

Abstract
In our study we characterised the immunophenotype of monocytes that migrated through an endothelial cell (EC) monolayer in vitro. We found that monocyte migration led to an enhanced expression of CD11a, CD33, CD45RO, CD54 [intercellular cell-adhesion molecule (ICAM)-1] and human leucocyte antigen-DR. The most striking increase was observed for ICAM-1 when ECs were activated with tumour necrosis factor-α and interleukin-1α. The results of our study indicate the following: (1) there is a characteristic immunophenotype on the surface of monocytes after transendothelial migration; (2) this phenotype seems to be induced by interactions between monocytes and ECs; and (3) this change is enhanced by the pretreatment of ECs with cytokines. Taken together, the results suggest that local cytokine production activating ECs is sufficient to enhance monocyte migration and that this, in turn, can induce changes consistent with an activated phenotype known to be interactive between antigen-presenting cells and T cells. These results have implications for our pathogenetic insights into rheumatoid arthritis.
Keywords: endothelium, inflammation, migration, monocyte, transendothelial migration
Synopsis
Introduction:
Acute and chronic inflammation are characterised by the enhanced migration of leucocytes from the blood vessels through the endothelium into the extravascular tissue. There are ample data on the transendothelial migration of lymphocytes [1,2,3,4,5,6]. In addition, T cells capable of transendothelial migration have a characteristic immunophenotype; for instance, migrating T cells express significantly higher amounts of CD29 (β1-integrin) than cells that do not interact with endothelial cells (ECs) [7]. In contrast, much less is known about the extravasation of cells of the monocyte/macrophage lineage. Under normal conditions, only a few monocytes are able to migrate from the bloodstream into healthy tissue, where they differentiate into macrophages [8]. In inflammatory diseases such as rheumatoid arthritis (RA), monocytes accumulate in the synovial membrane and contribute significantly to the pathogenesis of the disease, mainly by secreting cytokines such as tumour necrosis factor-α (TNF-α) and interleukin-1 (IL-1) [9]. Transendothelial migration of monocytes might also be an important step in the pathogenesis of non-inflammatory diseases, for example atherosclerosis [10,11,12].
Several pairs of receptors and counter-receptors that mediate the interaction of monocytes with ECs have been described [13,14,15,16,17], but the phenotype of monocytes capable of transendothelial migration was not defined. Previously we found an enhanced expression of CD54 [intercellular cell-adhesion molecule (ICAM)-1] on monocytes that had migrated through an unstimulated EC monolayer [18].
Aims:
In our study we characterised the immunophenotype of monocytes that migrated through an EC monolayer in an in vitro model. We found that monocyte migration led to an enhanced expression of CD11a, CD33, CD45RO, CD54 (ICAM-1) and human leucocyte antigen (HLA)-DR. The most striking increase was observed for CD54 when ECs were activated with TNF-α and IL-1α. Our findings of increased CD54 expression on monocytes that migrated through endothelium pretreated with TNF-α and IL-1α might be useful in elucidating the mode of action of therapy with antibodies against TNF-α and IL-1 in RA and thus might lead to new avenues of RA therapy in future.
Methods:
ECs were isolated from human umbilical cord veins by digestion with collagenase as described previously [7] and then cultured. ECs in the third to fifth passage were used. Peripheral blood mononuclear cells (PBMCs) were prepared from buffy coats of healthy blood donors by centrifugation over gradients of Ficoll-Hypaque. PBMCs were prepared immediately before starting the experiments.
Interactions with ECs in PBMC populations were examined on hydrated bovine collagen gels in the wells of 16 mm macrowell tissue culture plates, as described previously [7]. To form a confluent monolayer on the collagen gels, 5 ×105 ECs per well were incubated overnight.
To measure monocyte interaction with ECs, PBMCs (3 ×106) were resuspended in fresh culture medium, layered on top of collagen gels with and without ECs and incubated at 37°C. The range of the incubation period was 15 minutes to 24 hours. Nonadherent cells (NAD) were harvested by washing twice with culture medium. Cells bound to the surface (BND) were enriched by washing each well twice with warm (37°C) Puck's EDTA, twice with warm (37°C) EGTA [0.5 mM EGTA in phosphate-buffered saline (PBS)] and once with cold (4°C) Puck's EDTA. Finally, for the recovery of those cells that had migrated into the collagen gels (MIG), 0.7 ml of a solution containing 0.1% collagenase, 1% (v/v) fetal calf serum and 50 mM Hepes buffer was added per well. The collagen gels were then minced gently with a pipette and incubated for 60 minutes at 37°C, after which the migrated PBMCs were removed by washing the wells twice with PBS. Each population (NAD, BND and MIG) was washed, resuspended in culture medium and counted under a microscope. In some experiments we studied monocyte migration into plain collagen gels. In these experiments no ECs were layered on the collagen gels; in other respects the experiments were performed exactly as described above.
In some experiments the EC monolayer was preactivated by incubation with TNF-α, IL-1α, macrophage inflammatory protein (MIP)-1α or interferon-γ (IFN-γ). To this end, the medium in each well was removed and the ECs were incubated for 5 hours at 37°C with or without the respective cytokines or chemokines (100 IU/ml TNF-α, IL-1α or IFN-γ, or 50 ng/ml MIP-1α). After this 5-hour pretreatment, each well was washed thoroughly and the migration assay was performed as described above.
Staining of monocytes was performed with fluorescein isothiocyanate (FITC)-conjugated antibodies against CD11a, CD33, CD45RA, CD45RO, CD49d (α4-integrin), CD54 (ICAM-1), CD86, HLA-DR, CD45RB and CD62L (L-selectin) (functions and ligands of these surface markers are shown in Table 1). To distinguish monocytes from other immune cells, all samples were counterstained with a phycoerythrin-labelled anti-CD14 monoclonal antibody. Cells (3 × 105 to 4 × 105 per sample) were incubated at 4°C for 30 minutes. Cells were then pelleted and resuspended in 250 μl of PBS before analysis was performed on a flow cytometer. All results are expressed as the respective mean fluorescence intensity among CD14-positive cells. Because not only monocytes but also ECs express CD54 (ICAM-1), in the analyses of the expression of CD54 on monocytes by fluorescence-activated cell sorting, monocytes were defined by both the scatter profile and the expression of CD14. In addition, ECs, which are considerably larger, were excluded by size.
