

Abstract
Adenosine-to-inosine (A-to-I) RNA editing is a post-transcriptional modification of pre-mRNA catalysed by an RNA-specific adenosine deaminase (ADAR). A-to-I RNA editing has been previously reported in the pre-mRNAs of brain glutamate and serotonin receptors and in lung tissue during inflammation. Here we report that systemic inflammation markedly induces inosine-containing mRNA to approximately 5% of adenosine in total mRNA. Induction was the result of up-regulation of A-to-I RNA editing as both dsRNA editing activity and ADAR1 expression were increased in the spleen, thymus and peripheral lymphocytes from endotoxin-treated mice. Up-regulation of ADAR1 was confirmed in vitro in T lymphocytes and macrophages stimulated with a variety of inflammatory mediators including tumour necrosis factor-α and interferon-γ. A late induction of RNA editing was detected in concanavalin A-activated splenocytes stimulated with interleukin-2 in vitro. Taken together, these data suggest that a large number of inosine-containing mRNAs are produced during acute inflammation via up-regulation of ADAR1-mediated RNA editing. These events may affect the inflammatory and immune response through modulation of protein production.
Introduction
Adenosine to inosine (A-to-I) RNA editing is a modification of pre-messinger RNA (pre-mRNA) by RNA-specific adenosine deaminases that leads to the production of protein isoforms.1–3 This process appears to be well conserved during evolution as A-to-I RNA editing enzymes and homologous gene products were identified in a variety of species including mammals,4–8 Drosophila,9 zebrafish10 and Xenopus.11 In mammals, four A-to-I RNA editing enzymes, ADAR1, ADAR2, ADAR3 and ADAT1 have been cloned.4–8 ADAR1 and ADAR2 are widely expressed in many tissues and cells4,5,12 and are capable of both non-specific editing of double-stranded RNA (dsRNA) and site-specific editing of glutamate receptor subunit B (gluR-B) mRNA and serotonin receptor mRNA.1,13 ADAR3 was detected exclusively in the brain and its deaminase activity has not been demonstrated in vitro, possibly because of its substrate has not yet been identified.7 ADAT1, which was cloned from the human,8 mouse14 and Drosophila,15 specifically targets transfer RNA (tRNA).16
Only limited data exist regarding the functional consequences of A-to-I RNA editing. One function was demonstrated in the mammalian brain, where editing of gluR-B pre-mRNA by ADAR2 has been shown to alter the calcium permeability of excitatory neurons.17,18 Furthermore, studies in mice homozygous for a targeted functional null allele showed that ADAR2−/− animals displayed substantially reduced A-to-I RNA editing activity in diverse mRNAs, which was associated with seizures and mortality.19 In the Drosophila brain, disruption of the dADAR (a homologue of ADAR2) gene, completely abolished sodium (Para), calcium (Dmca1A), and chloride (DrosGluCl-alpha) channels.20–22 Mutants lacking dADAR exhibited extreme behavioural deficits and neuronal degeneration.22 In addition, an oxygen-sensitive dADAR mutant displayed prolonged recovery from anoxic stupor, vulnerability to heat shock, and increased O2 demands.21 Thus, editing of ion channel pre-mRNAs by dADAR appears to be critical for the integrity and function of the central nervous system.
The function of ADAR1 is also still obscure. Nevertheless, it has been shown that editing by ADAR1 is crucial for the embryonic development of erythrocytes as low editing activity in these cells affects their proliferation and differentiation.23 Moreover, studies in chimeric mouse embryos with a functional null allele for ADAR1 revealed a heterozygous embryonic lethal phenotype. Another function for ADAR1 could be protection of host cells against viral infections.24 This theory draws credence from several observations. First, ADAR1 edits and unwinds dsRNA and therefore can theoretically modulate dsRNA-dependent regulators containing dsRNA-binding domains. Second, editing by ADAR1 in cell extracts in vitro can be inhibited by synthetic VAI virus RNA.24,25
Very recently, ADAR1 has been reported to play a role in the development of acute, local lung inflammation induced by endotoxin in mice.26 In that report, A-to-I editing activity and ADAR1 mRNA expression were rapidly (2 hr) up-regulated in polymorphonuclear and epithelial cells from inflamed lungs. These events preceded the development of pulmonary oedema and leucocyte accumulation in lung tissue, and followed the local production of interferon-γ (IFN-γ), a known inducer of ADAR1 in other cell systems. The present study aimed to investigate further the role of ADAR1 in acute inflammation with emphasis on ADAR1 activity and expression in systemic inflammation and following stimulation of inflammatory cells in vitro.
Materials and methods
Materials
Endotoxin from Escherichia coli 0111:B4 (Sigma, St. Louis, MO) was used as a fresh solution in 0·9% NaCl. Recombinant mouse tumour necrosis factor-α (TNF-α) and IFN-γ (Sigma) were diluted to make 10 μg/ml and 106 U/ml, respectively. Concanavalin A (Con A, Sigma) was obtained commercially. The E. coli strain DH5α was incubated in Luria Broth at 37° until an optical density at 600 nm (OD600) of 1.0 was reached or 5 × 108 colony-forming units (CFU)/ml. Human adenovirus type V was prepared by infecting 293 cells [American Type Culture Collection (ATCC, Manassas, VA) #CRL-1573] with 10% fetal calf serum at 37°. The transfected cells and supernatant were harvested, cells were disrupted by ultrasonic manipulation, and the suspension was clarified by centrifugation (8000 g for 20 min). The supernatant was layered over 30% sucrose in STEU buffer [NaCl 0·1 m, Tris–HCl 0·01 m, ethylenediaminetetraacetic acid (EDTA) 0·001 m, and urea 1 m] and centrifuged at 100 000 g for 1 hr. The virus pellet was resuspended in phosphate-buffered saline (PBS) and frozen at − 70°. The virus titre was determined and adjusted to 1010 plaque-forming units (PFU)/ml.27
Mouse acute inflammation protocol
The model utilized was described previously.28–30 In brief, adult male C57BL/6 mice (6–8 week-old, 20–25 g; Charles River Laboratories, Cambridge, MA) were used. Endotoxin at 15 mg/kg (lethal dose 60%; LD60) was injected into the peritoneal cavity of conscious animals and tissues were harvested at several time-points for determination of editing activity and expression of editing enzymes. The Yale Animal Care and Use Committee approved the protocol.
