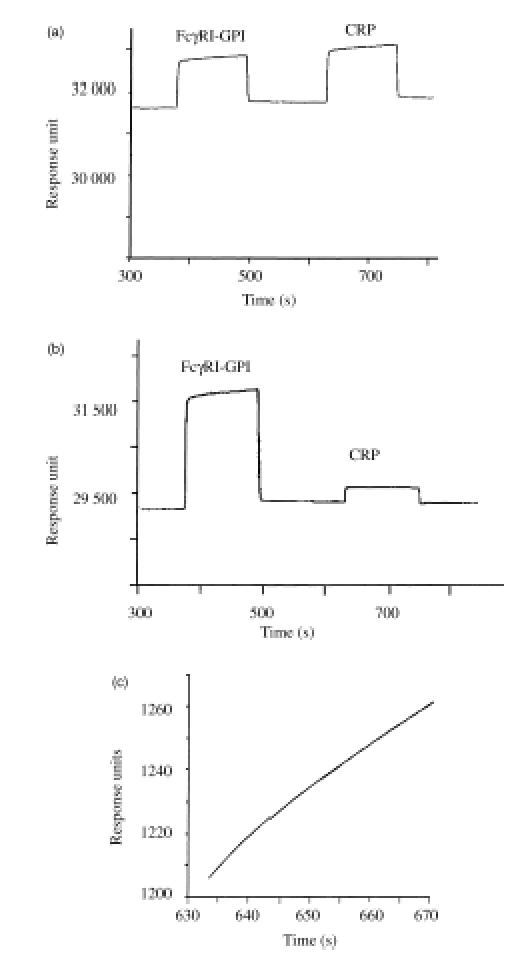
CRP binding to immobilized FcγRI on the BIAcore. (a) Sensogram showing sequential binding of FcγRI-GPI and CRP (1 mg/ml) to the GαM IgG1 and mouse anti-FcγRI (22/32) coated sensor chip surface. (b) Sensogram showing the same sequence as (a) in the absence of calcium, resulting in no binding of CRP to immobilized FcγRI. (c) The theoretical curve fit for the association of CRP for FcγRI (as determined by BIA evaluation 2·1 curve fit) directly overlays the actual curve from the sensogram in (a).
Table 1.
Representative kinetics of CRP binding to FcγRI immobilized on BIAcore at different concentrations
| Concn | Association constant (kass) | Dissociation constant (kdiss) | KD |
|---|---|---|---|
| 1·0 mg/ml | 9·72 × 106 | 4·53 × 10−3 | 4·7 × 10−10 |
| 0·75 mg/ml | 9·72 × 106 | 4·53 × 10−3 | 4·7 × 10−10 |
| 0·6 mg/ml | 7·81 × 106 | 5·01 × 10−3 | 6·4 × 10−10 |
| 0·5 mg/ml | 7·84 × 106 | 4·28 × 10−3 | 5·5 × 10−10 |
The mean (± SEM) kass and kdiss of CRP binding to FcγRI at 1 mg/ml were 6·10 × 106 (± 1·39 × 106) m−1 s−1 and 4·07 × 10−3 (± 0·41 × 10−3) s−1, respectively. The overall affinity calculated from these kinetics gave a mean KD of 0·81 × 10−9 (± 0·24 × 10−9) m.
In the absence of Ca2+, CRP no longer bound FcγRI (Fig. 1b) whereas the addition of 50 mm PC chloride did not affect FcγRI binding of CRP (data not shown). The observed rate of association matched that predicted for a simple A + B AB, type 1 association model with a χ2 of 0·015 (Fig. 1c) indicating that the stoichiometry matched one CRP binding to one Fc receptor. As a control, the homologous purified human SAP at equivalent concentrations and under equivalent conditions did not bind (data not shown). CRP did not bind to the control flow cell (with captured irrelevant antibody) under identical conditions.
