

Abstract
Optimal HIV vaccines should elicit CD8+ T cells specific for HIV proteins presented on MHC class I products, because these T cells contribute to host resistance to viruses. We had previously found that the targeting of antigen to dendritic cells (DCs) in mice efficiently induces CD8+ T cell responses. To extend this finding to humans, we introduced the HIV p24 gag protein into a mAb that targets DEC-205/CD205, an endocytic receptor of DCs. We then assessed cross-presentation, which is the processing of nonreplicating internalized antigen onto MHC class I for recognition by CD8+ T cells. Low doses of αDEC-gag, but not control Ig-gag, stimulated proliferation and IFN-γ production by CD8+ T cells isolated from the blood of HIV-infected donors. αCD205 fusion mAb was more effective for cross-presentation than αCD209/DC-SIGN, another abundant DC uptake receptor. Presentation was diverse, because we identified eight different gag peptides that were recognized via DEC-205 in 11 individuals studied consecutively. Our results, based on humans with highly polymorphic MHC products, reveal that DCs and DEC-205 can cross-present several different peptides from a single protein. Because of the consistency in eliciting CD8+ T cell responses, these data support the testing of αDEC-205 fusion mAb as a protein-based vaccine.
Keywords: CD205, CD209; cross-presentation; DC-SIGN; vaccine
Resistance to HIV is in part mediated by CD8+ T cells (1, 2), which recognize fragments of viral antigens presented on MHC class I products (3). HIV-specific, CD8+ T cells kill virus-infected targets in culture (4, 5) and produce antiviral chemokines (6, 7). CD8+ T cells also resist immunodeficiency viruses in vivo. In SIV-infected rhesus macaques, depletion of CD8+ T cells increases plasma viremia (8), including viremia due to attenuated vaccines (9). HIV mutants that escape recognition by CD8+ T cells in vivo also become more pathogenic (10–12). Therefore, effective protection against HIV will likely require vaccines that elicit strong and broad CD8+ T cell immunity.
Dendritic cells (DCs) are specialized antigen-presenting cells that capture infectious agents and tumors and initiate CD8+ T cell immunity (13, 14). DCs express a number of cytokines and membrane costimulators that drive the T cell response, and DCs “cross-present” antigens on MHC class I (15, 16). The cell biology underlying cross-presentation is not yet fully defined (17–19), but it allows DCs to extract peptides from nonreplicating internalized antigens for presentation to CD8+ T cells. Such peptides do not need to be synthesized in the DCs, but instead “cross” to their MHC I products from another source, e.g., from select proteins (20–22), tumor cells (23–25), inactivated virus or dying infected cells (26–28), immune complexes (29–31), and self-tissues (32). In contrast, the classical pathway for presentation on MHC I is to generate peptides from proteins produced during infection by replicating viruses (33, 34). The newly synthesized proteins, probably made as defective ribosomal initiation products (35), are degraded in the proteasome before transport into the rough endoplasmic reticulum, where there is binding of peptides to newly synthesized MHC I. In mice, DCs are the major cell type capable of cross-presentation in vivo (36–40). However, it has yet to be shown that DCs can cross-present peptides across a spectrum of MHC haplotypes, an essential requirement for protein-based vaccines in humans who are highly polymorphic at the MHC or HLA locus.
A recent strategy to explore and harness DC biology for vaccination is to target antigens to DCs in intact lymphoid organs by incorporating specific antigens into anti-DC mAbs (41). Among other advantages, the targeting of antigens in this way enhances the efficiency of antigen presentation to CD4+ and CD8+ T cells in vivo by 100-fold or more (41–44).
To extend these ideas to humans, we have selected a mAb to human DEC-205/CD205 (45). In mice, DEC-205 mediates cross-presentation (42–44). The receptor is also expressed on human monocyte-derived DCs along with other endocytic receptors (reviewed in ref. 46), such as the mannose receptor/CD206 and DC-SIGN/CD209 (47, 48). A potential advantage of CD205 over these other receptors is its high expression by DCs in the T cell areas of lymph nodes in the steady state, whereas CD206 and CD209 are abundant in macrophages in the medullary region of lymph nodes (49). This finding means that αCD205 mAb might provide superior targeting of vaccine antigens to DCs in lymphoid tissues, where the DCs are ideally positioned to select specific T cell clones from the repertoire. We now find that a fusion αCD205 mAb targets HIV gag for broad and efficient cross-presentation in HIV-infected individuals. The data provide a rationale for further testing of this vaccine approach in humans.