RNA editing activity in inflamed tissues
Tissues from six mice were collected at each time point and homogenized in four volumes of editing buffer (20 mm HEPES, pH 7·9, 100 mm KCl, 5 mm EDTA, 0·5% nonidet-40, 10% glycerol). The lysate was sonicated for 30 seconds and then centrifuged for 30 seconds at 4000 g. The supernatant was collected and the protein concentration was quantified and adjusted to 10 mg/ml. RNA editing activity was determined as previously described.12 In a typical dsRNA editing assay, 10 μl of cell extracts (100 μg total protein) were mixed with 1 μl of 32P-labelled dsRNA and incubated at 37° for 1 hr. The mixture was treated with an equal volume of PK buffer (10 mm Tris–HCl pH 8·0, 300 mm NaCl, 0·2% sodium dodecyl sulphate, 0·5 mg/ml proteinase K). The edited dsRNA was extracted with phenol, precipitated in ethanol, resuspended in 10 μl of RNase P1 mix (5 mm Tris–HCl pH 7·5, 10 mm NaCl, 1 unit of RNase P1) and incubated at 37° for 2 hr. After development in saturated (NH4)2SO4–isopropanol (95 : 5) solution, the converted inosine was analysed on thin-layer chromatography (TLC), visualized by autoradiography and quantified by scintillation.
RNA editing activity in cultured cells
Mouse cytotoxic T lymphocytes (CTLL-2, ATCC # TIB-214), alveolar macrophages (MH-S, ATCC # CRL-2019), fibroblasts (3T3, ATCC #CCL-92), or human umbilical vein endothelial cells (HUVEC) were cultured in triplets (n = 3) in proper culture media. Approximately 1 × 106 cells were stimulated with IFN-γ (1000 U/ml) or TNF-α (10 ng/ml) for several time periods, ranging from 0 to 4 hr. Macrophages were also cultured with escalating concentrations of E. coli (0–108 CFU/ml) or adenovirus [0–0·1 multiplicity of infection (MOI)] for 2 hr. For the RNA editing assay, cells were collected, resuspended in two volumes of editing buffer and sonicated as described above. Protein concentration was determined and equalized in each sample and editing activity was determined. To examine RNA editing activity in splenic cells activated by Con A and interleukin-2 (IL-2), spleens harvested from sham mice (n = 4), gently pressed through 40 µM Nylon Cell Strainer (Becton-Dickinson Labware, Franklin Lakes, NJ, USA). The primary cells were washed with PBS and activated in the presence of 500 units/ml of IL-2 and 2 μg/ml of Con A. Cells were collected after 0, 9, 30, 72 and 90 hr of incubation, washed with PBS and lysed by sonication in the editing buffer as described above. Protein concentration was determined and equalized in each sample and editing activity on dsRNA substrate was assayed (see above).
Northern blotting, Western blotting, reverse transcription–polymerase chain reaction (RT-PCR) and sequencing
For Northern blotting and RT-PCR, total RNA and mRNA were isolated from spleens (n = 6), thymuses (n = 60), or cultured cells (n = 4) using Trizol (Invitrogen, Carlsbad, CA) and Oligotex (Qiagen Inc., Valencia, CA) as described in the manufacturers' instructions. The quantity of the isolated RNA was measured using UV spectrometry. Equal amounts of mRNA (∼4 μg) were used for Northern blotting. ADAR1 and ADAR2 were detected by hybridization of the blots with 32P-labeled antisense probes (ADAR1, 1305–1265 and 3004–2996, GenBank accession number AF291050; ADAR2, 296–261, AF291049). Membranes were hybridized at 65° over night and washed with 0·1 × sodium saline citrate at 55° (final wash) for 10 min. For RT-PCR, 2 μg of total RNA was used for reverse transcription primed with poly(dT)12−18. ADAR1 mRNA was determined by semi-quantitative RT-PCR31 using primers that flank exons 5–8 (1975–2003 and 2436–2408, AF291050) or exons 1–2 (1–24 and 2436–2408, AF291050). PCR was performed at 60° for 30 seconds; 72° for 30 seconds; 94° for 30 seconds for 20, 22, 24, 26, 28 and 30 cycles. Only the results of cycle 26 are shown. PCR amplification of the mouse GAPDH gene in each sample was monitored as control for the semi-quantitative RT-PCR assay. For Western blotting, 60 μg total protein from splenic cells stimulated with Con A for different time periods was resolved on 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred on to polyvinyl difluoride membrane. The blot was detected using rabbit antiserum against mouse ADAR1 (from 2765 to 3459).