CRP does not bind FcγRI in the presence of human IgG1
As FcγRI binds human IgG1, the effect of receptor occupancy by immunoglobulin on CRP binding was determined. Following capture of FcγRI on the chip surface, human IgG1 (1 mg/ml) was added to the flow cell to occupy the receptor (65·2 response units bound). Subsequent addition of CRP (1 mg/ml) failed to bind to the receptor occupied by IgG1 as shown by no detectable positive reponse units. As a control, CRP was added to an additional flow cell where the same experimental protocol was used but human IgG1 was omitted and CRP bound (11·8 response units) with the expected association and dissociation kinetics (data not shown). Pre-binding of CRP to FcγRI reduced but did not prevent IgG binding (30·1 response units) and had no effect on the kinetics of IgG1 binding to the receptor (KD = 2·5 × 10−9 m).
These data imply that prebound IgG1 is able to prevent binding of CRP to FcγRI whereas prebinding of CRP does not greatly inhibit binding of human IgG1.
Extracellular domain 2 of FcγRI is important for CRP binding
Previous studies have shown that extracellular domain 2 (middle domain) of FcγRI is essential for high-affinity binding to human monomeric IgG. Using surface plasmon resonance, the role of this domain was determined using the receptor chimaera (I.II.I37). This chimaera has domains 1 and 3 from FcγRI extracellular domains and domain 2 from FcγRIIa and has ability to bind IgG with low affinity.37 The FcγRIIa R131 allotype was used which, as recently reported,14 is the form which binds CRP more avidly. Nevertheless, no binding to CRP was observed (data not shown).
CRP opsonization of erythrocytes for phagocytosis by human PBMCs
In order to demonstrate CRP opsonization and subsequent phagocytosis in vitro, sheep erythrocytes were labelled with PC (PCE) and added to adherent PBMCs obtained from eight normal donors. This resulted in a significantly increased uptake (P = 0·002) compared to unopsonized PCE, BSA-conjugated or unconjugated erythrocytes (Fig. 2a–c). This system was also used to determine that CRP at 2 μg/ml was sufficient to cause opsonization without generating aggregates of erythrocytes, which occurred at higher CRP concentrations. A significant increase in the number of erythrocytes per 100 cells was also observed (P = 0·003, Fig. 2d).
Figure 2.

Phagocytosis of erythrocytes (E) opsonized with CRP or IgG1 by PBMC adherent cells. Day 6 adherent PBMCs were incubated with (a) erythrocytes, (b) BSA-coupled erythrocytes (BSAE), or (c) PC-BSA-conjugated erythrocytes (PCE) which were unopsonized (open bars) or had been preopsonized with either 2 μg/ml CRP (filled bars) or rabbit IgG1 anti-stromal antibody (hatched bars). After lysing uninternalized cells, the percentage phagocytosis was determined using light micoscopy. Each bar represents the mean ±SEM of three independent experiments. The phagocytic index was expressed as the number of erythrocytes/100 cells for PCE conjugated erythrocytes (d). Each bar represents the mean ±SEM of eight independent experiments.
CRP opsonized erythrocytes bind to COS-7 cells transfected with human FcγRI resulting in increased rosetting and phagocytosis
When COS-7 cells were transfected with FcγRI/II chimaera (containing an ITAM in the FcγRIIa cytoplasmic domain), IgG1 opsonized PCE were shown to rosette 77% ± 2·7 of the cells (mean ±SEM, Fig. 3a). Following lysis of uninternalized erythrocytes, 42% ± 6·4 of the cells showed significantly increased phagocytic activity (P = 0·003, Fig. 3b). When analysed by flow cytometry for FcγRI expression, 60–70% of cells were routinely positive (data not shown). A negative control transfection was performed with cells expressing FcγRI-GPI and, although IgG1 opsonized PCE rosetted these cells consistent with the lack of ITAM, no internalization was observed (Fig. 3a,b).
Figure 3.