Table 1.
Description of the ligands and functions of the surface markers studied
| Surface marker | Designation | Ligand(s) | Function |
| CD11a | CD54, CD102 | Adhesion, T cell development | |
| CD33 | Sialylated glycoproteins? | Functions in haematopoesis | |
| CD45RA | Signal transduction | ||
| CD45RB | Signal transduction | ||
| CD45RO | Signal transduction | ||
| CD49d | α4-integrin | VCAM-1, fibronectin, MAdCAM-1, invasin | Adhesion, embryonic development |
| CD54 | ICAM-1 | LFA-1 (CD11a/CD18), Mac-1 (CD11b/CD18), CD43 | Adhesion, leucocyte transendothelial migration, signal transduction |
| CD62L | L-selectin | DNAd (CD34, GlyCAM-1, MAdCAM-1) | Adhesion; leucocyte homing, rolling and extravasation |
| CD86 | B 7-2 | CD28, CD152 | T cell interaction with dendritic cells and B cells, B cell co-stimulation |
All data are presented as means ± SD. Paired Student's t-tests were used for comparisons.
Results:
In initial experiments we studied the time course of PBMC migration into plain or EC-coated collagen gels, respectively. As shown in Fig. 1, the presence of an endothelium clearly facilitated the migration of PBMCs: after 30 minutes the percentage of PBMCs that had migrated was twice as high as that in the absence of ECs. After 2 hours, about 40% of the PBMCs could be recovered from collagen gels coated with an EC layer, whereas only 24% of PBMCs had migrated into plain collagen gels. Prolonging the incubation time to 24 hours allowed further migration of PBMCs only in the absence of ECs, but did not significantly increase the extent of EC-mediated migration.
Figure 1.
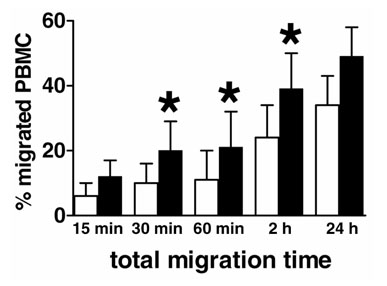
Endothelium enhances monocyte migration. The percentages of peripheral blood mononuclear cells (PBMCs) migrated in the absence (white columns) or presence (black columns) of endothelial cells are shown at different time points. Results are means ± SD for at least three independent experiments. Asterisks denote significant (P < 0.05) differences between the percentages of cells migrated in the absence of endothelium and in its presence.
The results and the statistical evaluation of the phenotypic analysis of monocytes recovered in various fractions of the migration assay are shown in Table 2. The expression of CD11a, CD33, CD45RO, CD54 and HLA-DR was significantly higher in MIG than in NAD. When compared with BND, these markers, and also CD45RB and CD62L, were significantly elevated in MIG. NAD, BND and MIG were incubated with collagenase for the same durations to control for possible cell activation by the collagenase treatment; the expression of adhesion molecules was similar to that on untreated cells.
Table 2.
Surface marker expression on different monocyte populations
| Marker | Initial | NAD | BND | MIG |
| CD11a | 312 ± 138 | 301 ± 154‡ | 273 ± 150 | 362 ± 187*† |
| CD33 | 138 ± 63 | 130 ± 49‡ | 118 ± 49 | 148 ± 70*† |
| CD45RA | 24 ± 15 | 25 ± 11 | 27 ± 11 | 46 ± 47 |
| CD45RB | 397 ± 149 | 367 ± 135‡ | 326 ± 134 | 375 ± 154† |
| CD45RO | 64 ± 35 | 80 ± 44 | 79 ± 50 | 111 ± 57*† |
| CD49d | 56 ± 10 | 53 ± 10 | 53 ± 12 | 65 ± 28 |
| CD54 | 41 ± 32 | 36 ± 12 | 35 ± 15 | 58 ± 33*† |
| CD62L | 22 ± 24 | 30 ± 24 | 29 ± 26 | 44 ± 33† |
| CD86 | 26 ± 19 | 36 ± 30 | 66 ± 103 | 26 ± 14 |
| HLA-DR | 174 ± 83 | 186 ± 89 | 207 ± 104 | 326 ± 254*† |
Data shown are fluorescence intensities (means ± SD) of the initial population and the nonadherent (NAD), bound (BND) and migrated (MIG) monocytes. In all 14 experiments performed, the migration period was 30 minutes; the endothelium was not pretreated. *Statistical significance (P < 0.05) MIG compared with NAD. †Statistical significance (P < 0.05) MIG compared with BND. ‡Statistical significance (P < 0.05) NAD compared with BND.
We also studied the capacity for monocyte migration into plain collagen gels, that is, in the absence of an endothelium. No significant difference in surface marker expression was observed when migrated monocytes were compared with any other fraction.
It has been reported that cytokines such as TNF-α, IL-1α and IFN-γ and also the chemokine MIP-1α can enhance the transendothelial migration of monocytes [19,20]. We were therefore interested to investigate whether pretreatment of the ECs with these factors would be sufficient to enhance the transendothelial migration of monocytes and/or to induce changes in their expression of surface markers. Figure 2 shows that pretreatment of the endothelium with any of the cytokines led to a consistent and significant increase in the number of migrated mononuclear cells in comparison with simultaneously performed control experiments in which the endothelium was not pretreated. Pretreatment with IL-1α was the most effective, resulting in a 132% increase in migrated cells (P < 0.001). The respective values for the other cytokines were as follows: MIP-1α, 194% (P = 0.043); TNF-α, 193% (P = 0.006); IFN-γ, 136% (P = 0.016).
Figure 2.
