

Abstract
Tumour antigen presentation by dendritic cells (DCs) to T cells in lymphoid organs is crucial for induction of anti-tumour immune responses. It has been previously reported that tumour necrosis factor-α (TNF-α) is required for DC activation and subsequent induction of optimal immune responses, and thus DCs for anti-tumour vaccination are often generated by culture in exogenous TNF-α. In the present study, we investigated the effect on anti-tumour immunity of vaccination with Mut1 tumour peptide-pulsed DCs engineered to express a TNF-α transgene. Our data shows that transfection of DCs with recombinant adenovirus AdV-TNF-α resulted in greater maturation of the DCs than occurred with control DCs cultured in exogenous TNF-α, as determined by up-regulated expression of pro-inflammatory cytokines (e.g. interleukins 1β and 18), chemokines [e.g. interferon-γ-inducible protein-10 and macrophage inflammatory protein-1β (MIP-1β)], the CC chemokine receptor CCR7, and immunologically important cell surface molecules (CD40, CD86 and intercellular adhesion molecule-1). These transgenic DCs stimulated stronger allogeneic T-cell responses in vitro and T-cell activation in vivo; displayed 2.4-fold enhanced chemotactic responses to the MIP-3β in vitro (P<0.05); and, perhaps most importantly, trafficked into the draining lymph nodes dramatically (seven-fold, P<0.01) more efficiently than the control DCs. Our data also demonstrate that vaccination of mice with Mut1 peptide-pulsed, AdV-TNF-α-transfected DCs stimulated more efficient in vitro Mut1-specific CD8+ cytotoxic T-cell responses and solid tumour immunity in vivo, when compared to the in vitro TNF-α-cultivated DCs. Thus, DCs engineered to secrete TNF-α may offer a new strategy in DC cancer vaccines.
Introduction
Cytotoxic T lymphocytes (CTLs) play a major role in the rejection of immunogenic tumours.1 Classically, CTLs target tumours through recognition of endogenous antigen peptides2 presented in the context of a major histocompatibility complex (MHC) class I molecule. However, there is evidence for an exogenous pathway whereby antigens that are not expected to gain access to the cytoplasm are presented on MHC class I molecules.3 A striking example of this is the in vivo phenomenon of cross-priming, whereby antigens from donor cells are acquired by host antigen-presenting cells (APCs) and presented on MHC class I molecules in an appropriate context of co-stimulation.4 Delivery of exogenous antigen to the endogenous MHC class I-restricted processing pathway of APCs is a critical challenge in cancer vaccine design.
Dendritic cells (DCs) are one of the most potent APCs. They migrate as precursors from the bone marrow into various organs, where they usually reside in an inactive state. However, during this regional residency, these cells efficiently endocytose and process antigens.5 Upon activation they undergo a differentiation process that down-regulates further antigen-processing capacity but enhances their expression of MHC, co-stimulatory, and other molecules important for successful antigen presentation, after which they migrate to the lymphoid organs to interact with or activate naive T cells.6 Because of the critical roles of DCs in the generation of primary immune responses, an important avenue of investigation is their potential for modulating immunological functions such as the induction of tolerance or tumour immunity. Recently, it has been shown that DCs pulsed with tumour-derived MHC class I-restricted peptides or tumour lysates are able to induce significant CTL-dependent anti-tumour immune responses both in vitro and in vivo.7,8 However, the therapeutic efficiency of these DC vaccine strategies has been quite limited, since they have protected against rechallenge with only small numbers of parental tumour cells or inhibited very early stage-established tumours. Thus, a strategic goal of current cancer vaccine research has become the induction of stronger tumour-specific CTL responses.
The maturational processes of DCs are efficiently regulated, such that these cells can achieve different states of activation/maturation and thereby different functional properties,8 depending on the precise nature of the signals they receive from their microenvironment. A number of cytokines that can be produced by DCs themselves (or by other cells within the local microenvironment) can significantly affect DC function at various levels, including their viability, morphology, migration, expression of MHC and accessory molecules, and binding and processing of antigen peptides.9 For example, granulocyte–macrophage colony-stimulating factor (GM-CSF), interleukin-4 (IL-4) and interferon-γ (IFN-γ), and inflammatory stimuli such as IL-1, IL-6 and tumour necrosis factor-α (TNF-α)9–11 are able to stimulate DCs to mature into cells with a strong T-cell stimulatory potential. Of perhaps more importance to cancer immunology, is the demonstration by Labeur et al. that the induction of anti-tumour immunity by DC vaccines is correlated with the maturation stage(s) of the DCs.12 In this respect, TNF-α appears to have profound effects on DC function, because it contributes to their activation,13 maturation,14 and migration to, and accumulation within, draining lymph nodes,15 and significantly reduces IL-10-mediated inhibition of DC development and function.16 Indeed, culture of DC with TNF-α prior to anti-tumour vaccination has been shown to induce a degree of anti-tumour immunity, although stronger anti-tumour responses were induced by treating the DC with ‘CpG’-containing oligonucleotides.14
Given these effects of TNF-α on DC, we wished to determine whether enforced expression of TNF-α by DCs that are carrying and presenting tumour peptides would further augment their ability to induce anti-tumour immunity in a mouse model system. Thus, we used adenovirus-mediated gene transfer to engineer DCs to secrete TNF-α, then pulsed the cells with an important MHC class I-restricted tumour peptide (Mut1) and compared the anti-tumour immunity induced by vaccination with these cells with that induced by DC that had been treated in vitro with TNF-α.
Materials and methods
Cell lines, antibodies, chemokines, peptides and animals
The 3LL cell line is a poorly immunogenic Lewis lung carcinoma line, while the EL4 cell line is a T-cell lymphoma line of C57BL/6 mouse (H-2Kb) origin; each was maintained in Dulbecco's modified Eagle's minimum essential medium (DMEM; Gibco, Gaithersburg, MD) supplemented with 10% fetal calf serum (FCS). Monoclonal rat anti-mouse H-2Kb, Iab, CD3, CD4, CD8, CD11b, CD11c, CD25, CD40, CD80, CD86 and intercellular adhesion molecule-1 (ICAM-1) antibodies, the fluorescein isothiocyanate (FITC) -conjugated anti-CD3 and phycoerythrin (PE) -conjugated anti-CD25 antibodies, and the recombinant soluble dimeric H-2Kb/immunoglobulin fusion protein were all purchased from Pharmingen (San Diego, CA). The FITC-conjugated goat anti-rat immunoglobulin G (IgG) and rabbit anti-mouse IgG1 antibodies were purchased from Bio/Can Scientific (Mississauga, Ontario, Canada). Recombinant mouse IL-2, IL-4, GM-CSF, macrophage imflammatory protein-3β (MIP-3β) and TNF-α were purchased from Endogen (Woburn, MA) or R & D Systems Inc. (Minneapolis, MN). The MHC class I-restricted Mut1 peptide (FEQNTAQP), comprising amino acids 52–59 of the mutated connexin 37 protein expressed by 3LL tumour cells,17 was synthesized by Multiple Peptide Systems (San Diego, CA). Female C57BL/6 (H-2Kb) and BALB/c (H-2Kd) mice were obtained from Charles River Laboratories (St Laurent, Quebec, Canada), and TNF-α knockout C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were housed in the animal facility of the Saskatoon Cancer Center; all animal experiments were carried out according to the guidelines of the Canadian Council for Animal Care.