Rosetting and phagocytosis of CRP- or IgG1-opsonized PCE by COS-7 cells transfected with FcγRI-II or FcγRI-GPI. COS-7 cells transfected with either FcγRI-II (open bars) or FcγRI-GPI (filled bars) were incubated with PCE or IgG1-opsonized PCE (a,b) and PCE or CRP-opsonized PCE (c,d). The percentage rosetted cells (a,c) and, after lysis of uninternalized erythrocytes, the percentage phagocytosis (b,d) were determined using light microscopy. In addition, the number of internalized erythrocytes per 100 cells was determined in FcγRI-II chimaera-transfected COS-7 cells after incubation with PCE, CRP-PCE and IgG1-PCE (e). The data represent the mean ±SEM of six independent experiments.
Upon the addition of CRP-opsonized PCE, similar significant increases in rosetting (34·7% ± 8·23) and phagocytosis (24% ± 4·1, P = 0·006) compared to PCE (17% ± 3·2 and 7% ± 2·6, respectively) were observed. Once again, CRP-opsonized PCE rosetted COS-7 cells expressing FcγRI-GPI but no internalization was observed (Fig. 3c,d). A significant increase in the number of erythrocytes per 100 cells was also observed (P = 0·025, Fig. 3e).
When cells were co-transfected with FcγRI and γ-chain similar results were obtained. Again the rosetting with CRP-opsonized PCE was less intense than that seen with IgG1-opsonised PCE, as was phagocytosis, suggesting that the limiting stage here was rosetting. However, we were limited to 2 μg/ml which would be suboptimal for CRP opsonization to PC-coated erythrocytes. Transfection of either component alone did not allow CRP-mediated phagocytosis, although rosetting was seen with cells expressing FcγRI alone for PCE opsonized with either CRP or IgG1 (Fig. 4a.c). A significant increase in phagocytic activity was observed with both CRP- and IgG1-opsonized PCE (P = 0·021, 0·021 and 0·043, Fig. 4b,d,e, respectively) compared to PCE alone.
Figure 4.

Rosetting and phagocytosis of CRP- or IgG1-opsonized PCE by COS-7 cells transfected with FcγRI ±γ-chain. COS-7 cells transfected with either FcγRI and γ-chain (open bars) or FcγRI alone (filled bars) were incubated with PCE or IgG1-opsonized PCE (a,b) and PCE or CRP-opsonized PCE (c,d). The percentage rosetted cells (a,c) and, after lysis of uninternalized erythrocytes, the percentage phagocytosis (b,d) were determined using light microscopy. In addition, the number of internalized erythrocytes per 100 cells was determined in FcγRI and γ-chain-transfected COS-7 cells after incubation with PCE, CRP-PCE and IgG1-PCE (e). The data represent the mean ±SEM of four independent experiments.
CRP binding to FcγRI-expressing U937 cells activates the PLD pathway
It has been reported that CRP binds to the surface of various human monocytic cell lines in addition to FcγRI/II transfected COS-7 cells. It is not known however, if on binding, CRP is able to cross-link FcγRI and activate these cells. Specific aggregation of FcγRI but not FcγRIIa in IFN-γ-primed U937 cells has recently been shown to activate PLD.38
IFN-γ-treated U937 cells were incubated with either human IgG1 followed by anti-mouse IgG1 to aggregate IgG-FcγRI complexes, or with CRP alone and PLD activity was measured.
Cells without either IgG1 or CRP gave a mean (± SEM) basal level of PLD activity of 0·28 (± 0·05)% [3H]phosphatidylbutanol/total [3H]palmitate incorporated). In cells following FcγRI aggregation, PLD activity rose to 0·53 (± 0·06)% (P = 0·006). Upon addition of CRP, basal levels were significantly increased to 0·58% (± 0·10, P = 0·032, Fig. 5), indicating that CRP not only binds to, but also aggregates FcγRI to activate the PLD pathway. To investigate whether human IgG1 blocks the cross-linking effect of CRP on these cells, as was seen in the BIAcore studies, CRP was added to cells loaded with monomeric IgG1. In the presence of IgG1, CRP was no longer able to activate PLD (0·23 ± 0·02%) suggesting that it was no longer able to bind to FcγRI (data not shown).