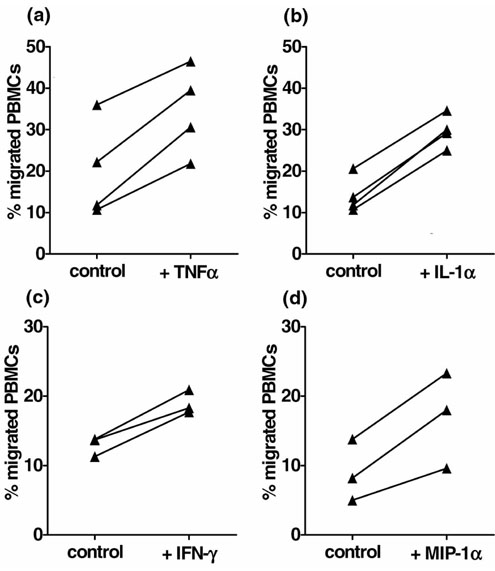
Monocyte migration is increased after the pretreatment of ECs with cytokines. Confluent monolayers of ECs that were formed on collagen gels were simultaneously incubated without cytokines (control) and with tumour necrosis factor-α (TNF-α) (a), interleukin-1α (IL-1α) (b), interferon-γ (IFN-γ) (c) or macrophage inflammatory protein-1α (MIP-1α) (d). The percentages of migrated peripheral blood mononuclear cells (PBMCs) after a migration period of 30 minutes are shown. Statistical significance: (a)P = 0.006; (b)P < 0.001; (c)P = 0.016; (d)P = 0.043.
Pretreatment of ECs with TNF-α led to a significant decrease in CD45RO and HLA-DR on migrated monocytes. In contrast, CD54 (ICAM-1) was significantly increased on monocytes that migrated through endothelium pretreated with TNF-α or IL-1α in comparison with migration through untreated endothelium (Figs 3 and 4, Table 3).
Figure 3.
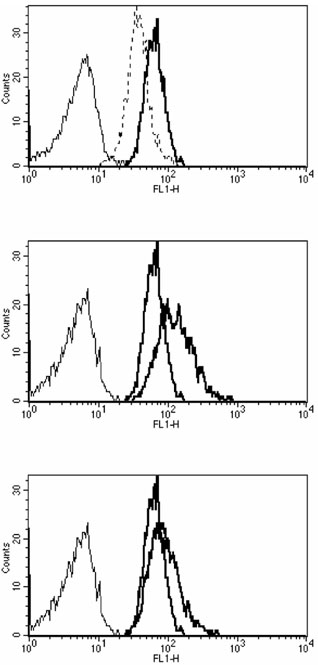
Migration through endothelium increases CD54 expression on monocytes. Histograms show the CD54 mean fluorescence intensity (mfi) of monocytes that migrated through untreated endothelium (grey line in each panel), or endothelium pretreated with tumour necrosis factor-α (black line in middle panel) or interleukin-1α (black line in bottom panel). CD54 mfi of the nonadherent monocyte fraction is shown by a dotted line (top panel), isotype controls are shown by a thin black line in each panel. The experiment shown is representative of three independent experiments.
Figure 4.
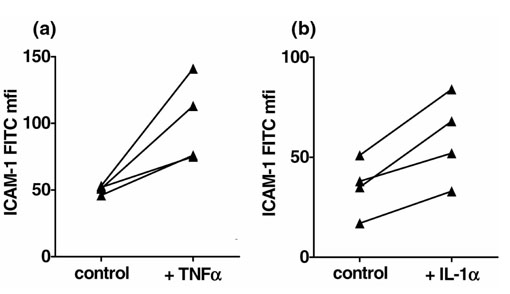
Cytokine-pretreated endothelium increases CD54 expression on monocytes. The fluorescein isothiocyanate (FITC) mean fluorescence intensity (mfi) of monocytes that migrated through endothelium pretreated with tumour necrosis factor-α (TNF-α) (a) or interleukin-1α (IL-1α) (b) is compared with the mfi of monocytes that simultaneously migrated through untreated endothelium (control). Statistical significance: (a)P = 0.043; (b)P = 0.019.
Table 3.
Changes in surface phenotypes of migrated monocytes
| Marker | TNF-α | IFN-γ | IL-1α | MIP-1α |
| CD11a | 96 ± 8 | 106 ± 16 | 76 ± 41 | 87 ± 9 |
| CD33 | 74 ± 49 | 105 ± 7 | 107 ± 16 | 165 ± 107 |
| CD45RA | 52 ± 53 | 134 ± 77 | 73 ± 65 | 63 ± 34 |
| CD45RB | 91 ± 7 | 86 ± 4 | 79 ± 44 | 84 ± 23 |
| CD45RO | 80 ± 9* | 98 ± 6 | 92 ± 8 | 78 ± 54 |
| CD49d | 92 ± 31 | 83 ± 26 | 81 ± 18 | 87 ± 21 |
| CD54 | 199 ± 55* | 111 ± 38 | 173 ± 27* | 76 ± 31 |
| CD62L | 88 ± 10 | 52 ± 39 | 105 ± 42 | 103 ± 16 |
| CD86 | 103 ± 25 | 219 ± 134 | 69 ± 25 | 90 ± 36 |
| HLA-DR | 86 ± 2* | 88 ± 18 | 87 ± 20 | 82 ± 52 |
The changes in individual surface markers after pretreatment of endothelial cells (ECs) with the indicated cytokines and chemokines are shown as percentages of control experiments with untreated ECs. All results were derived from at least three independent experiments for each cytokine or chemokine. HLA-DR, human leucocyte antigen-DR; IFN-γ, interferon-γ; IL-1α, interleukin-1α; MIP-1α, macrophage inflammatory protein-1α; TNF-α; tumour necrosis factor-α. *Statistically significant change (P < 0.05) compared with cells that migrated through untreated ECs.
When ECs were pretreated with IFN-γ or MIP-1α, no statistically significant change in monocyte surface markers was observed (Table 3).