Results
Characterization of HIV gag Fusion mAbs.
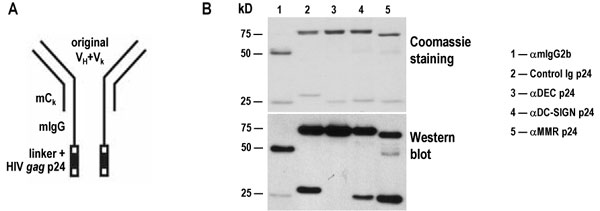
To deliver HIV antigens to human DCs, we cloned HIV gag p24 protein in frame into the carboxyl terminus of the heavy chain of mAbs to DEC-205, DC-SIGN and MMR, which are endocytic receptors expressed on monocyte-derived DCs; the heavy chain of an isotype-matched control Ig was also engineered as a negative control [supporting information (SI) Fig. 5A]. The fusion mAbs were produced by transient transfection in 293 T cells (≈1–2 mg/liter) and purified from culture supernatants by protein G-affinity chromatography. Purity was assessed after SDS/PAGE under reducing conditions, by both Coomassie staining and Western blot with a HRP-conjugated α-mouse IgG. The fusion mAbs were composed of a 75-kD heavy chain, as opposed to ≈50-kD heavy chain of an unconjugated mouse IgG2b, the same isotype as the αDEC-205 and αDC-SIGN mAbs (SI Fig. 5B). We also verified the functional integrity of the mAbs by binding to immature DCs, similar to that of the original unmodified mAbs (Fig. 1).
Fig. 1.

Binding of HIV gag p24 fusion mAbs to monocyte-derived DCs. Immature DCs were treated with 0.02, 0.2, and 2 μg/ml of αDEC p24, αDC-SIGN p24, and αMMR p24 and with 2 μg/ml control Ig p24. DCs were also treated with supernatant from each mAb clone as positive control and with nonreactive mAb as negative control, followed by incubation with an α-mIgG phycoerythrin-conjugated antibody.
Cross-Presentation of gag p24 Protein by αDEC-205 Fusion mAb.
To study the ability of αDEC p24 to mediate antigen presentation, we added fusion mAb to blood cells from HIV-infected individuals and measured proliferation and IFNγ production by bulk peripheral blood mononuclear cells (PBMCs) as well as cocultures of monocyte-derived DCs and T cells. We examined cells from treated chronically infected individuals as well as untreated long-term nonprogressors. All were clinically stable and had CD4+ T cell counts of >400 per microliter. The T cells were labeled with carboxyfluorescein succinimydl ester (CFSE) to follow their proliferation by successive halving of the amount of CFSE per cell with each division. Although we did not detect responses by CD4+ cells to gag antigen, the CD8+ T cells proliferated actively after stimulation with a pool of 55 peptides spanning the HIV gag sequence and to a low dose of αDEC p24 but not control Ig p24 (SI Fig. 6). The proliferating CD8+ T cells produced IFNγ if the cultures were rechallenged with p24 peptides for 6 h at the end of the 6- to 7-d expansion culture (SI Fig. 6 and Fig. 2). Results from five individuals are shown in Fig. 2 and indicate that the frequency of IFNγ+ CD8+ CFSElow T cells was greater after addition of the pool of gag peptides relative to αDEC p24. However, αDEC p24 was reliably a more efficient form of antigen than control Ig p24, which was comparable to the no-antigen or medium control. In each case, it was necessary to restimulate the proliferated T cells with gag peptides to detect their production of IFNγ (compare filled vs. open symbols in Fig. 2). Higher frequencies of responding T cells were noted in the DC–T cell cocultures than in bulk PBMCs (Fig. 2), which may reflect either the greater numbers or improved maturation of monocyte-derived DCs. Two seronegative donors did not show responses to αDEC p24 in these assays (data not shown). Together, these results demonstrate that low concentrations of αDEC p24 fusion mAb lead to antigen presentation on MHC I, inducing consistent expansion of CD8+ T cells capable of producing IFNγ from HIV-infected donors.
Fig. 2.

HIV gag p24-specific CD8+ T cell responses to αDECp 24. Summary of the frequencies of IFNγ-producing, proliferating, CD3+CD8+ T cells in response to medium, p24 peptides, αDEC p24, and control Ig p24 (1 μg/ml), with or without restimulation at the end of the 6- to 7-day culture with p24 peptides. Frequencies are shown for both PBMCs and cocultures of antigen-pulsed DCs and CFSE-labeled T cells.