Figure 5.

Induction of PLD activity by CRP or cross-linked IgG1 in IFN-γ treated U937 cells. IFN-γ-treated U937 cells were incubated with medium (basal), CRP (20 μg/ml) or IgG1 (40 μg/ml followed by F(ab)′2 anti-IgG). Following the addition of [3H]butanol, incorporation of phosphatidylbutanol was measured and expressed as a percentage of total [3H]palmitate incorporation. The bars represent the mean ±SEM of four independent experiments.
Discussion
Previous studies have shown that transfected cells expressing FcγRI or FcγRIIa are capable of binding CRP and that IgG could partially inhibit this binding. In this study we have extended these observations to show direct, high-affinity interaction between purified FcγRI and CRP. We have demonstrated binding of CRP to the extracellular portions of FcγRI using surface plasmon resonance (SPR) analysis, with an affinity that was higher than the affinity of IgG for FcγRI using the same system. Pre-loading FcγRI with IgG, inhibited CRP binding. This was observed previously by others and here in both cell-based and BIAcore experiments. IgG binding, on the other hand, was only slightly reduced by CRP binding. This suggests that IgG either masks the CRP-binding site or that IgG induces a conformational change in FcγRI which prevents further binding of CRP.
The presence of calcium in the CRP molecule alters configuration near the PC-binding face, however, other regions may also be affected and it has been demonstrated using infrared spectroscopy that considerable changes occur in the secondary structure.39 It was more recently shown in the crystal structure that removal of both calcium ions in a subunit led to the residues 140–150 forming a large loop away from the body of the molecule and the movement of several other loops in the region.3 Therefore, the calcium dependency of CRP binding to FcγRI is not too surprising, although there is no clear evidence of which region of CRP definitively binds to the Fc receptors nor whether this shows structural alteration in the presence or absence of calcium. It was also demonstrated that binding of PC did not alter structural features in the crystal structures,40 which may explain why PC did not alter the interaction between CRP and the Fc receptor. It does, however, indicate that the FcγRI-binding region is not close to the calcium-dependent site that interacts with PC containing micro-organisms. Whether higher affinity ligands, or ligands that can cross-link different subunits of CRP, would have any affect on the interaction is not clear. SAP was used as a control protein which is homologous to CRP and which, as a purified decamer structure at least, is not expected to interact with Fc receptors. Despite some evidence for Fc receptor binding, evidence for specific cell receptors for SAP is weak despite the demonstration of binding to a variety of carbohydrate structures.30 No binding of SAP to FcγRI on the BIAcore was observed. This observation should not necessarily be taken to mean that SAP in its physiological state in serum does not interact with Fc receptors since there is evidence that purified SAP may exist as a decamer while the serum form exists as a pentamer, exposing a potential Fc receptor-binding face.41
One of the major questions raised in this study was whether CRP could use FcγRI to induce phagocytosis. We were able for the first time to demonstrate that particles opsonized by CRP can be phagocytosed through binding to FcγRI and γ-chain and also result in the activation of PLD through this receptor. The uptake of opsonized erythrocytes was less impressive than that seen with IgG but the opsonization with CRP was limited to 2 μg/ml CRP, since higher levels caused aggregation of the erythrocytes. It is likely that higher concentrations of CRP might have increased the density of CRP opsonization and thus increased both rosetting and internalization. However, it is worth noting that CRP can act as an opsonin at normal serum concentrations. It would be misleading therefore to compare directly the relative abilities of CRP and IgG to cause phagocytosis from these studies.