Recombinant adenoviral vectors
The recombinant adenoviral vectors AdV-TNF-α, AdV-LacZ and AdV-pLpA were constructed as reported previously.18 AdV-TNF-α and AdV-LacZ express TNF-α and the Escherichia coliβ-galactosidase, respectively. The parental plasmid, AdV-pLpA (i.e. with no gene insert) was used as a control adenoviral vector. These E1-deleted replication-deficient recombinant adenoviruses were amplified in 293 cells, purified by caesium chloride ultracentrifugation and stored at −80°.
Generation of DCs
A slightly modified procedure from that described previously19 was used to generate DCs from bone marrow cultures. Briefly, bone marrrow cells prepared from the femurs and tibias of normal C57BL/6 mice were depleted of red blood cells with 0·84% ammonium chloride and plated in DC culture medium [DMEM plus 10% FCS, GM-CSF (10 ng/ml) and IL-4 (10 ng/ml)]. On day 3, the non-adherent granulocytes, and T and B cells were gently removed and fresh media were added, and 2 days later the loosely adherent proliferating DC aggregates were dislodged and replated. On day 7, the non-adherent, relatively mature DCs20 were harvested and used for in vitro AdV-TNF-α transfection or for in vitro cultivation in TNF-α-containing medium. The DCs generated in this manner displayed typical morphological features of DCs (i.e. numerous dendritic processes) and also significant expression of MHC class I (H-2Kb) and II (Iab) antigens, co-stimulatory molecules (CD40, CD80 and CD86) and adhesion molecules (ICAM-1, CD11b, and CD11c) (data not shown).
Adenoviral transfection/TNF-α treatments of DC
We transfected DCs with either E1-deleted control adenovirus (DCpLpA) or TNF-α gene recombinant E1-deleted adenovirus (DCAdVTNF-α). As a control for chromosomal TNF-α gene expression versus AdV-TNF-α transgene expression, in parallel we transfected DCs that had been generated from TNF-α knock-out mice (KODCAdVTNF-α). In order to test the amenability of DCs to adenoviral infection, serial dilutions of AdV-LacZ stock (2×1010 plaque-forming units/ml) were added to triplicate cultures of DCs in 96-well plates (1×105 cells/well) to form different multiplicities of infection (MOI). The cells were incubated with the adenovirus in 293 serum-free medium (Gibco) for 2 hr at 37°, then the medium was replaced with DMEM/10% FCS and the cells were incubated for an additional 24 h at 37°. To assess β-galactosidase expression17 the cells were fixed in formaldehyde/glutaraldehyde, then stained and counter-stained with X-gal and nuclear fast red, respectively. The proportions of positive (i.e. blue-staining) cells were determined from triplicate wells and taken as the percentage of transduction. Control DCs transfected with AdV-pLpA did not exhibit any intrinsic β-galactosidase activity or false-positive staining. We observed a dose-dependent response to the adenoviral infections, with maximal staining (82%) at a MOI of ≥100. Therefore, a MOI of 100 was selected for transfection of DCs with AdV-TNF-α in this study. To enact this, following viral adsorption for 1 h at 37° in six-well culture plates, the DC culture medium was replaced with DMEM/10% FCS and the cells were incubated for another 24 hr at 37°. The culture supernatants of the transfected DCs were assayed for TNF-α by enzyme-linked immunosorbent assay (ELISA). The means of TNF-α concentration in the supernatants of unstimulated DC and DCAdVTNF-α were 0·05 and 10 ng/ml, respectively, as determined by ELISA. Since 10 ng/ml TNF-α is commonly employed to stimulate DC maturation in vitro,11,21,22 and this was also the level of TNF-α secreted by our DCAdvTNF, we selected this dose of exogenous TNF-α for our DC treatments. DCs cultivated for one additional day in medium supplemented with 10 ng/ml recombinant TNF-α were termed DCTNF-α.
Immunophenotypic analysis
For phenotypic analyses by flow cytometry, DCs were stained for 30 min on ice with antibodies specific for H-2Kb, Iab, CD11b, CD11c, CD40, CD80, CD86 or ICAM-1 (each, 5 µg/ml), washed three times in phosphate-buffered saline (PBS), and then incubated for an additional 30 min on ice with FITC-conjugated goat anti-rat IgG antibody (1 : 60). After three more washes with PBS, the cells were analysed by flow cytometry. Isotype-matched monoclonal antibodies were used as controls.
RNAse protection assay
We used RNAse protection assays (RiboQuant Multi-Probe kit; Pharmingen) to examine the effects of TNF-α on the expression by the DCs of mRNA for multiple cellular markers. RNA was extracted from the cells using a commercial kit, and [32P]UTP (Amersham Canada Ltd, Oakville, Ontario, Canada) -labelled probes were generated by in vitro transcription of cytokine/chemokine-related multiprobe template sets (Pharmingen) using T7 RNA polymerase. The labelled probes were purified by phenol–chloroform extraction and ethanol precipitation and adjusted to 3×105 counts per minute (c.p.m.)/μl, then hybridized to the RNA samples (5 µg each). The reactions were subsequently digested with RNase, followed by Proteinase K treatment and phenol–chloroform extraction. After ethanol precipitation with 4 m ammonium acetate, the protected samples were resuspended in 1× loading buffer and realized on 5·7% acrylamide–bisacrylamide urea gels. The gels were absorbed to filter paper, dried under vacuum, and exposed to Kodak X-AR film with intensifying screens at −80°. The relative expressions of cytokine-, chemokine- and chemokine receptor-encoding mRNA were assessed by scanning densitometry (Molecular Dynamics, Sunnyvale, CA), using only autoradiograph signals within the linear signal density range of the film, and then normalized using the housekeeping gene value (GAPDH).
Peptide pulsing of transfected DCs
For peptide pulsing, 1×106–2×106 DCs were resuspended in 1 ml of DMEM containing 20 µmMut1 peptide. After 3 h incubation at 37° with gentle shaking every 30 min, the peptide-pulsed DCs were washed twice with PBS and resuspended in PBS for vaccination of the mice.
Allogeneic mixed lymphocyte reactions
Naive T cells were purified from BALB/c mouse splenocytes as nylon wool non-adherent cells.23 The primary mixed lymphocyte reactions (MLRs) were performed as previously described.24 Briefly, graded doses of irradiated DCs (3000 rads) including DCAdVTNF-α, untreated DCs and DCTNF-α cultivated with recombinant TNF-α (1, 10 and 20 ng/ml) for 1 and 3 days, respectively, were co-cultured in 96-well plates with constant numbers (1×105) of allogeneic T cells from BALB/c mice. After 3 days, T-cell proliferation was measured using an overnight [3H]thymidine (1 mCi/ml, Amersham Canada Ltd) uptake assay (1 µCi/well). The levels of [3H]thymidine incorporation into the cellular DNA were determined by liquid scintillation counting.