αDEC-205 Fusion mAb is Superior to αDC-SIGN mAb for CD8+ T Cell Responses.
To assess the consequences of targeting different receptors, we compared αDEC, αMMR, and αDC-SIGN p24 fusion mAbs. We found that αDEC p24 was reliably more effective than the other fusion mAbs in inducing proliferation and IFNγ production from CD3+CD8+ cells in PBMCs (Fig. 3 and SI Fig. 7A). We made similar findings in a limited study of DC–T cell cocultures from four individuals (e.g., SI Fig. 7B). These findings suggest that antigen delivery via DEC-205 is superior to the other endocytic receptors for expanding gag-specific CD8+ T cells.
Fig. 3.

Comparison of CD8+ T cell response after targeting of gag through different endocytic receptors. Frequency of IFNγ+ proliferating CD3+ CD8+ T cells measured in CFSE-labeled PBMCs stimulated for 6 days with medium, p24 peptides at 2 μg/ml and αDEC p24, αDC-SIGN p24, αMMR p24, and control Ig p24, all at 1 μg/ml. All samples were restimulated for the last 6 h with p24 peptides.
Broad CD8+ T Cell Responses After DEC-205 Targeting.
To document which gag peptides were presented by DEC-205 targeting, we studied 11 consecutive patients. All of them showed CD8+ T cell proliferation to the pool of HIV gag peptides and to the fusion αDEC p24 fusion mAb. Following expansion of the CFSE-labeled T cells in response to αDEC p24, we rechallenged the cultures for 8 h with five pools of 15-mer overlapping peptides that spanned the gag p24 sequence. In each donor, 1 or 2 pools of peptides were recognized, but when different donors were compared, all five pools could be recognized by one individual or another (Table 1). By further breaking down the reactive peptide pools, we found eight different “mimetope” peptides, i.e., the 15-mer peptides that mimic the actual peptide naturally processed by the DCs. Fig. 4 illustrates and Table 2 summarizes the identification of diverse peptides that could be presented from gag p24 by DEC-205 on DCs. The upper rows in Fig. 4 A–C show the identification of the active peptide pool, and the lower rows show the identification of the best peptide mimetope in the pool. After HLA typing and consultation with the Los Alamos database on known HIV gag peptides that are presented on specific MHC I products, we were able to identify the likely peptide sequences that were being presented after uptake, processing, and cross-presentation of αDEC p24. The data in Tables 1 and 2 indicate that the targeting of gag within αDEC-205 mAb allows DCs from highly polymorphic human MHC I products to cross-present at least 1 or 2 different peptides from this small protein.
Table 1.
IFNγ+ CFSElow CD8+ T cell frequencies after stimulation with gag peptides
| Patient | 6-d culture in medium |
6-d culture in peptides |
6-d culture in αhDEC-p24 |
|||||
|---|---|---|---|---|---|---|---|---|
| 6-h restim in p24 peptides | 6-h restim in peptides | 6-h restim in pool I | 6-h restim in pool II | 6-h restim in pool III | 6-h restim in pool IV | 6-h restim in pool V | ||
| LTNP | ||||||||
| LB01 PBMC | 0.02 | 3.1 | 1.4 | 0.25 | 0.34 | 1 | 0.38 | 0.29 |
| LB01 DC/T | 0.12 | 12 | 6.3 | 0.97 | 0.92 | 6.1 | ND | ND |
| LB02 PBMC | 0.04 | 6.3 | 3.8 | 3.2 | 0.07 | 0.14 | 0.64 | 0.11 |
| LB03 DC/T | 0.04 | 17 | 2.6 | 0.54 | 0 | 2.0 | 0.88 | 0.03 |
| LB04 PBMC | 0.02 | 16 | 2.1 | 1.5 | 0.13 | 0.27 | 0.36 | 0.37 |
| LB04 DC/T | 1.52 | 34.7 | 43.3 | 33.7 | 1.51 | 5.87 | 1.58 | 4.02 |
| LB05 DC/T | 0.55 | 37.6 | 26.4 | 1.27 | 0.45 | 16.3 | 19.1 | 0.44 |