The effect of substituting the proposed CRP binding R131 allotype14 domain 2 of FcγRIIa into FcγRI in place of the second domain was to prevent CRP binding completely, although binding was still seen to IgG at high concentrations.37 This loss of binding suggests that FcγRIIa is not a high-affinity ligand for CRP. It is possible, however, that CRP interaction with FcγRIIa domain 2 might be prevented by the presence of the extra FcγRI domain. Further studies using SPR to investigate CRP and Fc receptor interaction are vital to resolving such questions. We were able, however, to show that our erythrocytes coated with CRP form rosettes around cells transfected with FcγRIIa R131 but not when they were transfected with FcγRIIa H131 (not shown). These data are in agreement with those of Du Clos et al.15 who used zymosan particles. Further experiments are needed to address the question of whether other receptors are involved or whether CRP needs to be presented in a complexed form. It is probable, however, that CRP as well as IgG binds to the C'E loop of the Fc receptor family,12,42 since this loop appears to interact with IgG at several sites including the region 234–239 LLGGPS. This is homologous to the proposed Fc receptor interaction site on CRP: YLGG.11
Neutrophil phagocytosis of CRP-coated particles was demonstrated to occur only when the neutrophils were activated.43,44 Since neutrophils normally have low FcγRI and high FcγRIIa but increase FcγRI on activation, this suggests that FcγRIIa is poor or unable to mediate CRP-dependent phagocytosis and that FcγRI is responsible for neutrophil phagocytosis. It has also been reported recently that up-regulation of FcγRI improves phagocytic potential for IgG-opsonized bacteria by neutrophils.45 Studies should re-examine CRP-mediated neutrophil phagocytosis and in particular examine the role of IFN-γ (which also up-regulates neutrophil expression of FcγRI). Indeed CRP–PC–BSA complexes did not activate neutrophils in comparison with IgG–PC–BSA complexes,46 perhaps suggesting that the two complexes are internalized by different receptors.
The data obtained from the PLD assays, on cells maintained under conditions that only give PLD activation in response to IgG cross-linking of FcγRI and not FcγRIIa, demonstrate that CRP probably activates through FcγRI. It is also possible, however, that other receptors are involved. PLD activation due to CRP in the absence of cross-linking antibody is probably to be expected, since the pentameric structure of CRP is likely to result in the aggregation of FcγRI. This activation, however, was inhibited by the addition of monomeric IgG1. Control of direct effects of CRP on cellular responses may be at the level of competition between IgG and CRP. CRP activation of PLD, however, implies that responses to CRP may be similar to those observed with IgG. One consequence of PLD activation would also be activation of MAP kinases although in neutrophils CRP reduced p38 kinase activity following fMLP activation.47
Fc receptors play an important co-ordinating role in immune responses. A number of recent reports have highlighted important immunological functions for uptake through FcγRI and γ-chain. Ligation of FcγRI leads to reversal of pro-inflammatory responses of macrophages for example by the induction of interleukin-10 and inhibition of interleukin-12.48 There is no strong evidence that CRP influences cytokine synthesis, but this warrants reinvestigation in the light of improved knowledge of receptor interaction and the fact that FcγRI-mediated antigen uptake has been shown to direct strong presentation of antigen to T and B cells.21,22,49 Some of these properties are due to FcγRI chain involvement rather than γ-chain signalling, for example endocytosis and transfer to late endosome/lysosome compartments and improved presentation.49 Thus, investigation of the ability of CRP to induce endocytosis through receptors and receptor constructs would be interesting. Our results, however, have demonstrated that CRP may have an important role in helping the innate system direct the acquired immune system following phagocytosis and signalling through FcγRI/γ-chain.
Acknowledgments
We would like to thank the Wellcome Trust for supporting this project.