Given the surprisingly high value of ∼5% A conversion to I, it was essential to confirm this finding by direct cloning and sequencing of representative cDNAs from 0 and 24 hr samples. To that end, A/G changes in the high abundant COII gene and the constant region of T-cell receptor β (TCRβ) gene (cytochrome oxidase subunit II) from thymuses of endotoxin-challenged mice harvested at 0 or 24 hr were examined. COII and TCRβ cDNAs were amplified by RT-PCR, cloned into TA cloning vector, and sequenced.
Determination of the edited mRNA in inflamed thymic cells
Mice (n = 4) were challenged with endotoxin (intraperitoneally, 15 mg/kg) and thymuses were harvested at 0, 2, 4, 8, 20 and 24 hr. Poly(A)+ RNA was isolated using RNA Easy and OligoTex (Qiagen, Inc.) and contaminations of tRNA and rRNA were monitored on denatured agarose gel. Overloading of the purified poly(A) + RNA (25 μg) showed no visible bands of tRNA and rRNA, indicating contamination of <100 ng (0·4%). Thus, the maximal contamination from tRNA and rRNA could not exceed 0·1% even if all adenosines in the tRNA or rRNA were converted to inosine. Equal amounts of poly(A)+ RNA were completely digested with RNase T2 and labelled by T4 polynucleotide kinase (PNK). The 5′-, 3′-diphosphate nucleotides were treated with RNase P1 and sequentially analysed on TLC.1,12 Briefly, the position of 5′-monophosphate inosine was located in comparison with a standard 5′-[32P]monophosphate inosine, which was prepared by labelling 3′-monophosphate inosine by polynucleotide kinase followed by RNase P1 digestion. To confirm that PNK labels all nuclease generated nucleotides equally as efficiently, the same amount of 3′-monophosphate nucleotides, that are identical to the nucleotides generated with RNase T2, were labelled with PNK and analysed under the same conditions. The cellulose on the TLC plate together with 5′-phosphate inosine was recovered and extracted in water. Five microlitres of the extract was directly spotted on the second TLC plate. After autoradiography, inosines were analysed and quantified by phospherImager. The per cent of inosine was estimated by the radioactive count of scintillation or density of digitalized X-ray films in comparison with adenosine.
RNA editing on nuclear run-on transcripts
Nuclei were isolated from HeLa cells and applied to nuclear run-on transcription in the presence of α-[32P]ATP as previously described.32,33 The newly synthesized RNA with uniformly 32P-labelled adenosines was purified using Trizol and precipitated with ethanol. The labelled RNA (∼5000 counts per minute) was incubated with 20 μl of cell extracts (containing 50 μg total protein) from inflamed or sham mouse thymus (n = 6), or bovine serum albumin (10 μg/ml) at 37° for 2 hr. The edited RNA was recovered and inosines were analysed by a TLC assay as described above.
Results
Induction of A-to-I RNA editing in immune organs during endotoxin-induced inflammation
To study A-to-I RNA editing in immune organs during acute inflammation, tissue lysates were prepared from thymus and spleen harvested from endotoxin-challenged mice. Editing activity in these tissue lysates was measured by determining the ratio of A-to-I conversion in dsRNA.12 Tissues from sham animals (0 hr) displayed low baseline A-to-I conversion (less than 0·5%) (Fig. 1). In contrast, thymus and spleen from endotoxin-treated animals demonstrated increased editing activity (∼20% and ∼10% conversion, respectively) at 4 hr after stimulation (Fig. 1).
Figure 1.

Up-regulation of A-to-I RNA editing in mouse thymus (a) and spleen (b) during acute systemic inflammation. Spleens and thymuses were harvested (n = 6) at the indicated time-points after endotoxin-challenge and whole cell extracts were examined for editing activity. Editing activity was determined by measuring A-to-I conversion of dsRNA on TLC. The percentage of inosines was estimated in comparison with adenosines and plotted against the duration of endotoxin stimulation (c). pI, 5′-monophosphate inosine; pA, 5′-monophosphate adenosine. The ratio between pI and pA represents the percentage of adenosine that is converted to inosine.
Up-regulation of ADAR1 in immune organs during endotoxin-induced inflammation
Since ADAR3 is exclusively expressed in the brain5,7 and ADAT1 is specific to tRNA,8,14 it is the induction of ADAR1 and/or ADAR2 which conceivably accounts for the increased editing activity observed in endotoxin-induced inflammation. To determine which of the two latter editing enzymes is responsible for the increased editing activity, Northern blotting (Fig. 2) was performed on various tissues from sham and endotoxin-treated mice. Control animals had a baseline ADAR1 mRNA signal, which was rapidly (within 4 hr) up-regulated by endotoxin while no up-regulation of basal ADAR2 was observed (data not shown). It should be noted that ADAR1 mRNA expression following induction reached brain mRNA levels in sham animals previously reported to modulate gene function in cultured neuronal cells (data not shown). Taken together, these observations demonstrate that up-regulation of ADAR1 is responsible for the increased editing activity in systemic inflammation.
Figure 2.

Up-regulation of ADAR1 expression in acute systemic inflammation assayed by Northern blotting. (a) Mouse spleen, thymus, and lung were harvested (n = 6) at different time-points as indicated after endotoxin-challenge. Four micrograms of mRNA were isolated and analysed by Northern blotting. ADAR1 mRNA was detected using synthesized oligo complementary ADAR1 sequences from 1305 to 1265 and 3004 to 2996 (AF291050). Two transcripts, ADAR1L and ADAR1S, were observed in all organs. (b) ADAR1 mRNA was quantified by PhospherImager and normalized to the internal control β-actin. The ratios between ADAR1L and β-actin and of ADAR1S and β-actin were calculated to compare their relative expression. Note that the calculated ratios were similar prior to and early after induction of inflammation. ADAR1L became dominant later (>12 hr).