| LB06 PBMC | 0 | 9.73 | 6.09 | 1.8 | 1.38 | 4.94 | 1.8 | 1.58 |
| Chronic | ||||||||
| LB07exp1 PBMC | 0.02 | 4.6 | 2.8 | 0.28 | 0.83 | 1.2 | 0.54 | 3.7 |
| LB07 DC/T | 0.23 | 11 | 3.4 | 1.2 | 1.5 | 3.9 | 1.4 | 4.4 |
| LB07exp2 PBMC | 0.17 | 3.9 | 1.7 | 0.25 | 0.14 | 0.61 | 0.36 | 1.3 |
| LB08 DC/T | 0.18 | 4.1 | 1.5 | ND | ND | 0.21 | 0.46 | 1.7 |
| LB09 PBMC | 0.19 | 4.9 | 2.8 | 0.47 | 1.4 | 0.39 | 1.4 | 0.45 |
| LB10 PBMC | 0.022 | 1.28 | 1.22 | 0.09 | 1.42 | 0.09 | 0.09 | 0.05 |
| LB10 DC/T | 1.04 | 13.6 | 1.63 | 0.41 | 1.32 | 0.3 | 0.28 | 0.21 |
| LB11 PBMC | 0.006 | 10.6 | 5.96 | 0.39 | 5.22 | 0.55 | 1.06 | 0.57 |
Summary of the frequencies of IFNγ+ CFSE low CD8+ cells obtained from either PBMCs or DC/T cell cocultures stimulated for 6–7 days with medium, p24 peptides, and αDEC p24. The samples were then restimulated for 6–8 h with p24 peptides or with the individual peptide pools (see Materials and Methods). The bold data represent pools of peptides that yield responses that are three times more than the background of nonstimulated cells. LTNP, long-term nonprogressor.
Fig. 4.

Identification of mimetope peptides recognized by CD8+ T cells in response to α-DECp24. Results from patients LB04, LB06, and LB07 are shown in A–C, respectively. CFSE-labeled cells were stimulated with medium or αDEC p24 at 1 μg/ml for 6–7 d, whereupon the cells were restimulated with five different pools of gag peptides to detect IFNγ secretion from proliferated CFSElow, CD3+CD8+ T cells (Upper). The next day, parallel cultures were used to identify the individual mimetope peptides from the reactive peptide pools (Lower).
Table 2.
Identification of gag mimetope peptides presented via αhDECp24
| Patient | Reactive p24 peptides pool | Active p24 peptide in pool |
HLA class I | ||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Sequence | A1 | A2 | B1 | B2 | C1 | C2 | ||
| LB02 | I | 10 | FVEKAFSPEVIPMFSAL | 2010 | 3201 | 4001 | 5701 | 0304 | 0602 |
| LB03 | III | 5 | AGTTSTLQEQIGWMT | 0202 | 3601 | 5301 | 5701 | 0401 | 0401 |
| 6 | STLQEQIGWMTNNPP | ||||||||
| LB04 | I | 10 | EKAFSPEVIPMFSAL | 0301 | 1101 | 3501 | 5701 | 0401 | 0401 |
| LB05 | III | 5 | AGTTSTLQEQIGWMT | 3101 | 3303 | 5801 | 7801 | 0701 | 1601 |
| 6 | STLQEQIGWMTNNPP | ||||||||
| IV | 7 | FRDYVDRFYKTLRAE | |||||||
| 8 | VDRFYKTLRAEQASQ | ||||||||
| LB06 | III | 9 | NPPIPVGEIYKRWII | 2301 | 7401 | 0801 | 1801 | 0304 | 0202 |
| 10 | PVGEIYKRWIILGLN | ||||||||
| LB07 | III | 11 | IYKRWWIILGLNKIVR | 0301 | 1101 | 0702 | 0733 | 0702 | 0704 |
| V | 8 | EMMTACQGVGGPGHK | |||||||
| 9 | ACQGVGGPGHKARVL | ||||||||
| LB10 | II | 10 | AAEWDRLHPVHAGPI | 2402 | 6601 | 1503 | 3501 | 0401 | 0401 |
| 11 | DRLHPVHAGPIAPGQ | ||||||||
| LB11 | II | 9 | INEEAAEWDRLHPVH | 3201 | 3201 | 1402 | 4002 | 0802 | 0202 |
| 10 | AAEWDRLHPVHAGPI | ||||||||
As in Table 1 and Fig. 4, gag peptides were identified that were recognized by CD8+ T cells after proliferation in response to αDEC p24. We first identified a pool of gag peptides (column 2), and then the peptide pool was broken down into individual peptides to identify the optimal mimetope (columns 3 and 4). After typing for HLA class I alleles at HLA-A, B, and C loci for each patient, we were able to identify from the Los Alamos database (www.hiv.lanl.gov/content/index) known peptide sequences that are presented on a corresponding HLA product in bold and, in one case (patient LB07), a second peptide (underlined).