Abbreviations
- CRP
C-reactive protein
- FcγRI
high-affinity receptor for IgG
- FcγRIIa
low-affinity receptor for IgG
- GPI
glycosylphosphatidylinositol
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate-buffered saline
- PC
phosphorylcholine
- PC-BSA
phosphorylcholine coupled to bovine serum albumin
- PCE
phosphorylcholine-coupled erythrocytes
- PLD
phospholipase D
- SAP
serum amyloid P
References
- 1.Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today. 1994;15:81–8. doi: 10.1016/0167-5699(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 2.Murphy TM, Baum LL, Beaman KD. Extrahepatic transcription of human C-reactive protein. J Exp Med. 1991;173:495–8. doi: 10.1084/jem.173.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shrive AK, Cheetham GMT, Holden D, et al. Three dimensional structure of human CRP. Nature, Struct Biol. 1996;3:346–54. doi: 10.1038/nsb0496-346. [DOI] [PubMed] [Google Scholar]
- 4.Kindmark CO. In vitro binding of human CRP by some pathogenic bacteria and zymosan. Clin Exp Immunol. 1972;11:283–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Culley FJ, Harris RA, Kaye PM, McAdam KPW, Raynes JG. CRP binds to a novel ligand on Leishmania donovani and increases uptake into human macrophages. J Immunol. 1996;156:4691–6. [PubMed] [Google Scholar]
- 6.Kindmark CO. Stimulating effect of C-reactive protein on phagocytosis of various species of pathogenic bacteria. Clin Exp Immunol. 1971;8:941–8. [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson MD, Gray CA, Shankland GS. Opsonic effect of C-reactive protein on phagocytosis and intracellular killing of virulent and attenuated strains of Candida albicans by human neutrophils. FEMS, Microbiol Immunol. 1991;3:341–4. doi: 10.1111/j.1574-6968.1991.tb04259.x. [DOI] [PubMed] [Google Scholar]
- 8.Szalai AJ, Briles DE, Volanakis JE. Role of complement in C-reactive-protein-mediated protection of mice from Streptococcus pneumoniae. Infect Immun. 1996;64:4850–3. doi: 10.1128/iai.64.11.4850-4853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chudwin D, Artrip SG, Korenblit A, Schiffman G, Rao S. Correlation of serum opsonins with in vitro phagocytosis of Streptococcus pneumoniae. Infect Immun. 1985;50:213–17. doi: 10.1128/iai.50.1.213-217.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller H, Fehr J. Binding of C-reactive protein to human polymorphonuclear leukocytes: evidence for association of binding sites with Fc receptors. J Immunol. 1986;136:2202–7. [PubMed] [Google Scholar]
- 11.Marnell LL, Mold C, Volzer MA, Burlingame RW, Du Clos TW. CRP binds to FcγRI in transfected cos cells. J Immunol. 1995;155:2185–93. [PubMed] [Google Scholar]
- 12.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–73. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 13.Crowell RE, Du Clos TW, Montoya G, Heaphy E, Mold C. CRP receptors on the human monocytic cell line U-937. J Immunol. 1991;147:3445–51. [PubMed] [Google Scholar]
- 14.Stein MP, Edberg JC, Kimberley PP, Mangan EK, Bharadwaj D, Mold C, Du Clos TW. CRP binding to FcγRIIa on human monocytes and neutrophils is allele specific. J Clin Invest. 2000;105:369–76. doi: 10.1172/JCI7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du Clos TW, Mold C, Edberg JC, Kimberley RP. Reply to ‘Human CRP does not bind to FcγRIIa on phagocytic cells’. J Clin Invest. 2001;107:643. [Google Scholar]
- 16.Saeland E, van Royen A, Hendriksen K, Vile-Weekhout H, Rijkers GT, Sanders LAM, van der Winkel JGJ. C-reactive protein does not bind to FcγRIIa on pahgocytic cells. J Clin Invest. 2001;107:641–2. doi: 10.1172/JCI12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tebo JM, Mortensen RF. Characterisation and isolation of a CRP receptor from the human monocytic cell line U937. J Immunol. 1990;144:231–8. [PubMed] [Google Scholar]