Discussion:
Our experiments characterised the immuno-phenotype of monocytes that migrated through EC monolayers. Several surface molecules (CD11a, CD33, CD45RO, ICAM-1 and HLA-DR) were significantly increased on the migrated monocytes in comparison with the nonadherent population. The differences in the immunophenotype between migrating and nonadherent monocytes could be explained either by the preferential migration of a particular subset of monocytes or as a result of the interaction of monocytes with the endothelium. If the observed alterations in the immunophenotype of migrating monocytes had been due to the preferential migration of a particular subset, we would have expected a depletion of this subset in the nonadherent population. This did not occur; all surface markers studied were expressed to similar extents on the nonadherent and initial populations (see Table 2). The notion that the process of transendothelial migration leads to an upregulation of certain surface markers on monocytes is also supported by the fact that the expression of most markers was significantly higher in the migrated cells than in the bound cells (see Table 2). Taken together, our results suggest that transendothelial migration induces the activation or maturation of monocytes.
As described previously [19,20], we found that pretreatment of the endothelium with certain chemokines or cytokines enhanced transendothelial migration. The most striking phenotypic changes were seen for ICAM-1 expression, when ECs were pretreated with TNF-α and IL-1α (see Table 3 and Figs 3 and 4). In contrast, MIP-1α pretreatment did not change the monocyte phenotype investigated here. In the light of enhanced migration through MIP-1α-prestimulated endothelium, these results suggest a dichotomy of cytokine/chemokine effects on migration compared with surface marker expression: ECs activated with TNF-α and IL-1α seem to lead to an upregulation of both monocyte migration and surface marker expression, whereas MIP-1α only enhances migration, a finding that is compatible with the chemotactic chemokine nature of MIP-1α. Alternatively, MIP-1α could have been trapped in the collagen gel, acting as a chemotactic gradient directly on monocytes rather than via ECs. Thus TNF-α and IL-1α seem to mediate different proinflammatory events from those mediated by MIP-1α.
Our observations of increased expression of ICAM-1 on migrated monocytes after the pretreatment of ECs with TNF-α and IL-1α are especially remarkable because these cytokines are important in the pathogenesis of inflammation. In RA, TNF-α and IL-1 blockade showed an unequivocal therapeutic effect [21,22,23]. In addition, ICAM-1 and E-selectin levels of patients with RA who had received anti-TNF-α therapy decreased within a few days of the initiation of therapy [24].
Previous findings indicate that TNF-α and IL-1α induce an upregulation of the ICAM-1 counter-receptor on ECs [25]. This is consistent with the increase in cell migration found in our experiments and the altered expression of ICAM-1 on monocytes. Because the classic ICAM-1 counter-receptors LFA-1 and Mac-1 have not been detected on ECs, the existence of another ICAM-1 ligand, one that facilitates the transendothelial migration of monocytes, remains possible.
The reported changes indicate that, after migration, monocytes could become more liable to interact with T cells (which are known to enhance LFA-1 expression in the presence of TNF-α). This interaction might lead to a further mutual stimulation of T cells and macrophages. In fact the ligand-counterligand system consisting of LFA-1 and ICAM-1 also is one co-stimulatory pathway involved in interactions between antigen-presenting cells (APCs) and T cells [26]. Because our results demonstrate that both ICAM-1 and HLA-DR are upregulated on migrated monocytes, their function as APCs and possible ability to communicate with T cells, might be facilitated after transendothelial migration. Thus, this observation also supports previous notions on the importance of T cells in the pathogenesis of RA [27,28,29].
In summary, our findings indicate that monocyte migration is accompanied by changes in function-associated surface antigens and that TNF-α and IL-1α in particular increase the number of migrating monocytes and lead to an enhanced expression of certain surface markers involved in cell-cell interactions. These events might not only be partly responsible for the high inflammatory activity in RA synovium; they also suggest that ECs have a pivotal role in these processes and thus might constitute an important therapeutic target.
Introduction
Acute and chronic inflammation are characterised by an enhanced migration of leucocytes from the blood vessels through the endothelium into the extravascular tissue. There are ample data on the transendothelial migration of lymphocytes [1,2,3,4,5,6]. In addition, T cells capable of transendothelial migration have a characteristic immunophenotype; for instance, migrating T cells express significantly higher amounts of CD29 (β1-integrin) than cells that do not interact with endothelial cells (ECs) [7]. In contrast, much less is known about the extravasation of cells of the monocyte/macrophage lineage. Under normal conditions, only a few monocytes are able to migrate from the bloodstream into healthy tissue, where they differentiate into macrophages [8]. In inflammatory diseases such as rheumatoid arthritis (RA), monocytes accumulate in the synovial membrane and contribute significantly to the pathogenesis of the disease, mainly by secreting cytokines such as tumour necrosis factor-α (TNF-α) and interleukin-1 (IL-1) [9]. Transendothelial migration of monocytes might also be an important step in the pathogenesis of non-inflammatory diseases, for example atherosclerosis [10,11,12].
Several pairs of receptors and counter-receptors that mediate the interaction of monocytes with ECs have been described [13,14,15,16,17], but the phenotype of monocytes capable of transendothelial migration was not defined. Previously we found an enhanced expression of CD54 [intercellular cell-adhesion molecule (ICAM)-1] on monocytes that had migrated through an unstimulated EC monolayer [18].
It was the aim of the present study to perform a detailed characterisation of the immunophenotype of transendothelially migrated monocytes.
Materials and methods
Cell cultures
ECs were isolated from human umbilical cord veins by digestion with collagenase as described previously [7]. The culture medium consisted of MCDB-M 104 glutamine (Gibco, Paisley, UK) supplemented with 20% (v/v) fetal calf serum (FCS), 24 μg/ml EC growth supplement (TC Laevosan, Vienna, Austria), 50 IU/ml heparin, 2 mM L-glutamine (Gibco), penicillin (100 IU/ml; Gibco) and streptomycin (100 μg/ml; Gibco). ECs in the third to fifth passage were used.
Preparation of peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMCs) were prepared from buffy coats of healthy blood donors by centrifugation over gradients of Ficoll-Hypaque (Histopaque R, Sigma, Vienna, Austria). PBMCs were prepared immediately before starting the experiments.