Chemotaxis assay for CCR7 expression by DCs
We used microchemotaxis assays with recombinant MIP-3β to detect expression of the CCR7 by our DCs, essentially as reported previously.25 Briefly, MIP-3β (1–1000 ng/ml) was placed in the lower chambers of a modified Boyden apparatus and 50 µl of DCs (2×106 cells/ml DMEM plus 1% bovine serum albumin) was placed in the upper chamber of each well; the chambers were separated by 5-μm pore-sized polyvinylpyrrolidone-free polycarbonate membranes. After 2 hr incubation at 37°, the cells that had not migrated into the membranes were wiped from the upper surfaces and the membranes were fixed and stained using a Diff-Quik kit (American Scientific Products, McGraw Hill, IL). For each sample, the numbers of cells associated with the membranes were enumerated by direct counting of at least nine 40× objective fields per well. The results are expressed as the mean number of cells/40× field (±SD).
Assays for migration of DCs and T-cell activation in vivo
For measurement of DC migration toward the regional lymph nodes the DCs, DCAdVTNF-α and DCTNF-α cells were radiolabelled with 51Cr (36 mCi/ml; Amersham Canada Ltd) by culturing the cells for 1 hr in the presence of sodium 51Cr-chromate (50 µl/10×106 DCs in 0·5 ml DMEM) and then washing them three times with DMEM. Thereafter, 1×106 labelled cells were injected subcutaneously (s.c.) in 30 µl of PBS into the hind footpads of mice, and 1 day later the mice were killed. Both hind feet of each mouse were amputated 3 mm above the hairline and the regional draining lymph nodes (RDLNs) were recovered for measurement of radioactivity using a gamma-counter. The relative migration index of the DC was calculated using the formula: (c.p.m. in RDLNs)/(c.p.m. remaining in the footpad).
For measurement of T-cell activation, Mut1 peptide-pulsed DC, DCAdVTNF-α, or DCTNF-α cells (1×106 cells) were injected s.c. in 30 µl of PBS into the hind footpads of mice. Two days later, the regional lymph nodes were removed. The lymphocytes from these lymph nodes were harvested, stained with FITC-anti-CD3 and PE-anti-CD25 (activated T-cell marker) antibodies, and analysed by flow cytometry. In this experiment, naive spleen T cells, prepared as previously described,23 and activated 3LL tumour-specific T cells (below) were used as negative and positive controls, respectively, for CD25 expression.
Cytotoxic T lymphocyte assay
Red blood cell-depleted splenic lymphocytes from mice vaccinated with Mut1-pulsed DCs, DCAdVTNF-α, or DCTNF-α cells were co-cultured in 24-well plates with irradiated 3LL cells (20 000 rads), using 5×106 lymphocytes and 2×105 3LL cells per 2 ml of DMEM/10% FCS. After 4 days, the activated 3LL tumour-specific T cells were used as effector cells in a chromium-release assay against radiolabelled 3LL or EL4 target cells. The target cells (104/well) were incubated for 8 hr in triplicate cultures with effector cells at various effector to target ratios. The per cent specific lysis was calculated using the formula: [(experimental c.p.m. − spontaneous c.p.m.)/(maximal c.p.m. − spontaneous c.p.m.)]×100.
The spontaneous c.p.m. release in the absence of effector cells was less than 10% of specific lysis; maximal c.p.m. release was effected by adding 1% Triton X-100 to the cells.
Assay of T-cell receptor specificity for Mut1 peptides
We examined the T-cell receptor specificity of lymphocytes from our mice by assessing their abilities to bind Mut1 peptides that had been preloaded into a recombinant soluble H-2Kb/immunoglobulin fusion protein. Naive splenic T cells were generated from normal mice using nylon wool columns. Activated 3LL tumour-specific T cells were prepared from mice that had been immunized with DCAdVTNF-α, and were activated by co-culture with irradiated 3LL tumour cells, as described above. T cells from naive C57BL/6 mice were stimulated in medium containing concanavalin A (2 µg/ml) and IL-2 (20 units/ml) for 7 days and used as a negative control population. For Mut1 peptide loading, soluble dimeric H-2Kb/immunoglobulin fusion protein (8 µg) was incubated at 4° for 48 hr with Mut1 peptide (5·3 µg) in PBS (pH 7·2) according to the supplier's recommendation. To probe the T cells, 106 cells in 50 µl PBS/1% FCS were incubated for 60 min at 4° with 2 µg of peptide-loaded fusion protein, then washed and stained for 60 min on ice with FITC-conjugated rabbit anti-mouse IgG1 antibody. After three washes with PBS, cells were analysed by flow cytometry. For phenotypic analysis, the cells were also stained with anti-CD3, -CD4, -CD8 and -CD25 antibodies.
DC-peptide vaccination of mice
For evaluation of tumour prevention, C57BL/6 mice were vaccinated s.c. with 0·5×106Mut1-pulsed untreated DCs, DCpLpA, DCAdVTNF-α, DCTNF-α, or KODCAdVTNF-α. Ten days later, the mice (n=8–10 per group) were challenged by s.c injection of low (0·5×105) or high (3×105) numbers of 3LL tumour cells. Animal mortality and tumour growth were monitored daily for up to 10 weeks; for humanitarian reasons, all mice with 1·5-cm diameter tumours were killed.
Results
Transfection of mouse DCs with AdV-TNF-α enhances DC maturation
We compared the phenotypic changes induced within DCs of relative maturity, generated by culture in GM-CSF and IL-4,20 of treatment with exogenous TNF-α (DCTNF-α) versus that induced by endogenous expression of equivalent levels of cytokine via a TNF-α transgene (DCAdVTNF-α). RNAse protection assays indicated that the DCTNF-α expressed moderately increased levels of mRNA for IL-12, ΙL-1β, MIP-1β and IFN-γ-inducible protein-10 (IP-10), and the chemokine receptor CCR7 relative to the untreated GM-CSF/IL-4 DCs (Fig. 1). On the other hand, TNF-α transgene expression within the DCAdVTNF-α led to a marked further (three- to five-fold) up-regulation of TNF-α, ΙL-1β, regulated upon activation, normally T-cell-expressed and presumably secreted (RANTES) and IP-10 mRNA in these cells, in addition to a more moderate augmentation of IL-12, IL-18 and MIP-1β expression (Fig. 1). On the contrary, adenoviral control vector transfection by itself had only mild effects on the expression by DCpLpA of ΙL-1β and MIP-1β as well as endogenous TNF-α. These data clearly indicate that while adenoviral infection alone or in vitro treatments with TNF-α can moderately affect DC cytokine responses, TNF-α transgene expression was associated with very substantially increased cytokine responses.
Figure 1.