It should be noted that long and short transcripts measuring 7 and 5 kb, respectively, were detected in all tissues from both sham and challenged animals. The two transcripts were differentially regulated because a late induction of the long but not of the short transcript was observed after 16 hr of stimulation.
Up-regulation of ADAR1 in inflammatory cells
In situ hybridization studies were performed to identify the cellular localization of the up-regulated ADAR1 during inflammation. This technique demonstrated increased ADAR1 signal in inflammatory cells such as lung monocytes and macrophages26 as well as peripheral lymphocytes from endotoxin-treated mice (Fig. 3). Since both TNF-α and IFN-γ are induced during endotoxaemia and as their induction precedes the up-regulation of ADAR1,28,34 we hypothesized that ADAR1 up-regulation in inflammatory cells is mediated by pro-inflammatory cytokines. To test this notion, macrophage (MH-S) and lymphocyte (CTLL-2) cell lines were stimulated with a variety of inflammatory agents and pathogens including TNF-α, IFN-γ and live E. coli and adenovirus, and were evaluated for their ADAR1 mRNA expression by RT-PCR. As predicted, up-regulation of ADAR1 mRNA was identified in lymphocytes stimulated with IFN-γ and TNF-α as well as in macrophages stimulated with IFN-γ (Fig. 4a). This result is consistent with the previously reported induction of ADAR1 by IFN-γ in U-cells25 and the identification of IFN-response element in the promoter region of the human ADAR1.35 Live E. coli and adenovirus, both pathogens of systemic infection and inflammation,36,37 stimulated ADAR1 expression in macrophages (Fig. 4b). No significant induction was observed in endothelial cells (HUVEC) or fibroblasts (3T3) incubated with either IFN-γ or TNF-α (data not shown). Induction of ADAR1 in inflammatory cells by IFN-γ, TNF-α and pathogenic bacteria suggests that ADAR1-mediated RNA editing is involved in the development of inflammation.38
Figure 3.
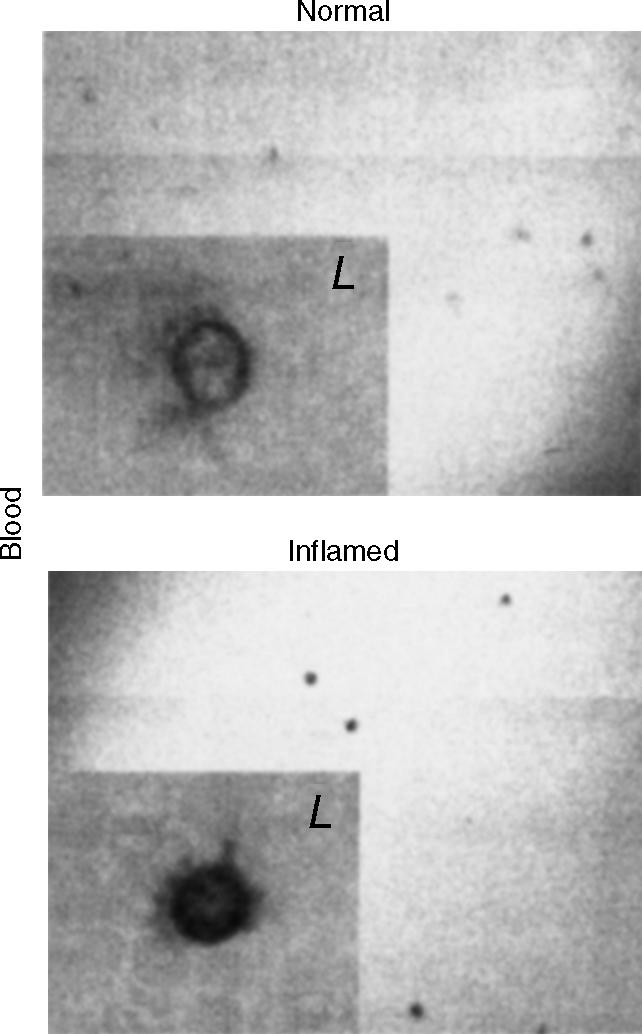
Cellular localization of ADAR1 in peripheral blood during acute systemic inflammation by in situ hybridization. In situ hybridization was applied on mouse peripheral blood smear for the detection of ADAR1 mRNA using digoxigenin-labelled antisense ADAR1 RNA probes (1305–1265 and 3014–2972, AF291050). Positive signals were identified in lymphocytes (L). Similar observations were uniformly seen in all animals (n = 6).
Figure 4.

Induction of ADAR1 by inflammatory mediators (a) and pathogens (b) in vitro. (a) Cytotoxic T cells (CTLL-2) and macrophages (MH-S) were stimulated with IFN-γ (1000 U/ml) or TNF-α (10 ng/ml). In both cell types RT-PCR analysis indicated a marked increase in ADAR1 after 4 hr of stimulation. (b) Macrophages (5 × 106 MH-S cells) were stimulated with live E. coli (104−109 CFU) or adenovirus (0·01 and 0·1 MOI) for 2 hr. ADAR1 was up-regulated by both pathogens. Each gel represents a single experiment. Similar observations were uniformly seen in all experiments (n = 3). The primers for RT-PCR cover exons 5–8. Two RT-PCR products were seen in all cases because of alternative splicing in exon 7 (AF291050 and AF291876).