- 18.Zhong W, Zen Q, Tebo J, Schlottmann K, Coggeshall M, Mortensen RF. Effect of human CRP on chemokine and chemotactic factor-induced chemotaxis and signalling. J Immunol. 1998;161:2533–40. [PubMed] [Google Scholar]
- 19.Ravetch JV, Kinet JP. Fc receptors. Annu Rev Immunol. 1991;9:457–92. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 20.Gosselin EJ, Wardell K, Gosselin DR, Alter N, Fisher JL, Guyre PM. Enhanced antigen presentation using FcγR (monocyte/macrophage) specific immunogens. J Immunol. 1999;149:3477–81. [PubMed] [Google Scholar]
- 21.Heijnen IAFM, van Vugt MJ, Fanger NA, et al. Antigen targeting to myeloid specific human FcγRI /CD64 triggers enhanced Antibody responses in transgenic mice. J Clin Invest. 1996;97:331–8. doi: 10.1172/JCI118420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Goldstein J, Grazian RF, He J, O'Shea JK, Deo Y, Guyre GM. FcγRI targetted fusion proteins result in efficient presentation by human monocytes of antigenic and antagonistic T cell epitopes. J Clin Invest. 1996;98:2001–7. doi: 10.1172/JCI119004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heller T, Gesture JE, Schmidt RE, Los A, Bausch W, Kohl J. Fc receptor type I for IgG on macrophages and complement mediate the inflammatory response in immune complex peritonitis. J Immunol. 1999;162:5657–61. [PubMed] [Google Scholar]
- 24.Indik ZK, Park JG, Hunter S, Schreiber AD. Structure/function relationships of Fcγ receptors in phagocytosis. Semin Immunol. 1995;7:45–54. doi: 10.1016/1044-5323(95)90007-1. [DOI] [PubMed] [Google Scholar]
- 25.Allen JM, Seed B. Isolation and expression of functional high-affinity Fc receptor complementary DNAs. Science. 1989;243:378–81. doi: 10.1126/science.2911749. [DOI] [PubMed] [Google Scholar]
- 26.Davis W, Harrison PT, Hutchinson MJ, Allen JM. Two distinct regions of FcγRI initiate separate signalling pathways involved in endocytosis and phagocytosis. EMBO J. 1995;14:432–41. doi: 10.1002/j.1460-2075.1995.tb07019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daëron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–34. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 28.Mold C, Gresham D, Du Clos TW. Serum amyloid P component and C-reactive protein mediate phagocytosis through murine FcγRs. J Immunol. 2001;166:1200–5. doi: 10.4049/jimmunol.166.2.1200. [DOI] [PubMed] [Google Scholar]
- 29.Volanakis JE, Clements WL, Schrohenloher RE. CRP purification by affinity chromatography and physiochemical characterisation. J Immunol Methods. 1978;23:285–95. [Google Scholar]
- 30.Loveless RW, O'Sullivan G, Raynes JG, Yuen CT, Feizi T. Human SAP is a multispecific adhesive protein whose ligands include 6-phosphorylated mannose and the 3-sulphated saccharides galactose, N-acetylgalactosamine and glucuronic acid. EMBO J. 1992;11:813–19. doi: 10.1002/j.1460-2075.1992.tb05118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Böyum A. Separation of lymphocytes, lymphocyte subgroups and monocytes: a review. Lymphology. 1977;10:71–6. [PubMed] [Google Scholar]
- 32.Seed B, Aruffo A. Molecular cloning of the CD2 antigen, the T-cell erythrocyte receptor, by a rapid immunoselection procedure. Proc Natl Acad Sci USA. 1987;84:3365–9. doi: 10.1073/pnas.84.10.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Küster H, Thompson H, Kinet JP. Characterization and expression of the gene for the human Fc receptor gamma subunit. Definition of a new gene family. J Biol Chem. 1990;265:6448–52. [PubMed] [Google Scholar]