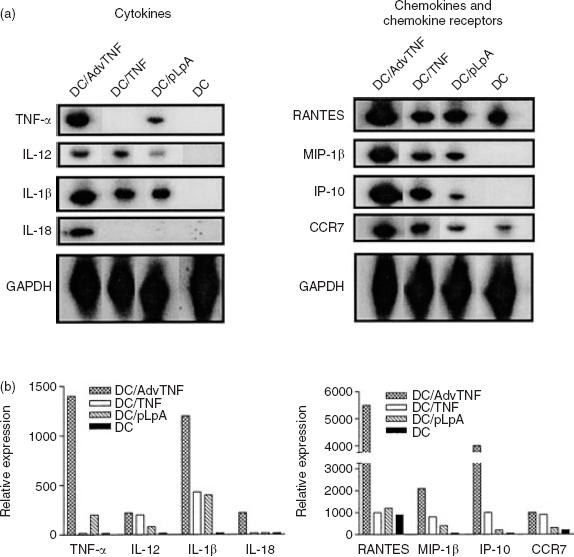
TNF-α transgene expressing dendritic cells (DC) display enhanced levels of mRNA for a panel of DC-associated cytokines, chemokines and chemokine receptors. DCs were either left untreated, cultured with 10 ng/ml TNF-α (DCTNF), or transfected with a control adenoviral vector (DCpLpA) or a TNF transgene-expressing adenoviral vector (DCAdvTNF), and then assessed by RNAse protection assay for their expression of the indicated markers. (a) Autoradiograph of the RNAse protection assay gels for each population of cells. (b) Graphic presentation of the data in (a), showing the relative expression of each transcript within the DCAdVTNF-α, DCTNF-α, DCpLpA and the uninfected control DC (shaded bars).
It has been reported that pro-inflammatory cytokines, including TNF-α, have maturation-promoting effects on DCs.9,11,13,14 Thus, we also analysed the impact of exogenous vs. endogenous (i.e. transgene-derived) TNF-α on the expression of a number of maturation markers in GM-CSF/IL-4 DCs. The addition of recombinant TNF-α, or control virus transfections, led to very mild (i.e. <20%) up-regulation of CD40, CD86 and ICAM-1 expression. The DCAdVTNF-α that expressed the TNF-α gene in trans, were more markedly affected, with 2·3- to 3·2-fold greater expression of CD40, CD86 and ICAM-1 being evident in these cells, relative to the untreated DCs (Fig. 2). The expression of MHC class I and II antigens, CD11c and CD80 remained unchanged on these DCs (data not shown).
Figure 2.

Comparison of the phenotypic changes induced in DC cultured in TNF-α-containing medium versus cell infected with an adenoviral TNF-α transgene. The cell populations were generated as in Figure 1. The expression of CD40, CD86 and ICAM-1 in DCAdVTNF-α, DCTNF-α, DCpLpA and uninfected DCs (solid lines) was analysed by FACS using FITC-labelled antibodies. The values of mean fluorescence intensity are shown in the right upper corners. Isotype-matched monoclonal antibodies (dotted lines) were used as controls. One representative experiment of three is shown.
These data suggested that the expression of TNF-α in the AdV-TNF-α-transfected cells markedly altered the physiology of these cells, increasing their expression of a panel of cytokines/chemokines and activation/maturation markers. These effects could be attributable to the cytokine product of the TNF-α transgene, to an up-regulated expression of the chromosomal TNF-α genes in these cells, or to a combination of these. In order to distinguish unequivocally between these possibilities, we repeated our experiments using DCs generated from TNF-α knockout mice. The phenotype of the KODCAdVTNF-α cells from these mice, as determined by RNAse protection and fluorescence-activated cell sorter (FACS) analyses, was indistinguishable from that of the DCAdVTNF-α (data not shown), indicating that the TNF-α transgene product was responsible for the augmented maturational effects observed in the DCAdVTNF-α. Taken together, these results clearly indicate that AdV-TNF-α transfection very substantially augments the maturational responses of DC relative to cells treated in vitro with levels of exogenous TNF-α protein equivalent to those found in the culture supernatants of the TNF-α transgene-expressing cells.
DCAdVTNF-α display increased chemotactic and trafficking responses
MIP-3β can be an important signalling molecule for DCs, which recognize this chemokine via the CCR7 receptor. MIP-3β acts as a homing signal for cells attaining peripheral lymphoid organs.26 We used microchemotaxis assays to determine whether the enhanced expression of CCR7 mRNA observed above in the AdV-TNF-α-transfected DCAdVTNF-α cells (Fig. 1) would translate into an increased responsiveness of the cells to MIP-3β. We found that each population of DCs (DCAdVTNF-α, DCTNF-α and untreated DCs) responded to this chemokine in vitro in a dose-dependent fashion (data not shown). As might be expected, the in vitro responses of the high CCR7 mRNA-expressing DCAdVTNF-α were 2·4-fold (P<0·05) greater than those of the uninfected DCs, though only 10% greater than the DCs cultured with exogenous TNF-α (Table 1).
Table 1.
Enhanced capability of DCs transfected with AdV-TNF-α to migrate in vitro and in vivo
| Lymph node homing‡[c.p.m. in tissue (% of total c.p.m.)] | |||
|---|---|---|---|
| Cell population* | In vitro response to MIP-3β† | Injection site | Draining lymph node |
| DC | 182±8 | 22303±7767 (87%) | 3411±1270 (13%) |
| DCTNF-α | 390±37 | 4487±760 (14%) | 27569±3134 (86%) |
| DCAdVTNF-α | 432±15§ | 3825±264 (9%) | 22514±1156¶ (91%) |
DC and DCAdVTNF-α represent uninfected and AdV-TNF-α-infected DCs, respectively. DCTNF-α represent the DCs that were cultured in medium containing levels of exogenous recombinant TNF-α equivalent to those found in the culture supernatants of DCAdvTNF.
In an in vitro chemotaxis assay, 1×105 DCs in DMEM and MIP-3β (500 ng/ml) were added in triplicate to top and lower wells, respectively, of modified Boyden chambers. After incubation for 2 hr at 37°, the membranes were stained and the cells associated with the membranes were enumerated by direct counting. The results are expressed as the mean number of cells/40× field (±SD). One representative experiment of two is shown.
In an in vivo migration assay, 1×10651 Cr-labelled DCs in 30 µl of PBS were injected into the hind footpads of the mice (n=8/group). One day later, the mice were killed and the levels of 51Cr associated with the injection sites and regional lymph nodes were assessed by gamma-counting. One representative experiment of two is shown.
DCAdVTNF-α versus DCTNF-α (P>0.05) and uninfected DC (P<0.05) (Student's t-test).
DCAdVTNF-α versus DCTNF-α (P>0.05) and uninfected DC (P<0.01) (Student's t-test).
Of further importance to the initiation of immune responses is the ability of the APC to traffic from the site of antigen entry or delivery to the site of antigen presentation (i.e. the regional lymph nodes). Thus, while we demonstrated that the DCAdVTNF-α cells displayed evidence of an advanced maturational state (Figs 1, 2), it was critical to also demonstrate that they migrated efficiently to lymph nodes adjacent to sites of tumour challenge/development. We injected 51Cr-labelled DCs into the footpads of our mice and tracked their migration to the draining lymph nodes over the next 24 hr (Table 1). We found that 87% of untreated DCs remained at the injection sites after 24 hr, while the DCAdVTNFα trafficked very efficiently (91% migration) to the draining lymph nodes. The DCAdVTNF-α were approximately seven-fold (P<0·01) more efficient in this response than uninfected DCs. There were no significant differences between DCAdVTNF-α and DCTNF-α cells in migration into the regional lymph nodes, which accurately reflects the relative levels of CCR7 mRNA expressed in these two populations.