Monocyte-EC binding and transendothelial migration
Interactions with ECs in PBMC populations were examined on hydrated bovine collagen gels in the wells of 16 mm macrowell tissue culture plates, as described previously [7]. In brief, the collagen gels were made of 50% (v/v) bovine collagen (Vitrogen 100; Collagen Biomaterials, Palo Alto, California, USA), 7% (v/v) NaOH, 10% (v/v) 10 × phosphate-buffered saline (PBS; Gibco) and 33% (v/v) distilled water. To form a confluent monolayer on the collagen gels, 5 × 105 ECs per well were incubated overnight.
To measure monocyte interaction with ECs, PBMCs (3 × 106) were resuspended in fresh culture medium, layered on top of collagen gels with and without ECs and incubated at 37°C. The range of the incubation period was 15 minutes to 24 hours. Nonadherent cells (NAD) were harvested by washing twice with culture medium. Cells bound to the surface (BND) were enriched by washing each well twice with warm (37°C) Puck's EDTA, twice with warm (37°C) EGTA (0.5 mM EGTA in PBS) and once with cold (4°C) Puck's EDTA. Finally, for the recovery of those cells that had migrated into the collagen gels (MIG), 0.7 ml of a solution containing 0.1% collagenase (Sigma), 1% (v/v) FCS and 50mM Hepes buffer (Gibco) was added per well. The collagen gels were then gently minced with a pipette and incubated for 60 minutes at 37°C, after which the migrated PBMCs were removed by washing the wells twice with PBS. Each population (NAD, BND and MIG) was washed, resuspended in culture medium and counted by microscope. In some experiments monocyte we studied migration into plain collagen gels. In these experiments no ECs were layered on the collagen gels; in other respects the experiments were performed exactly as described above.
Pretreatment of ECs
In some experiments the EC monolayer was preactivated by incubation with TNF-α (Pharma Biotechnology, Hannover, Germany), IL-1α, macrophage inflammatory protein-1α (MIP-1α) or interferon-γ (IFN-γ) (all purchased from Serotec, Oxford, UK). To this end, the medium in each well was removed and the ECs were incubated for 5 hours at 37°C with or without the respective cytokines or chemokines (100 IU/ml TNF-α, IL-1α or IFN-γ, or 50 ng/ml MIP-1α). After this 5-hour pretreatment, each well was washed thoroughly and the migration assay was performed as described above.
Analysis of monocyte surface markers by dual colour flow cytometry
Staining of monocytes was performed with fluorescein isothiocyanate (FITC)-conjugated antibodies against CD11a, CD33, CD45RA, CD45RO, CD49d (α4-integrin), CD54 (ICAM-1), CD86, HLA-DR (all purchased from Serotec), CD45RB (Dako, Glostrup, Denmark) and CD62L (L-selectin) (Becton-Dickinson, San Jose, California, USA) (functions and ligands of these surface markers are shown in Table 1). To distinguish monocytes from other immune cells, all samples were counterstained with a phycoerythrin-labelled anti-CD14 monoclonal antibody. Cells (3 × 105 to 4 × 105 per sample) were incubated at 4°C for 30 minutes. Cells were then pelleted and resus-pended in 250 μl of PBS before analysis was performed on a flow cytometer (FACScan; Becton-Dickinson). All results are expressed as the respective mean fluorescence intensity among CD14-positive cells. Because not only monocytes but also ECs express CD54 (ICAM-1), in the analyses of the expression of CD54 on monocytes by fluorescence-activated cell sorting, monocytes were defined by both the scatter profile and the expression of CD14. In addition, ECs, which are considerably larger, were excluded by size.
Statistics
All data are presented as means ± SD. Paired Student's t-tests were used for comparisons.
Results
Transendothelial migration of PBMCs
In initial experiments we studied the time course of PBMC migration into plain or EC-coated collagen gels, respectively. As shown in Fig. 1, the presence of an endothelium clearly facilitated the migration of PBMCs: after 30 minutes the percentage of PBMCs that had migrated was twice as high as that in the absence of ECs. After 2 hours, about 40% of the PBMCs could be recovered from collagen gels coated with an EC layer, whereas only 24% PBMCs had migrated into plain collagen gels. Prolonging the incubation time to 24 hours allowed further migration of PBMCs only in the absence of ECs, but did not significantly increase the extent of EC-mediated migration.
Phenotypic analysis of monocytes capable of transendothelial migration
The results and the statistical evaluation of the phenotypic analysis of monocytes recovered in various fractions of the migration assay are shown in Table 2. The expression of CD11a, CD33, CD45RO, CD54 and HLA-DR was significantly higher in MIG than in NAD. When compared with BND, these markers, and also CD45RB and CD62L, were significantly elevated in MIG. NAD, BND and MIG were incubated with collagenase for the same durations to control for possible cell activation by the collagenase treatment; the expression of adhesion molecules was similar to that on untreated cells.
Phenotypic analysis of monocytes migrated into plain collagen gels
We also studied the capacity for monocyte migration into plain collagen gels, that is, in the absence of an endothelium. No significant difference in surface marker expression was observed when migrated monocytes were compared with any other fraction.
Effect of pretreatment of ECs on the transendothelial migration of monocytes
It has been reported that cytokines such as TNF-α, IL-1α and IFN-γ and also the chemokine MIP-1α can enhance the transendothelial migration of monocytes [19,20]. We were therefore interested to investigate whether pretreat-ment of the ECs with these factors would be sufficient to enhance the transendothelial migration of monocytes and/or to induce changes in their expression of surface markers. Figure 2 shows that pretreatment of the endothelium with any of the cytokines led to a consistent and significant increase in the number of migrated mononuclear cells in comparison with simultaneously performed control experiments in which the endothelium was not pretreated. Pretreatment with IL-1α was the most effective, resulting in a 132% increase in migrated cells (P < 0.001). The respective values for the other cytokines were as folows: MIP-1α, 194% (P = 0.043); TNF-α, 193% (P = 0.006); IFN-γ, 136% (P = 0.016).
Effect of pretreatment of ECs on the immunophenotype of migrated monocytes
Pretreatment of ECs with TNF-α led to a significant decrease in CD45RO and HLA-DR on migrated monocytes. In contrast, CD54 (ICAM-1) was significantly increased on monocytes that migrated through endothelium pretreated with TNF-α or IL-1α in comparison with migration through untreated endothelium (Figs 3 and 4, Table 3).