Association of RNA editing with lymphocyte proliferation
Non-specific dsRNA RNA editing was monitored in activated spleens from sham mice (n = 6). Activation was induced by the commonly used combination of IL-2 and Con A, which is known to stimulate both cell growth and proliferation.36 Editing activity, which was undetectable in non-stimulated lymphocytes, increased at 30 hr and peaked at 72 hr (Fig. 5a). In agreement with the late induction of editing activity, up-regulation of ADAR1 protein was demonstrated by Western blotting in splenic cells stimulated with Con A at 24 and 48 hr (Fig. 5b). Thus, in addition to the early induction of ADAR1 expression and activity, a later response occurs in activated lymphocytes. As the time course of this late response parallels that of lymphocytic growth, proliferation and function, A-to-I RNA editing activity may play a role in lymphocyte-dependent immunity.
Figure 5.

Late induction of RNA editing in activated splenic cells. (a) Splenic cells were harvested from sham mice (n = 6) and activated with Con A (2 μg/ml) and IL-2 (2 μg/ml) for the times indicated. Whole cell extracts were prepared for editing activity. (b) Splenic cells were harvested from sham mice (n = 6) and activated with Con A (2 µg/ml) for the times indicated. Whole cell extracts were prepared for Western blotting as described above. Note that induction of editing was observed at 30 hr in correlation with cell proliferation. On the Western blots, two ADAR1 variants (p150 and p115) were detected. Induction of the short form corresponded with the increased RNA editing activity.
It should be noted that at least two ADAR1 protein variants with molecular weights of 115 000 and 150 000 were detected in this experiment. Following activation, the 115 000 MW ADAR1 was up-regulated whereas the 150 000 MW protein was down-regulated. These findings are supported by the identification of two ADAR1 variants in human cells. Nevertheless, the human 110 000 MW ADAR1 is constitutively expressed whereas the 150 000 MW ADAR1 human variant was induced in U-cells after IFN-γ stimulation.25
Production of inosine-containing mRNA in thymic lymphocytes in response to inflammation
Since inosine-containing mRNA is the product of A-to-I RNA editing, the demonstration of increased levels of I-mRNA would support the hypothesis that editing is induced during inflammation. Thus, the number of inosines in thymic mRNA from sham or endotoxin-treated mice was analysed by two sequential TLC after RNA T2 digestion, polynucleotide kinase labelling, and RNase P1 treatment. The induction of inosine in thymic mRNA was detected on the first TLC compared with the standard 3′-pI (Fig. 6a). The increase in inosine level was further confirmed when the inosines were recovered and analysed on the second TLC plate (Fig. 6b). The ratio between inosine and adenosine in thymic mRNA, calculated by comparing the radioactivity of each spot, was increased from a baseline of approximately 0·5–5% at 24 hr (Fig. 6c). Although inosines are present in tRNA,40 their high level was mostly the result of editing of the thymic poly(A) + RNA because the contamination tRNA and rRNA in the purified poly(A) + mRNA was estimated to be less than 0·1%. To exclude the possibility that the high level of inosine in mRNA was generated by incorporation of 5′-inosine triphosphate during transcription, a newly synthesized nuclear RNA prepared by nuclear run-on transcription was subjected to editing in the thymic cell extracts. The detection of high levels of inosine only in endotoxin-stimulated thymic cell extracts (data not shown) confirmed that these inosines were generated after RNA was transcribed. Therefore, inosines in the newly transcribed RNA are derived from adenosines as a result of inflammation-induced editing activity. Note that PNK labelled all nuclease generated nucleotides equally as efficiently (Fig. 6d).
Figure 6.

Induction of inosines in thymic mRNA during acute systemic inflammation. (a) The first TLC assay. Inosine-containing mRNA was analysed in thymuses from endotoxin-stimulated mice (n = 4) at 0–24 hr. Total poly(A)+ RNA (tRNA and rRNA were not visible when monitored on denature agarose gels) from each time-point was isolated and a synthetic RNA was made by in vitro transcription using a plasmid with T7 promoter. Poly(A)+ RNA was digested with RNase T2, labelled with polynucleotide kinase and digested again with RNase P1. The inosine-labelled mRNA was significantly increased. pA, pG, pI, pC and pU are 5′-monophosphate nucleotides; I, the standard 5′-[32P]monophosphate inosine. (b) The second TLC assay. After the first TLC, inosine and adenosine were recovered and analysed on a new TLC. In agreement with the first TLC, inosine-labelled mRNA was significantly increased. S, synthetic RNA. (c) Quantification of the increased level of inosine-containing mRNA. Per cent of inosine was calculated using radioactive counting or an NIH Image software. Results were plotted against the duration of stimulation. Note the increase from a baseline of approximately 0·5–5% at 24 hr. (d) Labelling efficiency of 3′-monophosphate nucleotides. PNK equally labelled all 3′-monophosphate nucleotides (pA, pG, pC, pU and pI).
The high level of A-to-I conversion in the inflamed thymic tissue was confirmed by direct cloning and sequencing of representative cDNAs from the COII and the TCRβ genes. While five A/G changes were identified in the cDNAs from challenged thymuses only one was identified in normal thymuses.
Discussion
The primary finding of the present study is the robust production during endotoxin-induced inflammation of inosine-containing mRNA through up-regulation of A-to-I RNA editing. Induction of A-to-I RNA editing and ADR1 expression occurred in immune organs as early as 2 hr after endotoxin stimulation, which peaked at 4–10 hr and was localized to lymphocytes and macrophages. The expression of ADAR1 mRNA was also confirmed in cultured lymphocytes and monocytes stimulated with a variety of pro-inflammatory mediators and pathogens. The up-regulation of ADAR1-mediated RNA editing by various mediators suggests an important role for A-to-I RNA editing in acute inflammation.