DCAdVTNF-α induce enhanced T-cell proliferation in vitro and activation in vivo
DCs are potent stimulators of primary MLRs and are able to induce the proliferation of allogeneic CD8+ T cells in vitro.27 Just as stimulation of antigen-specific T-cell responses by DCs is strongly affected by their maturational status,12 so too is stimulation of primary MLRs by these cells. Thus, we next compared the abilities of our DC populations to stimulate primary MLRs among allogeneic CD8+ T cells. We found that DCAdVTNF-α induced stronger allogeneic T-cell proliferative responses in vitro than untreated DC and DCTNF-α cultivated with different concentrations of recombinant TNF-α for 1 and 3 days (Fig. 3).
Figure 3.

DCs expressing a TNF-α transgene induce stronger mixed lymphocyte reactions than DC grown in TNF-α-containing medium. The DC populations were generated as in Figure 1. Irradiated DCAdVTNF-α (•), untreated DCs (□) and DCTNF-α cultivated with 1 ng/ml (▴), 10 ng/ml (○) and 20 ng/ml (▵) of recombinant TNF-α for 1 day (a) and 3 days (b) (1×104 cells/well) and reciprocal dilutions thereof, were co-cultured for 3 days with 1×105 allogeneic BALB/c T cells. The overnight [3H]thymidine uptake seen on day 4 is expressed as the mean of three determinations. Background proliferation of DCs or T cells alone was always below 2000 c.p.m. One experiment of two is shown.
We then examined the abilities of Mut1 peptide-pulsed DC to stimulate T-cell activation in vivo. We confirmed that the splenic lymphocytes of naïve mice comprised CD3− CD25− cells (B cells), and CD3+ CD25− cells (quiescent T cells; Fig. 4a), while activated T cells displayed both CD3 and CD25 (IL-2 receptor; Fig. 4b). Immunization of mice with DCAdVTNF-α stimulated greater T-cell activation (i.e. CD25 expression) in the regional lymph nodes than either DCTNF or normal (i.e. GM-CSF/IL-4) DCs. In one experiment representative of two, CD25+ T cells comprised 7·34%, 5·16%, or 3·99% of the lymphocytes draining sites of vaccination with DCAdVTNF, DCTNF-α, or uninfected DCs, respectively.
Figure 4.

Vaccination of naïve mice with Mut1 tumour peptide-pulsed TNF transgene-expressing DCs leads to augmented T-cell activation in the draining lymph nodes. Mice were vaccinated with various DC populations (Figure 1) that had been pulsed with Mut1 tumour peptide, and 2 days later the regional draining lymph node CD3-positive cells (T cells) were assessed for their expression of the CD25 activation marker by FACS. Control cells for the FACS included naive splenic T cells (a) and activated SP2/0 tumour-specific T cells (b). The assay populations included T cells from the regional lymph nodes of naive mice (c) or of mice immunized with Mut1 peptide-pulsed GM-CSF/IL-4 DCs (d), DCTNF-α (e) or DCAdVTNF-α (f). One experiment of two is shown.
DCAdVTNF-α significantly enhance tumour-specific T-cell responses in vitro
Next, we addressed the specific anti-tumour effector functions induced by vaccination of the mice with Mut1-pulsed DCAdVTNF-α, assessing the CTL activities against 51Cr-labelled 3LL target cells of splenocytes from the vaccinated animals (Fig. 5). T cells from mice vaccinated with Mut1-pulsed DCAdVTNF-α cells displayed substantially (44%) enhanced CTL activity relative to analogous cells from animals vaccinated with the peptide-pulsed TNF-α-treated DCTNF-α and 73% enhanced activity relative to the peptide-pulsed untreated DCs. This CTL activity was immunologically specific, in as much as none of these populations showed cytotoxic activities against the irrelevant EL4 tumour cells, and T cells from naive mice had no activity against the 3LL cells (data not shown). To confirm the specificity of the T-cell receptors on these effector cells, we assessed their abilities to bind Mut1 peptide presented in the context of a soluble MHC class I-peptide complex (i.e. Mut1 peptide-loaded H-2Kb/immunoglobulin fusion protein). As shown by the flow cytometric analyses in Fig. 6, Mut1 peptide-loaded H-2Kb/immunoglobulin protein complexes bound specifically to the T-cell receptors of CD25+ (i.e. activated) CD8+ T cells of vaccinated mice, but not to concanavalin A/IL-2-stimulated CD25+ CD4+ or CD25+ CD8+ T cells of naive mice. Thus, our data indicate that vaccination with DCAdVTNF-α that had been pulsed with Mut1 peptide is able to stimulate CD8+ CTLs with T-cell receptors specific for Mut1 peptide. This CTL response against 3LL tumours is more efficient than that induced by vaccination with DCTNF-α or untreated DCs.
Figure 5.

DCAdVTNF, which express a TNF-α transgene, induce more robust tumour-specific cytotoxic T-cell responses than DCTNF, grown in TNF-α-containing medium. Splenic lymphocytes were harvested from mice that had been vaccinated with Mut1 peptide-pulsed DCAdVTNF-α (○), DCTNF-α (▵) or uninfected DCs (•) and co-cultured with irradiated 3LL cells (20 000 rads) for 4 days, then used as effector cells in a chromium release assay with 51Cr-labelled 3LL tumour cells. To confirm that the T-cell cytotoxicity was 3LL tumour-specific, we also included EL4 cells (▴) as a target control with activated T cells from DCAdVTNF-α immunized mice as effector cells. Each point represents the mean of three replicates. This experiment was repeated once with equivalent results.
Figure 6.

The activated T cells from mice vaccinated with Mut1 peptide-pulsed DCAdVTNF-α display Mut1-specific T-cell receptors. The activated T cells from vaccinated mice and concanavalin A/IL-2-stimulated T cells from naïve mice were assessed by FACS for expression of CD3, CD4, CD8, CD25, or Mut1-specific T-cell receptors, using specific antibodies or a soluble Mut1/H-2Kb/immunoglobulin complex. The primary antibodies or ligand were detected using FITC-conjugated goat anti-rat IgG (solid lines) or rabbit anti-mouse IgG1 control antibody (dotted lines). One experiment of two is shown.
DCAdVTNF-α strongly induce protective tumour-specific immunity in vivo
To examine whether DCAdVTNF-α cells were also capable of inducing enhanced anti-tumour immunity in vivo, we vaccinated mice with peptide-pulsed DCs from each population and 10 days later challenged the animals with 3LL tumour cells. Both low-dose (0·5×105 cells) and high-dose (3×105 cells) 3LL tumour cell challenges were invariably lethal within 3–4 weeks post-implantation for the PBS treatment control mice (Fig. 7). Prior vaccination with Mut1 peptide-pulsed untreated DCs was sufficient to protect the mice from tumour growth after challenge with low doses (10/10 protected at 10 weeks), but not high doses (10/10 dead at 5 weeks) of 3LL cells (Fig. 7). In fact, vaccination with any of the peptide-pulsed DC populations was sufficient to induce protection against low-dose tumour cell challenge (Fig. 7a). Thus, the real test of the protective effects of the different DC populations was only apparent in the higher dose tumour cell challenge experiments. Here, as expected, adenoviral infection by itself did not significantly enhance the efficiency of DC vaccination, since all mice vaccinated with Mut1 peptide-pulsed DCpLpA died within 6 weeks of inoculation with 3×105 tumour cells. On the other hand, use of Mut1 peptide-carrying DCTNF-α, generated by culture in TNF-α-containing medium, enhanced anti-tumour immunity, with four of 10 mice protected from challenge with 3×105 3LL tumour cells at 10 weeks (Fig. 7b). Most importantly, vaccination with peptide-pulsed DCAdVTNF-α was a much more effective therapy, protecting all mice from high-dose tumour cell challenge until 10 weeks. To confirm the importance of the TNF-α transgene expression in this DC vaccination, as opposed to endogenous (i.e. chromosomal) TNF-α gene expression, we also immunized mice with peptide-pulsed TNF-α knock-out KODCAdVTNF-α cells, and here too found 100% (10/10 mice) protection against challenge with high doses of cells (Fig. 7b). Thus, our data very clearly demonstrate that vaccination using Mut1-pulsed DCAdVTNF-α cells, even if they secrete only transgene-derived TNF-α, induced a much more robust anti-tumour immunity than similar vaccination using Mut1-pulsed DC that had been cultured in vitro with TNF-α (i.e. DCTNF-α cells). No side-effects or toxicity were observed in these animal studies.