When ECs were pretreated with IFN-γ or MIP-1α, no statistically significant change in monocyte surface markers was observed (Table 3).
Discussion
Our experiments characterised the immunophenotype of monocytes that migrated through EC monolayers. Several surface molecules (CD11a, CD33, CD45RO, ICAM-1 and HLA-DR) were significantly increased on the migrated monocytes in comparison with the nonadherent population. The differences in the immunophenotype between migrating and nonadherent monocytes could be explained either by the preferential migration of a particular subset of monocytes or as a result of the interaction of monocytes with the endothelium. If the observed alterations in the immunophenotype of migrating monocytes had been due to the preferential migration of a particular subset, we would have expected a depletion of this subset in the non-adherent population. This did not occur; all surface markers studied were expressed to similar extents on the nonadherent and initial populations (see Table 2). The notion that the process of transendothelial migration leads to an upregulation of certain surface markers on monocytes is also supported by the fact that the expression of most markers was significantly higher in the migrated cells than in the bound cells (see Table 2). Taken together, our results suggest that transendothelial migration induces the activation or maturation of monocytes.
The fact that monocytes that migrated into collagen gels without an endothelium failed to change their immunophenotype indicates that monocyte differentiation might be signalled not by the migration step itself or by the presence of collagen but by interactions between monocytes and ECs. Additional control experiments proved that collagenase itself did not lead to any of the observed changes. Recent observations with lymphocytes indicate that at least some of the changes could be due to the transfer of surface molecules from ECs to monocytes [30]. It is not known at present whether this is also true of monocytes. For T cells a preferential migration of a CD45RO-positive subset is already known [2,7]. Here we found an upregulation of CD45RO in the whole population of migrated monocytes; the significance of this finding is not clear and also merits further elucidation.
CD11a on monocytes, and its ligand ICAM-1 on ECs, are known to be important for adhesion in monocyte migration [31]; our observation of upregulated CD11a on monocytes after transendothelial migration is in line with these data.
Audran et al [32] compared the difference in the expression of certain adhesion molecules between monocytes and differentiated macrophages. They found that ICAM-1 increased during differentiation and showed stronger expression on macrophages than on monocytes. Moreover, monocytes and macrophages that were recovered from inflammatory sites, such as from the synovial fluid of patients with RA [33], from bronchial biopsies of patients with asthma [34] or from peritoneal fluid of patients with peritonitis [35], had a high expression of ICAM-1. These findings and our results therefore suggest that the immunophenotype of macrophages or monocytes recovered from inflammatory lesions is determined, at least in part, by the process of transendothelial migration.
As described previously [19,20], we found that pretreatment of the endothelium with certain chemokines or cytokines enhanced transendothelial migration. The most striking phenotypic changes were seen for ICAM-1 expression, when ECs were pretreated with TNF-α and IL-1α (see Table 3 and Figs 3 and 4). In contrast, MIP-1α pretreatment did not change the monocyte phenotype investigated here. In the light of enhanced migration through MIP-1α-prestimulated endothelium, these results suggest a dichotomy of cytokine/chemokine effects on migration compared with surface marker expression: ECs activated with TNF-α and IL-1α seem to lead to an upregulation of both monocyte migration and surface marker expression, whereas MIP-1α only enhances migration, a finding that is compatible with the chemotactic chemokine nature of MIP-1α. Alternatively, MIP-1α could have been trapped in the collagen gel, acting as a chemotactic gradient directly on monocytes rather than via ECs. Thus TNF-α and IL-1α seem to mediate different proinflammatory events from those mediated by MIP-1α.
Generally, the influence of ECs on monocytes could be explained in two ways: either such signals are provided by cell-cell interaction via pairs of receptors and counter-receptors during the process of migration, or, alternatively, ECs could secrete chemoattractants. However, in the latter case we would instead expect similar effects on the bound subpopulation as well as a higher percentage of EC-bound monocytes.
Our observations of increased expression of ICAM-1 on migrated monocytes after the pretreatment of ECs with TNF-α and IL-1α are especially remarkable because these cytokines are important in the pathogenesis of inflammation. In RA, TNF-α and IL-1 blockade showed an unequivocal therapeutic effect [21,22,23]. In addition, ICAM-1 and E-selectin levels of patients with RA who had received anti-TNF-α therapy decreased within a few days of the initiation of therapy [24].
Previous findings indicate that TNF-α and IL-1α induce an upregulation of the ICAM-1 counter-receptor on ECs [25]. This is consistent with the increase in cell migration found in our experiments and the altered expression of ICAM-1 on monocytes. Because the classic ICAM-1 counter-receptors LFA-1 and Mac-1 have not been detected on ECs, the existence of another ICAM-1 ligand, one that facilitates the transendothelial migration of monocytes, remains possible.
The reported changes indicate that, after migration, monocytes could become more liable to interact with T cells (which are known to enhance LFA-1 expression in the presence of TNF-α). This interaction might lead to a further mutual stimulation of T cells and macrophages. In fact the ligand-counterligand system consisting of LFA-1 and ICAM-1 also is one co-stimulatory pathway involved in interactions between antigen-presenting cells (APCs) and T cells [26]. Because our results demonstrate that both ICAM-1 and HLA-DR are upregulated on migrated monocytes, their function as APCs and possible ability to communicate with T cells, might be facilitated after transendothelial migration. Thus, this observation also supports previous notions on the importance of T cells in the pathogenesis of RA [27,28,29].
We also observed an upregulation of CD33 on migrated monocytes. In this respect it is noteworthy that Randolph et al. reported monocyte differentiation into dendritic cells after reverse transmigration [36]; furthermore, it was shown that dendritic cells isolated from RA synovial fluid expressed high levels of CD33 [37]. Because it is known that the synovial lesions in RA are enriched in dendritic cells [37], it is tempting to speculate that some CD33 high monocytes might differentiate into dendritic cells in inflammatory sites.