- 34.Hutchinson MJ, Harrison PT, Floto RA, Allen JM. FcγR-mediated phagocytosis requires tyrosine kinase activity and is ligand independent. Eur J Immunol. 1995;25:481–7. doi: 10.1002/eji.1830250226. [DOI] [PubMed] [Google Scholar]
- 35.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J Exp Med. 1997;185:1977–85. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrison PT, Campbell IW, Allen JM. Use of GPI-anchored proteins to study biomolecular interactions by surface plasmon resonance. FEBS Lett. 1998;422:301–6. doi: 10.1016/s0014-5793(98)00027-1. [DOI] [PubMed] [Google Scholar]
- 37.Harrison PT, Allen JM. High affinity IgG binding by FcγRI (CD64) is modulated by two distinct IgSF domains and the transmembrane domain of the receptor. Protein Engineering. 1998;11:225–32. doi: 10.1093/protein/11.3.225. [DOI] [PubMed] [Google Scholar]
- 38.Melendez A, Floto RA, Gillooly DJ, Harnett MH, Allen JM. FcγRI coupling to phospholipase D initiates sphingosine kinase-mediated calcium mobilisation and vesicular trafficking. J Biol Chem. 1998;273:9393–402. doi: 10.1074/jbc.273.16.9393. [DOI] [PubMed] [Google Scholar]
- 39.Dong A, Caughey WS, Du Clos TW. Effects of calcium, magnesium, and phosphorylcholine on secondary structures of human C-reactive protein and serum amyloid P component observed by infrared spectroscopy. J Biol Chem. 1994;269:6424–30. [PubMed] [Google Scholar]
- 40.Thompson D, Pepys MB, Wood SP. The physiological structure of human CRP and its complex with phosphocholine. Structure. 1999;7:169–77. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 41.Sörensen IJ, Andersen O, Nielsen EH, Svehag SE. Native human serum amyloid P component is a single pentamer. Scand J Immunol. 1995;41:263–7. doi: 10.1111/j.1365-3083.1995.tb03562.x. [DOI] [PubMed] [Google Scholar]
- 42.Maxwell KF, Powell MS, Hulett MD, Barton PA, McKenzie IFC, Garrett TPJ, Hogarth PM. Crystal structure of the human leukocyte receptor FcγRIIa. Nature Structural Biol. 1999;6:437–42. doi: 10.1038/8241. [DOI] [PubMed] [Google Scholar]
- 43.Kilpatrick JM, Volanakis JE. Opsonic properties of human CRP. Stimulation by PMA enables human neutrophils to phagocytose CRP coated cells. J Immunol. 1985;134:3364–70. [PubMed] [Google Scholar]
- 44.Kilpatrick JM, Gresham HD, Griffin FM, Jr, Volanakis JE. Peripheral blood mononuclear leukocytes release a mediator (s) that induces phagocytosis of C-reactive protein-coated cells by polymorphonuclear leukocytes. J Leuk Biol. 1987;41:150–5. doi: 10.1002/jlb.41.2.150. [DOI] [PubMed] [Google Scholar]
- 45.Fjaertoft G, Hakansson L, Ewald U, Foucard T, Venge P. Neutrophils from term and preterm newborn infants express the high affinity FcγRI (CD64) during bacterial infections. Pediatr Res. 1999;45:871–6. doi: 10.1203/00006450-199906000-00016. [DOI] [PubMed] [Google Scholar]
- 46.Romero IR, Morris C, Rodriguez M, Du Clos TW, Mold C. Inflammatory potential of C-reactive protein complexes compared to immune complexes. Clin Immunol Immunopathol. 1998;87:155–62. doi: 10.1006/clin.1997.4516. [DOI] [PubMed] [Google Scholar]
- 47.Heuertz RM, Trocomi SM, Ezekiel UR, Webster RO. CRP inhibits chemotactic peptide induced p38 MAP kinase activity and human neutrophil movement. J Biol Chem. 1999;274:17968–74. doi: 10.1074/jbc.274.25.17968. [DOI] [PubMed] [Google Scholar]
- 48.Sutterwala FS, Noel GJ, Salgame P, Mosser DA. Reversal of proinflammatory responses by ligating the macrophage FcγRI. J Exp Med. 1998;188:217–22. doi: 10.1084/jem.188.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Vugt M, Kleijmeer MJ, Keler T, Zeelenberg I, van Dijk MA, Leussen JHW, Geuze HJ, van der Winkel JGJ. The FcγRIa (CD64) ligand binding chain triggers MHCII antigen presentation independently of its associated FcR γ chain. Blood. 1999;94:808–17. [PubMed] [Google Scholar]