Discussion
Improved presentation of antigens by DCs offers the potential of increased vaccine efficacy. DCs are potent inducers of T cell-mediated immunity and memory and have the potential to cross-present antigens from safe forms of vaccines to generate protective CD8+ T cells. Nevertheless, the prior literature on cross-presentation has emphasized the study of single peptides presented on single MHC I proteins. In mice, research is dominated by the presentation of one peptide from ovalbumin on H-2Kb, in part because the binding of this peptide to MHC class I is of such high affinity that it becomes easier to detect cross-presentation. In humans, select peptides have been defined that can be presented on HLA-A2.1, the MHC I molecule that has dominated the literature.
Using the DEC-205/CD205 receptor to deliver antigens to monocyte-derived DCs in vitro, we established that numerous peptides from the HIV protein, gag p24, can be processed and presented by DCs on many allelic forms of human MHC I. Our data indicate that DEC-205 accesses the cross-presenting pathway for many human HLA haplotypes.
Interestingly, antibodies to other receptors expressed on the same DCs, DC-SIGN/CD209 or mannose receptor/CD206, were less effective than αCD205. All three mAbs bound comparably to monocyte-derived DCs and were internalized by DCs (data not shown), so we suspect that the DEC-205 receptor better allows access to the cross-presentation pathway. This pathway typically requires that portions of the gag protein gain access to the cytoplasm, followed by proteosome-mediated degradation and transport into the rough endoplasmic reticulum (reviewed in refs. 18, 50, and 51). Another advantage of CD205 targeting is that it is expressed on many DCs in the T cell areas of human lymphoid tissues, whereas CD206 and CD209 are abundant on macrophages in the lymph node medulla (49). This positioning would allow the CD205-targeted vaccine to efficiently select specific clones of T cells that recirculate from the blood through lymphoid organs.
Although HIV-infected individuals exhibit CD4+ T cell responses that were too weak for us to study here, other research in mice shows that DEC-205 targeting is a powerful means for inducing CD4+ T cell immunity (43, 44) as well as antibody responses (43). Together, the results with αDEC-205 targeting provide preclinical support to develop this strategy as a means to induce strong T cell immunity in humans.
Materials and Methods
Patients.
Peripheral blood (40–100 ml) was obtained from HIV-1 infected individuals who were recruited to the St. Vincent's Hospital Comprehensive Clinic and The Rockefeller University Hospital according to institutional guidelines and after obtaining informed consent. Patient information is summarized in SI Table 3. Blood samples from two healthy seronegative subjects were analyzed as negative controls.
Preparation of Dendritic Cells and T Cells.
PBMCs were isolated from heparinized blood on Ficoll density gradients. Monocytes were enriched by using CD14 microbeads (Miltenyi Biotec, Auburn, CA) and then cultured for 5 d with IL-4 (10 ng/ml) and GM-CSF (100 units/ml) in RPMI 5% human serum to generate immature DCs (52). The CD14− fraction was used as source of bulk T cells and cryopreserved in freezing medium (GIBCO) before use.
Cloning and Production of Fusion HIV gag mAbs.
mAbs for the human receptors DEC-205, DC-SIGN, and MMR as well as a control mAb to mouse I-Ak were cloned from total RNA from the MG38.2 (45), 25B9G8 (49), 3.29 (47) (kindly provided by A. Lanzavecchia, Bellinzona, Switzerland), and 10–2.16 (TIB93; American Type Culture Collection, Manassas, VA) hybridomas. The variable regions were produced with 5′-RACE PCR kit (GIBCO-BRL, Carlsbad, CA) by using primers for the 3′ ends of mouse IgG2b (DEC-205 and DC-SIGN mAbs) or mouse IgG1 (MMR), and Ig λ (DEC-205) or Ig κ (DC-SIGN, MMR). To obtain full-length heavy and light chain Ig cDNA, the V regions were cloned in-frame with a signal peptide and the respective mouse Ig heavy and light constant domains (41 7753). DNA coding for the BH10 clade B HIV-1 gag p24 (NIH AIDS Reference Reagent), amino acid 133-363 was cloned in-frame into the carboxyl terminus of the heavy chains. Fusion HIV gag p24 mAbs were produced by transient transfection (calcium-phosphate) in 293 T cells in serum-free DMEM supplemented with Nutridoma SP (Roche, Indianapolis, IN), purified on protein G columns (GE Healthcare), and characterized by SDS/PAGE and Western blot analysis (41). The integrity of the mAbs was further characterized on immature DCs by FACS using a phycoerythrin-conjugated goat α-mouse IgG (Jackson ImmunoResearch, West Grove, PA).