Figure 7.

Vaccination of mice with Mut1-pulsed DCAdvTNF-α or KODCAdVTNF-α induces stronger anti-tumour immunity than vaccination with DCTNF-α. C57BL/6 mice were vaccinated with Mut1 peptide-pulsed DCAdVTNF-α, DCTNF-α, DCpLpA, or uninfected DCs, then challenged subcutaneously 10 days later with (a) low (0·5×105 cells; n=8 mice/group) or (b) high (3×105 cells; n=10 mice/group) numbers of 3LL tumour cells. Control mice were treated with PBS (▵) or vaccinated with 0·5×106 TNF transgenic DCs generated from TNF knock-out C57BL/6 mice [KODCAdVTNF-α; (b) only]. This control group confirms that the effector TNF secretion product of the DCs used for the treatments was from the cell's TNF transgene, and not from the cell's endogenous (chromosomal) TNF gene. The survival time of each mouse was monitored daily. The animal experiment was repeated once with similar results.
Discussion
DC are the most potent stimulators of primary immune responses currently known4 and, as such, have been recognized as potentially important tools for immunotherapy and vaccine strategies, especially in the case of tumour therapies.6–8 Unfortunately, tumour cells can interfere with the host DC maturation and function.28 As a circumvention strategy aimed at this problem, investigators have begun to employ successfully cytokine-stimulated DCs in both animal models and clinical trials.29,30 Indeed, antigen-specific CTL responses have been induced in vivo following vaccination with DCs pulsed in vitro with antigen.6–8 As a further strategy to enhance their abilities to induce anti-tumour responses, the effects of DCs genetically engineered to express cytokines such as IL-7, IL-12 and GM-CSF have also been tested. As such, intratumoral administration of adenoviral IL-7 gene-modified DCs can augment specific anti-tumour immunity and induce tumour eradication.31 In both mouse and human studies, IL-12 expression by engineered DCs can augment priming to antigens delivered in a variety of ways to the cells, such that these DCs were powerful catalysts for the induction of tumour-specific CD4+ T helper cells and CD8+ CTLs.32,33 Engineering DCs to express GM-CSF also reportedly increases therapeutic anti-tumour immunity in vivo.24 However, in none of these reports have the potential mechanisms involved in observed enhancement of anti-tumour immunity been addressed. Furthermore, the impact of genetically modifying DCs with the TNF-α gene has not been studied. As noted above, TNF-α is known to have very substantial effects on DCs both in vitro and in vivo.13–16 We have demonstrated now also that vaccination of mice with Mut1 peptide-pulsed DCs that had been transfected with AdV-TNF-α induced more efficient Mut1-specific CD8+ CTL cytotoxicity in vitro and substantially more effective anti-tumour immunity in vivo than similar vaccination with DCs simply generated by culture in exogenous TNF-α.
The degree of differentiation or maturation of a population of DCs largely legislates their functional capabilities. In general, antigen processing is maximal with immature populations, while T-cell sensitization is more effective with mature DCs, which express enhanced levels of MHC class II, CD40, co-stimulatory and adhesion molecules. In this study, the in vitro TNF-α-cultivation induced some, but relatively mild, up-regulation of immunogenes such as CD40, CD86 and ICAM-1, as previously reported.13,14 The transfection of DCs with the control vector (AdV-pLpA) slightly, but not significantly, altered the phenotype of the DCs, which is also consistent with previous reports.24,34 However, transfection of DCs with AdV-TNF-α significantly augmented their maturation, with higher level expression by these cells, relative to DCpLpA or DCTNF, of adhesion molecule ICAM-1 and co-stimulatory molecules CD40 and CD86, possibly due to the synergistic effects on DC maturation from both TNF-α stimulation and adenoviral infection itself. The enhanced expression of the cell adhesion (ICAM-135) and T-cell co-stimulatory (CD40, CD8636,37) molecules by DCAdVTNF-α could feasibly have played a significant role in the enhanced antigen presentation to, and activation of, T cells observed in our experiments.
In this study, we also demonstrated that transfection of DCs with AdV-TNF-α up-regulated their expression of numerous cytokines (ΙL-1β, TNF-α, IL-12 and IL-18) and chemokines (RANTES, IP-10 and MIP-1β), many of which affect anti-tumour responses in other systems. For example, TNF-α is an immunoregulatory cytokine with the ability to activate both T cells38 and DCs13,14 and to ameliorate IL-10-mediated inhibition of DC development and function.16 The IL-12 and IL-18 expressed by DCAdVTNF-α could also affect T helper type 1-directed immune responses, which are known to be associated with anti-tumour cytotoxic T-cell responses.39,40 The chemokines expressed by DCAdVTNF-α (IP-10, RANTES, MIP-1β and CD86) are each chemotactic for T cells and macrophages.41 Therefore, up-regulation of the above cytokines and chemokines may play some role in the enhanced anti-tumour immunity observed in this study. Especially, the secretion of significant amounts of TNF-α by DCAdVTNF-α cells may play an important role in enhanced anti-tumour immunity by continuously attracting and activating T cells, and by continuously stimulating DCs in an autocrine fashion as they migrate into the regional lymph nodes. The importance of transgene TNF-α expression was further confirmed in our animal studies using AdV-TNF-α-transfected DCs derived from TNF-α knockout mice. Vaccination of mice with these Mut1-pulsed KODCAdVTNF-α cells, secreting similar amounts of transgene TNF-α as DCAdVTNF-α, also resulted in 100% (10/10) immune protection against high-dose challenge with 3LL tumour cells.