Clinically, endotoxaemia triggers the production of multiple pro- and counter-inflammatory mediators,26,34,41 which lead to a systemic inflammatory response (SIRS) and multiple organ failure. Our observation that a variety of inflammatory mediators and pathogens can trigger ADAR1 induction in vitro and in vivo, suggests that ADAR1 may play a role in these clinical conditions. Specifically, TNF-α and IFN-γ, both induced by endotoxin in mice,26,34,41 triggered early induction of ADAR1 in inflammatory cells in vitro. The possibility that these two mediators affect RNA editing during inflammation is supported by recent studies showing TNF-α and IFN-γ expression prior to ADAR1 induction in a mouse model of acute lung inflammation.26,28 TNF-α and IFN-γ may also be involved in the induction of RNA editing by live E. coli and adenovirus as these mediators are rapidly produced during macrophage activation,42 indicating that editing by ADAR1 may be also involved in bacterial or viral infection.24 Taken together, these data suggest that RNA editing is induced early in the inflammatory cascade, possibly through de novo produced inflammatory mediators.
Although ADAR1 was induced early following inflammatory stimulation, a later induction was also noticed in splenic and thymic cells from endotoxin-challenged mice (24 hr) and in IL-2 and Con A activated lymphocytes from the spleen (90 hr). As IL-2 and/or Con A stimulate cell proliferation in a time course that resembles this late induction, it is possible that RNA editing during inflammation is bimodal. ADAR1 is induced early through IFN-γ or TNF-α mediation and later in parallel to lymphocyte growth and proliferation.
The induction of RNA editing by proliferative mediators suggests a potential role for ADAR1 in the proliferation and differentiation of leucocytes during inflammation. This notion is supported by a recent study that demonstrated a critical role for ADAR1 in the proliferation and differentiation of embryonic erythrocytes.23 In this study, chimeric embryos with a functional null allele for ADAR1 died before embryonic day 14 with defects in the haematopoietic system, suggesting that under-editing by ADAR1 is detrimental to normal embryonic erythropoiesis. Additional studies demonstrated that C-to-U editing in B lymphocytes modulates class switch recombination and somatic hypermutation during B-cell maturation.43–47 Taken together, these observations point to a potential role for ADAR1 in the proliferation and differentiation of leucocytes during inflammation. Furthermore, because Con A stimulates antigen-dependent T-cell proliferation, it can be postulated that the inflammation-induced editing activity correlates with antigen-dependent clonal expansion of lymphocytes.
Two ADAR1 mRNA transcripts, ADAR1L and ADAR1S, were observed in all tested tissues. These transcripts could be transcribed from different promoters31 or from the same promoter by alternative splicing. At the RNA level, the ratio between ADAR1L/β-actin and ADAR1S/β-actin changes after endotoxin stimulation. In the thymus, ADAR1L and ADAR1S expression was similar in sham animals and equally up-regulated during the early induction. Later, ADAR1L became the dominant form. At the protein level, two distinct ADAR1 forms with 150 000 and 115 000 MW were identified in activated splenic lymphocytes in vitro. Interestingly, the ratio between the larger and smaller protein isoforms decreased following inflammatory stimulation because of both decreased production of the larger form and increased expression of the smaller form. Two ADAR1 protein products, which respond differently to inflammatory stimulation, were also identified in human cells.48 While both the long (150 000) and short (110 000) ADAR1 forms are present in sham conditions, only the long form of the human ADAR1 is inducible when U-cells are stimulated by IFN-γ. Taken together, these data suggest that long and short ADAR1 isoforms are induced in the human and mouse, which may be the result of different transcripts and/or alternative splicing of a single pre-mRNA.
The functional role of A-to-I RNA editing in acute inflammation is still obscure. Nevertheless, the dramatic increase of inosine-containing mRNA to approximately 5% of adenosines in the poly(A) + RNA isolated from endotoxin-challenged thymus suggests that ADAR1 could exert a modulatory effect on protein production during inflammation. As inflammation involves the production of multiple proteins, A-to-I RNA editing could be a key player in the pathogenesis of this process. Since inosine is recognized as guanosine during transcription, translation and base-pairing interaction, it is possible that during inflammation these inosines produce more mutations and variants at both the mRNA and protein levels. Therefore, it is suggested that during inflammation the cDNA sequence may not accurately represent the transcripts of genomic sequences anticipated based upon the Watson–Crick model. A significant fraction of codons in mRNA could be switched by RNA editing. If inosines generated by the editing process are located in the first or second codon positions, the identity encoded for amino acids is switched with the resultant production of protein variants (Fig. 7). Although editing at the third codon position or outside the coding region does not change the encoded protein sequences, this ‘silent’ editing may still affect mRNA processing by coupling with alternative splicing49 or by promoting mRNA decay through inosine-specific RNase.50,51
Figure 7.

Codon switch by A-to-I RNA editing. Editing in the coding region of mRNA could switch the codon identity. The arrows indicate <0·1% codon switch in normal conditions and 5% during acute inflammation. If editing occurs at the first or second codon positions, it switches the encoded amino acids. Editing at the third codon position or outside of the coding region of inosine-containing mRNA is termed ‘silent’ editing.