HIV gag Peptides.
A library of overlapping (staggered by 4 aa) 15-mer peptides was obtained from the NIH AIDS Reference Reagent Program. This library contained 55 peptides, covering the entire gag p24 region (amino acids 133–363), that were pooled and resuspended at 1 mg/ml of each peptide in 100% DMSO. The library was also divided into five pools of 9–12 single peptides, spanning amino acids 133–183 (pool I), amino acids 173–231 (pool II), amino acids 221–279 (pool III), amino acids 269–327 (pool IV), and amino acids 317–363 (pool V) of gag p24.
Expansion of Antigen-Specific T Cells in PBMCs.
PBMCs were labeled with 1 μM CFSE (Molecular Probes, Eugene, OR), and 106 cells were plated in 96-well deep-well plates in 500 μl of RPMI 5% HS. The cells were left unstimulated (negative control) or stimulated with SEB (Sigma, St. Louis, MO) at 20 ng/ml (positive control), HIV gag peptides at 2 μg/ml, and with αDEC p24, αDC-SIGN p24, αMMR p24, and control Ig p24 at 0.1 to 10 μg/ml. After 6–7 d of culture, samples were restimulated for 8 h with or without p24 peptides (2 μg/ml), either all 55 peptides or individual pools of peptides, in the presence of 0.5 μg/ml of αCD28 and αCD49d (clones L293 and L25; BD Biosciences), adding BFA (Sigma) at 10 μg/ml for the last 6 h to block cytokine secretion. The cells were fixed, permeabilized and stained with a combination of fluorochrome-conjugated antibodies: perCP-CD3, APC-CD8, and PE-IFNγ or its respective isotype control. Cells were analyzed on a FACS-Calibur II using CELLQuest software, collecting 50,000–100,000 high-CD3+ events. Most of the data were displayed as two-color dot plots (FL1 vs. FL2) to measure CFSE dilution and IFNγ production in CD3+ CD8+ cells.
Expansion of Antigen-Specific T Cells in DC/T Cell Coculture.
To directly assess the function of DCs in presenting the gag fusion mAbs, we used, in parallel to the PBMCs assay above, monocyte-derived DCs. Immature cells at day 5 of monocyte culture in GM-CSF and IL-4 were collected and pulsed overnight with medium, p24 peptides (2 μg/ml), or αDEC p24, αDC-SIGN p24, αMMR p24, and control Ig p24 at 1 μg/ml. The antigen-pulsed DCs were matured by adding γ-irradiated CD40L-expressing cells (kindly provided by J. Banchereau, Dallas, TX) at a ratio of 1:5 (CD40L:DC) for 48 h. CD40 ligation enhances cross-presentation by DCs (53). Syngeneic CD14− cells were thawed, rested for 2 h at 37°C before CFSE labeling and plating in 48-well plates at 1.5 to 2 × 106 per ml and cultured with the antigen-pulsed mature DCs at a DC/T ratio 1:30. The cocultures were incubated at 37°C for 6–7 d. Each sample was then restimulated for the last 8 h with or without p24 peptides (2 μg/ml) in the presence of costimulator mAbs (0.5 μg/ml). Cells were analyzed for CFSE dilution and IFNγ secretion as above. Where necessary, data comparing the frequency of CFSElow proliferating, IFNγ secreting, CD3+ CD8+ T cells were compared by using a paired two-tail t test.
Supplementary Material
Acknowledgments
We thank the patients from the St. Vincent's Hospital Comprehensive Clinic and The Rockefeller University Hospital for their cooperation and interest, J. Adams for help with the graphics, and Drs. C. Münz and M. Dhodapkar for discussions. This work was by supported by National Institutes of Health (NIH) Grant AI40874, by the Grand Challenges in Global Health initiative, and by Direct Effect. This project has been funded in part by federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400 and the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- CFSE
carboxyfluorescein succinimydl ester
- DC
dendritic cell
- PBMCs
peripheral blood mononuclear cells.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610383104/DC1.