The capacity of DC to migrate into T-cell areas of lymph nodes is key to the successful induction of protective immunity.42 Recent studies have demonstrated that chemokines play critical roles in DC migration. The migratory capacity of DCs is dictated by their changing responsiveness to various chemokines during their development and maturation.26,43,44 Immature DCs respond to inflammatory chemokines (e.g. MCP-1, MIP-1α, MIP-3α and RANTES) via their CCR1, CCR2, CCR5, CCR6 and CXCR1 receptors, whereas mature DCs respond to MIP-3β and SLC via the CCR7 receptor.43 Cumberbatch and Kimber have previously demonstrated that TNF-α is required for optimal DC migration to, and accumulation in, regional lymph nodes, although the molecular mechanism(s) responsible for this migration was not elucidated.15 In the present study, we showed that DCAdVTNF-α displayed augmented expression of the CCR7 receptor and demonstrated enhanced migratory responses for the CCR7 ligand MIP-3β in chemotaxis assays in vitro. Furthermore, DCAdVTNF-α showed seven-fold more efficient in migration toward lymph nodes than non-transfected DCs, suggesting that the enhanced expression of CCR7 of DCAdVTNF-α promoted the augmented migration into the regional lymph nodes in vivo. Our data are thus consistent with a recent report by Hirao et al. in which up-regulation of the CCR7 (induced by phagocytosis of apoptotic tumour cells) was found to play a critical role in migration of DCs from tumour sites to draining lymph nodes.45 The DCAdVTNF-α and DCTNF-α expressed equivalent levels of CCR7 mRNA and migrated more or less equally well in vitro and in vivo, but they had decidedly differing abilities to induce protective immunity. This clearly indicates that an enhanced migratory capacity alone does not translate in a straightforward fashion into enhanced induction of immune responses.
In summary, we have demonstrated that AdV-TNF-α-transfected DCs express heightened levels of multiple maturation markers, including expression of the CCR7, which correlated with the AdV-TNF-α-transfected facilitated trafficking of DCs from the interstitial tissues to the regional lymph nodes. As a whole, these DC features are associated with the development of marked protective immune responses directed against the tumour epitopes delivered by the DC and apparently mediated by augmented CTL activity. We did not investigate the molecular mechanisms responsible for these effects. Nevertheless, it is clear that vaccination with Mut1 peptide-pulsed, engineered DCAdVTNF-α induces substantially stronger anti-tumour immunity, even against poorly immunogenic tumour cells, than vaccination with similarly pulsed, uninfected or in vitro TNF-α-cultivated DCs. These results indicate that DC AdVTNF-α transfection is a superior approach for induction of DC maturation, T-cell activation and anti-tumour immunity than is culture of the DCs in TNF-α-containing medium. Therefore, our study suggests that the principle of using DCs engineered to secrete TNF-α could or should perhaps be considered for clinical testing within DC cancer vaccines.
Acknowledgments
This work was supported by research grants (ROP15151 and MT11861) from the Canadian Institutes of Health Research and the Medical Research Council of Canada, respectively.
Abbreviations
- APC
antigen-presenting cell
- CCR1, 2, 5 and 7
CC chemokine receptors 1, 2, 5 and 7
- c.p.m.
count per minute
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- DMEM
Dulbecco's modified Eagle's medium
- ELISA
enzyme-linked immunosorbent assay
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- IFN-γ
interferon-γ
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- ICAM
intercellular adhesion molecule
- IP-10
interferon-γ-inducible protein-10
- IL-1
interleukin-1
- MIP
macrophage inflammatory protein (e.g. MIP-1α)
- MHC
major histocompatibility complex
- MLR
mixed lymphocyte reaction
- MOI
multiplicity of infection
- PBS
phosphate-buffered saline
- RDLN
regional draining lymph node
- RANTES
regulated upon activation, normally T-cell-expressed and presumably secreted
- TNF-α
tumour necrosis factor-α
References
- 1.Melief D. Tumor eradication by adoptive transfer of cytotoxic T lymphocytes. Adv Cancer Res. 1992;58:143–75. doi: 10.1016/s0065-230x(08)60294-8. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell J, Bennink J. Cell biology of antigen processing and presentation to MHC class I molecule restricted T lymphocytes. Adv Immunol. 1992;52:1–23. doi: 10.1016/s0065-2776(08)60875-5. [DOI] [PubMed] [Google Scholar]
- 3.Watts C. Capture and processing of exogenous antigens for presentation on MHC molecules. Annu Rev Immunol. 1997;15:821–50. doi: 10.1146/annurev.immunol.15.1.821. [DOI] [PubMed] [Google Scholar]
- 4.Parra E, Wingren A, Hedlund G, Kalland T, Dohlsten M. The role of B7-1 and LFA-3 in costimulation of CD8 T cells. J Immunol. 1997;158:637–42. [PubMed] [Google Scholar]
- 5.Steinman R, Bancherear J. Dendritic cells and the control of immunity. Nature. 1988;392:245–152. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 6.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9:10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 7.Nestle F, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nature Med. 1998;4:328–32. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 8.Nair S, Snyder D, Rouse B, Gilbao E. Regression of tumors in mice vaccinated with professional antigen-presenting cells pulsed with tumor extracts. Int J Cancer. 1997;70:706–15. doi: 10.1002/(sici)1097-0215(19970317)70:6<706::aid-ijc13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Jonuleit H, Knop J, Enk A. Cytokines and their effects on maturation, differentiation and migration of dendritic cells. Arch Dermatol Res. 1996;289:1–8. doi: 10.1007/s004030050144. [DOI] [PubMed] [Google Scholar]
- 10.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GMCSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–69. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 11.Jonuleit H, Muller K, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk A. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–42. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 12.Labeur M, Roters B, Pers B, Mehling A, Luger T, Schwarz T, Grabbe S. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol. 1999;162:168–75. [PubMed] [Google Scholar]
- 13.Rieser C, Bock G, Klocker H, Bartsch G, Thumher M. Prostaglandin Ea and tumor necrosis factor alpha cooperate to activate human dendritic cells: synergistic activation of interleukin 12 production. J Exp Med. 1997;186:1603–8. doi: 10.1084/jem.186.9.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunner C, Seiderer J, Schlamp A, et al. Enhanced dendritic cell maturation by TNF-α or cytidine-phosphate-guanosine DNA drives T cell activation in vitro and therapeutic antitumor immune responses in vivo. J Immunol. 2000;165:6278–86. doi: 10.4049/jimmunol.165.11.6278. [DOI] [PubMed] [Google Scholar]
- 15.Cumberbatch M, Kimber I. Tumor necrosis factor-α is required for accumulation of dendritic cells in draining lymph nodes and for optimal contact sensitization. Immunol. 1995;84:31–5. [PMC free article] [PubMed] [Google Scholar]