ADAR1 is known to edit dsRNA. Thus, it is possible that as yet unidentified mRNAs with long stretches of double-stranded structure are its main substrates. Therefore, disruption of dsRNA or mRNA with a long stretch of dsRNA structure may affect the function of dsRNA-dependent proteins. One such protein is the key modulatory enzyme dsRNA-dependent protein kinase (PKR), which modulates both transcription and translation through phosphorylation of I-κB52 and eIF-2α,53, respectively.
In summary, this study supports a functional role for RNA editing by ADAR1 during acute inflammation. This observation can serve as a basis for future studies on the involvement of A-to-I RNA editing in acute inflammatory diseases.
Acknowledgments
We thank Michael Centrella and Alfred Bothwell for their discussion. This work was supported by NIH Grants GM-60426 to Dr Yang and HL57963-01 to Dr Rabinovici. It is also partially supported by Natural Science Foundation of China Grant 39870418, 39870176 and 30021002 and a Hellman Fellowship to Dr Yang.
References
- 1.Yang JH, Sklar P, Axel R, Maniatis T. Editing of glutamate receptor subunit B pre-mRNA in vitro by site-specific deamination of adenosine. Nature. 1995;374:77–81. doi: 10.1038/374077a0. [DOI] [PubMed] [Google Scholar]
- 2.Melcher T, Maas S, Higuchi M, Keller W, Seeburg PH. Editing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor-B pre-mRNA in vitro reveals site-selective adenosine to inosine conversion. J Biol Chem. 1995;270:8566–70. doi: 10.1074/jbc.270.15.8566. [DOI] [PubMed] [Google Scholar]
- 3.Rueter SM, Burns CM, Coode SA, Mookherjee P, Emeson RB. Glutamate receptor RNA editing in vitro by enzymatic conversion of adenosine to inosine. Science. 1995;267:1491–4. doi: 10.1126/science.7878468. [DOI] [PubMed] [Google Scholar]
- 4.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci USA. 1994;91:11457–61. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–4. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 6.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;271:31795–8. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 7.Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains [In Process Citation] RNA. 2000;6:755–67. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maas S, Gerber AP, Rich A. Identification and characterization of a human tRNA-specific adenosine deaminase related to the ADAR family of pre-mRNA editing enzymes. Proc Natl Acad Sci USA. 1999;96:8895–900. doi: 10.1073/pnas.96.16.8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA. 2000;6:1004–18. doi: 10.1017/s1355838200000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slavov D, Clark M, Gardiner K. Comparative analysis of the RED1 and RED2 A-to-I RNA editing genes from mammals, pufferfish and zebrafish. Gene. 2000;250:41–51. doi: 10.1016/s0378-1119(00)00174-8. [DOI] [PubMed] [Google Scholar]
- 11.Hough RF, Bass BL. Analysis of Xenopus dsRNA adenosine deaminase cDNAs reveals similarities to DNA methyltransferases. RNA. 1997;3:356–70. [PMC free article] [PubMed] [Google Scholar]
- 12.Yang JH, Sklar P, Axel R, Maniatis T. Purification and characterization of a human RNA adenosine deaminase for glutamate receptor B pre-mRNA editing. Proc Natl Acad Sci USA. 1997;94:4354–9. doi: 10.1073/pnas.94.9.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing [see comments] Nature. 1997;387:303–8. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 14.Maas S, Kim YG, Rich A. Sequence, genomic organization and functional expression of the murine tRNA-specific adenosine deaminase ADAT1. Gene. 2000;243:59–66. doi: 10.1016/s0378-1119(99)00562-4. [DOI] [PubMed] [Google Scholar]
- 15.Keegan LP, Gerber AP, Brindle J, Leemans R, Gallo A, Keller W, O'Connell MA. The properties of a tRNA-specific adenosine deaminase from Drosophila melanogaster support an evolutionary link between pre-mRNA editing and tRNA modification. Mol Cell Biol. 2000;20:825–33. doi: 10.1128/mcb.20.3.825-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerber A, Grosjean H, Melcher T, Keller W. Tad1p, a yeast tRNA-specific adenosine deaminase, is related to the mammalian pre-mRNA editing enzymes ADAR1 and ADAR2. EMBO J. 1998;17:4780–9. doi: 10.1093/emboj/17.16.4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lomeli H, Mosbacher J, Melcher T, et al. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–13. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 18.Feldmeyer D, Kask K, Brusa R, et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi M, Maas S, Single FN, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2 [In Process Citation] Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 20.Hanrahan CJ, Palladino MJ, Ganetzky B, Reenan RA. RNA editing of the Drosophila para Na (+) channel transcript. Evolutionary conservation and developmental regulation. Genetics. 2000;155:1149–60. doi: 10.1093/genetics/155.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma E, Gu XQ, Wu X, Xu T, Haddad GG. Mutation in pre-mRNA adenosine deaminase markedly attenuates neuronal tolerance to O2 deprivation in Drosophila melanogaster. J Clin Invest. 2001;107:685–93. doi: 10.1172/JCI11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000;102:437–49. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 23.Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science. 2000;290:1765–8. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 24.Lei M, Liu Y, Samuel CE. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology. 1998;245:188–96. doi: 10.1006/viro.1998.9162. [DOI] [PubMed] [Google Scholar]