Previous studies investigated the transendothelial migration of lymphocytes in particular T cells. For both monocytes and T cells the presence of an endothelium facilitated the migration into collagen gels; however, in contrast with the monocyte situation, the capacity of T cells is an intrinsic ability of certain subpopulations, for example CD4-positive memory cells [7]. Interestingly, the activation of ECs by IL-1 and IFN-γ shows the same migration-enhancing effect on T cells as seen here for monocytes [7,38]. Oppenheimer-Marks et al [2,39] reported that ICAM-1 on T cells promoted binding and migration through non-activated endothelium. This conclusion is consistent with our results: ICAM-1 seems to be a pivotal adhesion molecule in the transendothelial migration of both monocytes and T cells. Moreover, in a study in vivo it was observed that the application of monoclonal antibodies against ICAM-1 induced T cell hyporesponsiveness in patients with RA [40] and led to a clinical benefit in patients with early or subacute RA.
In summary, our findings indicate that monocyte migration is accompanied by changes in function-associated surface antigens and that TNF-α and IL-1α in particular increase the number of migrating monocytes and lead to an enhanced expression of certain surface markers involved in cell-cell interactions. These events might not only be partly responsible for the high inflammatory activity in RA synovium; they also suggest that ECs have a pivotal role in these processes and thus might constitute an important therapeutic target.
Abbreviations
APC = antigen-presenting cell; BND = population of cells bound to the surface; EC = endothelial cell; ICAM = intercellular cell-adhesion molecule; IFN-γ = interferon-γ; IL = interleukin; MIG = population of cells migrated into collagen gel; MIP = macrophage inflammatory protein; NAD = non-adherent cell population; PBMC = peripheral blood mononuclear cells; PBS = phosphate-buffered saline; RA = rheumatoid arthritis; TNF-α = tumour necrosis factor-α.
Acknowledgments
Acknowledgements
This work was supported in part by a research grant from Solvay Pharma, Klosterneuburg, Austria.
References
- Vachula M, Van Epps D. In vitro models of lymphocyte transendothelial migration. Invasion Metastasis. 1992;12:66–81. [PubMed] [Google Scholar]
- Oppenheimer-Marks N, Lipsky PE. The adhesion and transendothelial migration of human T-lymphocyte subsets. Behring Inst Mitt. 1993;92:44–50. [PubMed] [Google Scholar]
- Ziff M. Pathways of mononuclear cell infiltration in rheumatoid synovitis. Rheumatol Int. 1989;9:97–103. doi: 10.1007/BF00271865. [DOI] [PubMed] [Google Scholar]
- Azzali G, Orlandini G, Gatti R. The migration of lymphocytes and polymorphonuclear leukocytes across the endothelial wall of the absorbing peripheral lymphatic vessel. J Submicrosc Cytol Pathol. 1990;22:543–549. [PubMed] [Google Scholar]
- Pankonin G, Reipert B, Ager A. Interactions between interleukin-2-activated lymphocytes and vascular endothelium: binding to and migration across specialized and non-specialized endothelium. Immunology. 1992;77:51–60. [PMC free article] [PubMed] [Google Scholar]
- Lidington E, Nohammer C, Dominguez M, Ferry B, Rose ML. Inhibition of the transendothelial migration of lymphocytes but not monocytes by phosphodiesterase inhibitors. Clin Exp Immunol. 1996;104:66–71. doi: 10.1046/j.1365-2249.1996.d01-660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann P, Cush J, Lipsky P, Oppenheimer-Marks N. Identification of subsets of human T cells capable of enhanced transendothelial migration. J Immunol. 1992;149:1170–1178. [PubMed] [Google Scholar]
- van Furth R. Monocyte production during inflammation. Comp Immunol Microbiol Infect Dis. 1985;8:205–211. doi: 10.1016/0147-9571(85)90045-1. [DOI] [PubMed] [Google Scholar]
- Arend WP, Dayer JM. Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum. 1990;33:305–315. doi: 10.1002/art.1780330302. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of artherosclerosis: a perspective study for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Masuyama J, Ikeda I, Kasahara T, Kitigawa S, Takahashi Y, Shimeda K, Kano S. Induction of monocyte chemoattractant protein-1 synthesis in human monocytes during transendothelial migration in vitro. Circ Res. 1995;76:750–757. doi: 10.1161/01.res.76.5.750. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis - an inflammatory disease. New Engl JMed. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Chuluyan HE, Issekutz AC. VLA-4 integrin can mediate CD-11/CD-18-independent transendothelial migration of human monocytes. J Clin Invest. 1993;92:2768–2777. doi: 10.1172/JCI116895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerschaert JA, Furie MB. Monocytes use either CD-11/CD-18 or VLA-4 to migrate across human endothelium in vitro. J Immunol. 1994;152:1915–1926. [PubMed] [Google Scholar]
- Meerschaert JA, Furie MB. The adhesion molecules used by monocytes for migration across endothelium include CD-11a/CD-18, CD-11b/CD-18, and VLA-4 on monocytes and ICAM-1, VCAM-1 and other ligands on endothelium. J Immunol. 1995;154:4099–4112. [PubMed] [Google Scholar]
- Shang XZ, Issekutz AC. Contribution of CD 11a/CD 18, CD 11b/CD 18, ICAM-1 (CD 54), and -2 (CD 102) to human monocyte migration through endothelium and connective tissue fibroblast barriers. Eur J Immunol. 1998;28:1970–1979. doi: 10.1002/(SICI)1521-4141(199806)28:06<1970::AID-IMMU1970>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Shang XZ, Lang BJ, Issekutz AC. Adhesion molecule mechanisms mediating monocyte migration through synovial fibroblast and endothelium barriers: role for CD-11/CD-18, very late antigen-4 (CD-49d/CD-29), very late antigen-5 (CD-49e/CD-29), and vascular cell adhesion molecule-1 (CD-106). J Immunol. 1998;160:467–474. [PubMed] [Google Scholar]