- 16.Brossart P, Zobywalski A, Grunebach F, Behnke L, Stuhler G, Reichardt V, Kanz L, Brugger W. Tumor necrosis factor α and CD40 ligand antagonize the inhibitory effects of interleukin 10 on T cell stimulatory capacity of dendritic cells. Cancer Res. 2000;60:4485–92. [PubMed] [Google Scholar]
- 17.Mandelboim O, Berke M, Fridkin M, Feldman M, Eisenstein M, Eisenbach L. CTL induction by a tumor-associated antigen octapeptide derived from a murine lung carcinoma. Nature. 1994;369:67–72. doi: 10.1038/369067a0. [DOI] [PubMed] [Google Scholar]
- 18.Wright P, Braun R, Babiuk L, Drunen Littel-van den Hurk S, Moyana T, Zheng C, Chen Y, Xiang J. Adenovirus-mediated TNF-α gene transfer induces significant tumor regression in mice. Cancer Biother Radiopharm. 1999;14:49–57. doi: 10.1089/cbr.1999.14.49. [DOI] [PubMed] [Google Scholar]
- 19.Song W, Kong H, Carpenter H, Torii H, Granstein R, Rafii S, Moore M, Crystal R. Dendritic cells genetically modified with adenovirus vector encoding the cDNA for a model antigen induce protective and therapeutic antitumor immunity. J Exp Med. 1997;186:1247–56. doi: 10.1084/jem.186.8.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamada N, Katz S. Generation of mature dendritic cells from a CD14 cell line (XS52) by IL-4, TNF-α, IL-1β and agonistic anti-CD40 monoclonal antibody. J Immunol. 1999;163:5331–7. [PubMed] [Google Scholar]
- 21.Chen Z, Dehm S, Bonham K, Kamencic H, Juurlink B, Zhang X, Gordon J, Xiang J. DNA array and biological characterization of the impact of the maturation status of mouse dendritic cells on their phenotype and antitumor vaccination efficacy. Cellular Immunol. 2002;290:66–72. doi: 10.1006/cimm.2001.1883. [DOI] [PubMed] [Google Scholar]
- 22.Brossart P, Zobywalski A, Grunebach F, Behnke L, Stuhler G, Reichardt V, Kanz L, Brugger W. Tumor necrosis factor α and CD40 ligand antagonize the inhibitory effects of IL-10 on T cell stimulatory capacity of dendritic cells. Cancer Res. 2000;60:4485–92. [PubMed] [Google Scholar]
- 23.Xiang J, Moyana T. Regression of engineered tumor cells secreting cytokines is related to a shift in host cytokine profile from type 2 to type 1. J Interferon Cytokine Res. 2000;20:349–54. doi: 10.1089/107999000312270. [DOI] [PubMed] [Google Scholar]
- 24.Curiel-Lewandrowski C, Mahnke K, Labeur M, Roters B, Schmidt W, Granstein R, Luger T, Grabbe S. Transfection of immature murine bone marrow-derived dendritic cells with the granulocyte-macrophage colony-stimulating factor gene potently enhances their in vivo antigen-presenting capacity. J Immunol. 1999;163:174–83. [PubMed] [Google Scholar]
- 25.Cross A, Richardson V, Ali S, Palmer I, Taub D, Rees R. Migration responses of human monocytic cell line to α- and β-chemokines. Cytokine. 1997;9:521–8. doi: 10.1006/cyto.1996.0196. [DOI] [PubMed] [Google Scholar]
- 26.Sozzani S, Allavena P, D'Amico G, et al. Differential regulation of chemokine receptors during dendritic cell maturation: a model for their traficking properties. J Immunol. 1998;161:1083–6. [PubMed] [Google Scholar]
- 27.Inaba K, Young J, Steiman R. Direct activation of CD8 cytotoxic T lymphocytes by dendritic cells. J Exp Med. 1987;166:182–94. doi: 10.1084/jem.166.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabrilovech D, Chen H, Girgis K, Cunningham H, Meny G, Nadaf S, Kavanaugh D, Carbone D. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nature Med. 1996;2:1096–103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 29.Miller P, Sharma S, Stolina M, Chen K, Zhu L, Paul R, Dubinett S. Dendritic cells augment granulocyte-macrophage colony-stimulating factor (GM-CSF)/herpes simplex virus thymidine kinase-mediated gene therapy of lung cancer. Cancer Gene Ther. 1998;5:380–9. [PubMed] [Google Scholar]
- 30.Nestle F, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nature Med. 1998;4:328–32. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 31.Miller P, Sharma S, Stolina M, et al. Intratumoral administration of adenoviral interleukin-7 gene-modified dendritic cells augments specific antitumor immunity and achieves tumor eradication. Hum Gene Ther. 2000;11:53–65. doi: 10.1089/10430340050016157. [DOI] [PubMed] [Google Scholar]
- 32.Tuting T, Wilson C, Martin D, et al. Autologous human monocyte-derived dendritic cells genetically modified to express melanoma antigens elicit primary cytotoxic T cell responses in vitro: enhancement by cotransfection of genes encoding the Th1-biasing cytokines IL-12 and IFN-α. J Immunol. 1998;160:1139–47. [PubMed] [Google Scholar]
- 33.Nishioka Y, Hirao M, Robbins PD, Lotze MT, Tahara H. Induction of systemic and therapeutic antitumor immunity using intratumoral injection of dendritic cells genetically modified to express interleukin 12. Cancer Res. 1999;59:4035–41. [PubMed] [Google Scholar]
- 34.Zhong L, Granelli-Piperno A, Choi Y, Steinman R. Recombinant adenovirus is an efficient and non-perturbing genetic vector for human dendritic cells. Eur J Immunol. 1999;29:964–72. doi: 10.1002/(SICI)1521-4141(199903)29:03<964::AID-IMMU964>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 35.Lub M, van Kooyk Y, Figdor C. Competition between lymphocyte function-associated antigen 1 (CD11a/CD18) and Mac-1 (CD11b/CD18) for binding to intercellular adhesion molecule-1 (CD54) J Leukoc Biol. 1996;59:648–55. doi: 10.1002/jlb.59.5.648. [DOI] [PubMed] [Google Scholar]
- 36.Bennett S, Carbone F, Karamalis F, Flavell R, Miller J, Heath W. Help for cytotoxic-T-cell responses is mediated by CD40 signaling. Nature. 1998;393:478–83. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 37.Yang G, Hellstrom K, Hellstrom I, Chen L. Antitumor immunity elicited by tumor cells transfected with B7–2, a second ligand for CD28/CTLA-4 costimulatory molecules. J Immunol. 1995;154:2794–800. [PubMed] [Google Scholar]
- 38.Robinet E, Branelec D, Termijtelen A, et al. Evidence for tumor necrosis factor involvement in the optimal induction of class I allospecific cytotoxic T cells. J Immunol. 1990;144:4555–61. [PubMed] [Google Scholar]
- 39.Tsung K, Meko J, Peplinski G, Tsung Y, Norton J. IL-12 induces T helper 1-directed antitumor response. J Immunol. 1997;158:3359–65. [PubMed] [Google Scholar]
- 40.Kohno K, Kataoka J, Ohtsuki T, Suemoto Y, Okamoto I, Usui M, Ikeda M, Kurimoto M. IFN-γ-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol. 1997;158:1541–50. [PubMed] [Google Scholar]
- 41.Sozzani S, Locati M, Allavena P, Van Damme J, Mantovani A. Chemokines: a superfamily of chemotactic cytokines. Int J Clin Lab Res. 1996;26:69–82. doi: 10.1007/BF02592349. [DOI] [PubMed] [Google Scholar]
- 42.Banchereau J, Steinman R. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 43.Dieu M, Vanbervliet B, Vicari A, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–86. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kellermann S, Hudek S, Oldham E, Liu Y, McEvoy L. The CC chemokine receptor 7 ligands and MIP-3β are potent chemoattractants for in vitro and in vivo-derived dendritic cells. J Immunol. 1999;162:3859–64. [PubMed] [Google Scholar]
- 45.Hirao M, Onai N, Hiroishi K, Watkins S, Matsushima K, Robbins P, Lotze M, Tahara H. CC chemokine receptor 7 on dendritic cells is induced after interaction with apoptotic tumor cells: critical role in migration from the tumor site to draining lymph nodes. Cancer Res. 2000;60:2209–17. [PubMed] [Google Scholar]