- 25.Patterson JB, Thomis DC, Hans SL, Samuel CE. Mechanism of interferon action: double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons. Virology. 1995;210:508–11. doi: 10.1006/viro.1995.1370. [DOI] [PubMed] [Google Scholar]
- 26.Rabinovici R, Kabir K, Chen M, Su Y, Zhang D, Luo X, Yang JH. ADAR. 1 is involved in the development of microvascular lung injury. Circ Res. 2001;88:1066–71. doi: 10.1161/hh1001.090877. [DOI] [PubMed] [Google Scholar]
- 27.Reuman PD, Keely SP, Schiff GM. Rapid recovery in mice after combined nasal/oral immunization with killed respiratory syncytial virus. J Med Virol. 1990;32:67–72. doi: 10.1002/jmv.1890320112. [DOI] [PubMed] [Google Scholar]
- 28.Kabir K, Gelinas JP, Chen M, Chen DF, Zhang D, Yang JH, Carter KC, Rabinovici R. Characterization of a murine model of endotoxin-induced acute lung injury. Shock. 2002;17:300–3. doi: 10.1097/00024382-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 29.Guglielmotti A, Aquilini L, Rosignoli MT, Landolfi C, Soldo L, Coletta I, Pinza M. Benzydamine protection in a mouse model of endotoxemia. Inflamm Res. 1997;46:332–5. doi: 10.1007/s000110050197. [DOI] [PubMed] [Google Scholar]
- 30.Hotchkiss RS, Karl IE. Dantrolene ameliorates the metabolic hall-marks of sepsis in rats and improves survival in a mouse model of endotoxemia. Proc Natl Acad Sci USA. 1994;91:3039–43. doi: 10.1073/pnas.91.8.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci USA. 1999;96:4621–6. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenberg ME, Bender TP. Identification of newly transcribed RNA. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocol in Molecular Biology. Cambridge, MA: John Wiley & Sons Inc.; 1997. pp. 4.10.11–14.10.11. [Google Scholar]
- 33.Hirayash K, Liz JT. Nuclear run-on assays: assessing transcription by measuring density of engorged RNA polymerases. Methods Enzymol. 1999;304:351–62. doi: 10.1016/s0076-6879(99)04021-5. [DOI] [PubMed] [Google Scholar]
- 34.Horgan MJ, Palace GP, Everitt JE, Malik AB. TNF-alpha release in endotoxemia contributes to neutrophil-dependent pulmonary edema. Am J Physiol. 1993;264:H1161–5. doi: 10.1152/ajpheart.1993.264.4.H1161. [DOI] [PubMed] [Google Scholar]
- 35.George CX, Samuel CE. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon- inducible ADAR1 promoter. Gene. 1999;229:203–13. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- 36.Dinarello CA. The proinflammatory cytokines interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J Infect Dis. 1991;163:1177–84. doi: 10.1093/infdis/163.6.1177. [DOI] [PubMed] [Google Scholar]
- 37.Ginsberg HS, Moldawer LL, Sehgal PB, Redington M, Kilian PL, Chanock RM, Prince GA. A mouse model for investigating the molecular pathogenesis of adenovirus pneumonia. Proc Natl Acad Sci USA. 1991;88:1651–5. doi: 10.1073/pnas.88.5.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michie HR, Manogue KR, Spriggs DR, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318:1481–6. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]
- 39.Nitta T, Konno-Ejiri H, Okumura S, Ozawa A, Nakano M. Role of interleukin 2 on enhancement of concanavalin A-induced human peripheral blood lymphocyte proliferation by murine B cell mitogens. Microbiol Immunol. 1985;29:441–9. doi: 10.1111/j.1348-0421.1985.tb00845.x. [DOI] [PubMed] [Google Scholar]
- 40.Haumont E, Fournier M, de Henau S, Grosjean H. Enzymatic conversion of adenosine to inosine in the wobble position of yeast tRNAAsp: the dependence on the anticodon sequence. Nucl Acids Res. 1984;12:2705–15. doi: 10.1093/nar/12.6.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wysocka M, Kubin M, Vieira LQ, Ozmen L, Garotta G, Scott P, Trinchieri G. Interleukin-12 is required for interferon-gamma production and lethality in lipopolysaccharide-induced shock in mice. Eur J Immunol. 1995;25:672–6. doi: 10.1002/eji.1830250307. [DOI] [PubMed] [Google Scholar]
- 42.Fadok VA, McDonald PP, Bratton DL, Henson PM. Regulation of macrophage cytokine production by phagocytosis of apoptotic and post-apoptotic cells. Biochem Soc Trans. 1998;26:653–6. doi: 10.1042/bst0260653. [DOI] [PubMed] [Google Scholar]
- 43.Longacre A, Storb U. A novel cytidine deaminase affects antibody diversity. Cell. 2000;102:541–4. doi: 10.1016/s0092-8674(00)00075-1. [DOI] [PubMed] [Google Scholar]
- 44.Neuberger MS, Scott J. Immunology. RNA editing AIDs antibody diversification? Science. 2000;289:1705–6. doi: 10.1126/science.289.5485.1705. [DOI] [PubMed] [Google Scholar]
- 45.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 46.Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper–IgM syndrome (HIGM2) Cell. 2000;102:565–75. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 47.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–6. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 48.Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376–88. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 50.Scadden AD, Smith CW. A ribonuclease specific for inosine-containing RNA. a potential role in antiviral defence? EMBO J. 1997;16:2140–9. doi: 10.1093/emboj/16.8.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scadden AD, Smith CW. Specific cleavage of hyper-edited dsRNAs. EMBO J. 2001;20:4243–82. doi: 10.1093/emboj/20.15.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci USA. 1994;91:6288–92. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Haro C, Mendez R, Santoyo J. The eIF-2alpha kinases and the control of protein synthesis. FASEB J. 1996;10:1378–87. doi: 10.1096/fasebj.10.12.8903508. [DOI] [PubMed] [Google Scholar]