- Pietschmann P, Stohlawetz P, Brosch S, Steiner G, Smolen JS, Peterlik M. The effect of alendronate on cytokine production, adhesion molecule expression, and transendothelial migration of human peripheral blood mononuclear cells. Calcif Tissue Int. 1998;63:325–330. doi: 10.1007/s002239900535. [DOI] [PubMed] [Google Scholar]
- Issekutz AC, Issekutz TB. Quantitation and kinetics of blood monocyte migration to acute inflammatory reactions, and IL-1α, tumor necrosis factor-1α, and IFN-γ. J Immunol. 1993;151:2105–2115. [PubMed] [Google Scholar]
- Chuluyan HE, Schall TJ, Yoshimura T, Issekutz AC. IL-1 activation of endothelium supports VLA-4 (CD-49d/CD-29)-mediated monocyte transendothelial migration to C5a, MIP-1α, RANTES, and PAF but inhibits migration to MCP-1: a regulatory role for endothelium-derived MCP-1. J Leukoc Biol. 1995;58:71–79. doi: 10.1002/jlb.58.1.71. [DOI] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H, Woody JN. Randomised double blind comparsion of chimeric monoclonal antibody to tumor necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, Weaver AL, Keystone EC, Furst DE, Mease PJ, Rudermann EM, Horwitz DA, Arkfed DG, Garrison L, Burge DJ, Blosch CM, Lange ML, McDonnell ND, Weinblatt ME. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130:478–486. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- Bresnihan J, Alvaro-Gracia JM, Cobby M, Doherty M, Domljan Z, Emery P, Nuki G, Pavelka K, Rau R, Rozman B, Watt I, Williams B, Aitchison R, McCabe D, Musikic P. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–2204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Paleolog EM, Hunt M, Elliott MJ, Feldmann M, Maini RN, Woody JN. Deactivation of vascular endothelium by monoclonal anti-tumour necrosis factor α antibody in rheumatoid arthritis. Arthritis Rheum. 1996;39:1082–1091. doi: 10.1002/art.1780390703. [DOI] [PubMed] [Google Scholar]
- Stratowa C, Audette M. Transcriptional regulation of the human intercellular adhesion molecule-1 gene: a short overview. Immunobiology. 1995;193:293–304. doi: 10.1016/S0171-2985(11)80558-9. [DOI] [PubMed] [Google Scholar]
- Zuckerman LA, Pullen L, Miller J. Functional consequences of costimulation by ICAM-1 on IL-2 gene expression and T cell activation. J Immunol. 1998;160:3259–3268. [PubMed] [Google Scholar]
- Smolen JS, Tohidast-Akrad M, Gal A, Kunaver M, Eberl G, Zenz P, Falus A, Steiner G. The role of T-lymphocytes in rheumatoid arthritis. Scand J Rheumatol. 1996;25:1–4. doi: 10.3109/03009749609082660. [DOI] [PubMed] [Google Scholar]
- Panayi GS. Targeting of cells involved in the pathogenesis of rheumatoid arthritis. Rheumatology. 1999;38 (suppl 2):8–10. [PubMed] [Google Scholar]
- Weyand CM. New insights into the pathogenesis of rheumatoid arthritis. Rheumatology. 2000;39 (suppl 1):3–8. doi: 10.1093/oxfordjournals.rheumatology.a031491. [DOI] [PubMed] [Google Scholar]
- Brezinschek R, Oppenheimer-Marks N, Lipsky PE. Activated T cells aquire endothelial cell surface determinants during transendothelial migration. J Immunol. 1999;162:1677–1684. [PubMed] [Google Scholar]
- Beekhuizen H, Blokland I, van Furth R. Cross-linking of CD14 molecules results in a CD11/CD18- and ICAM-1-dependent adherence to cytokine-stimulated human endothelial cells. J Immunol. 1993;150:950–959. [PubMed] [Google Scholar]
- Audran R, Lesimple T, Delamaire M, Picot C, van Damme J, Toujas L. Adhesion molecule expression and response to chemotactic agents of human monocyte-derived macrophages. Clin Exp Immunol. 1996;103:155–160. doi: 10.1046/j.1365-2249.1996.d01-4.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köller M, Aringer M, Kiener H, Erlacher L, Machold K, Eberl G, Graninger W, Smolen J. Expression of adhesion molecules on synovial fluid and peripheral blood monocytes in patients with inflammatory joint disesease and osteoarthritis. Ann Rheum Dis. 1999;58:709–712. doi: 10.1136/ard.58.11.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset P, Tillie-Leblond I, Janin A, Copin MC, Wallaert B, Tonnel AB. Expression of E-selectin, ICAM-1 and VCAM-1 on bronchial biopsies from allergic and non-allergic asthma patients. Int Arch Allergy Immunol. 1995;106:69–77. doi: 10.1159/000236892. [DOI] [PubMed] [Google Scholar]
- Faull RJ, Wang J, Stavros W. Changes of the expression of adhesion molecules as peripheral blood monocytes differentiate into peritoneal macrophages. Nephrol Dial Transplant. 1996;11:2037–2044. doi: 10.1093/oxfordjournals.ndt.a027093. [DOI] [PubMed] [Google Scholar]
- Randolph GA, Beaulieu S, Lebeque S, Steinmann RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. [PubMed] [Google Scholar]
- Thomas R, Quinn C. Functional differentiation of dendritic cells in rheumatoid arthritis: role of CD86 in the synovium. J Immunol. 1996;156:3074–3086. [PubMed] [Google Scholar]
- Ding Z, Xiong K, Issekutz TB. Regulation of chemokine-induced transendothelial migration of T lymphocytes by endothelial activation: differential effects on naive and memory T cells. J Leukoc Biol. 2000;67:825–833. doi: 10.1002/jlb.67.6.825. [DOI] [PubMed] [Google Scholar]
- Oppenheimer-Marks N, Davis LS, Bogue DT, Ramberg J, Lipsky PE. Differential utilisation of ICAM-1 and VCAM-1 during the adhesion and transendothelial migration of human T-lymphocytes. J Immunol. 1991;147:2913–2921. [PubMed] [Google Scholar]
- Davis LS, Kavanaugh AF, Nichols LA, Lipsky PE. Induction of persistent T cell hyporesponsiveness in vivo by monoclonal antibody to ICAM-1 in patients with rheumatoid arthritis. J Immunol. 1995;154:3525–3537. [PubMed] [Google Scholar]