References
- 1.Letvin NL. Annu Rev Med. 2005;56:213–223. doi: 10.1146/annurev.med.54.101601.152349. [DOI] [PubMed] [Google Scholar]
- 2.McMichael AJ. Annu Rev Immunol. 2006;24:227–255. doi: 10.1146/annurev.immunol.24.021605.090605. [DOI] [PubMed] [Google Scholar]
- 3.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Nature. 1987;329:512–518. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- 4.Walker CM, Moody DJ, Stites DP, Levy JA. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 5.Yang OO, Kalams SA, Rosenzweig M, Trocha A, Jones N, Koziel M, Walker BD, Johnson RP. J Virol. 1996;70:5799–5806. doi: 10.1128/jvi.70.9.5799-5806.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Science. 1996;270:1811–1816. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 7.Wagner L, Yang OO, Garcia-Zepeda EA, Ge Y, Kalams SA, Walker BD, Pasternack MS, Luster AD. Nature. 1998;391:908–911. doi: 10.1038/36129. [DOI] [PubMed] [Google Scholar]
- 8.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, et al. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 9.Metzner KJ, Jin X, Lee FV, Gettie A, Bauer DE, Di Mascio M, Perelson AS, Marx PA, Ho DD, Kostrikis LG, et al. J Exp Med. 2000;191:1921–1932. doi: 10.1084/jem.191.11.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price DA, Goulder PJ, Klenerman P, Sewell AK, Easterbrook PJ, Troop M, Bangham CR. Proc Natl Acad Sci USA. 1997;94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MBA, et al. Nat Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 12.Jones NA, Wei X, Flower DR, Wong M, Michor F, Saag MS, Hahn BH, Nowak MA, Shaw GM, Borrow P. J Exp Med. 2004;200:1243–1256. doi: 10.1084/jem.20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palucka AK, Banchereau J. Curr Opin Immunol. 2002;14:420–431. doi: 10.1016/s0952-7915(02)00365-5. [DOI] [PubMed] [Google Scholar]
- 14.Pulendran B. Immunol Rev. 2004;199:227–250. doi: 10.1111/j.0105-2896.2004.00144.x. [DOI] [PubMed] [Google Scholar]
- 15.Albert ML, Sauter B, Bhardwaj N. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 16.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 17.Touret N, Paroutis P, Terebiznik M, Harrison RE, Trombetta S, Pypaert M, Chow A, Jiang A, Shaw J, Yip C, et al. Cell. 2005;123:157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Ackerman AL, Giodini A, Cresswell P. Immunity. 2006;25:607–617. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Imai J, Hasegawa H, Maruya M, Koyasu S, Yahara I. Int Immunol. 2005;17:45–53. doi: 10.1093/intimm/dxh184. [DOI] [PubMed] [Google Scholar]
- 20.Norbury CC, Chambers BJ, Prescott AR, Ljunggren HG, Watts C. Eur J Immunol. 1997;27:280–288. doi: 10.1002/eji.1830270141. [DOI] [PubMed] [Google Scholar]
- 21.Singh-Jasuja H, Toes RE, Spee P, Munz C, Hilf N, Schoenberger SP, Ricciardi-Castagnoli P, Neefjes J, Rammensee HG, Arnold-Schild D, et al. J Exp Med. 2000;191:1965–1974. doi: 10.1084/jem.191.11.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Accapezzato D, Visco V, Francavilla V, Molette C, Donato T, Paroli M, Mondelli MU, Doria M, Torrisi MR, Barnaba V. J Exp Med. 2005;202:817–828. doi: 10.1084/jem.20051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nouri-Shirazi M, Banchereau J, Bell D, Burkeholder S, Kraus ET, Davoust J, Palucka KA. J Immunol. 2000;165:3797–3803. doi: 10.4049/jimmunol.165.7.3797. [DOI] [PubMed] [Google Scholar]
- 24.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhodapkar MV, Krasovsky J, Olson K. Proc Natl Acad Sci USA. 2002;99:13009–13013. doi: 10.1073/pnas.202491499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachmann MF, Lutz MB, Layton GT, Harris SJ, Fehr T, Rescigno M, Ricciardi-Castagnoli P. Eur J Immunol. 1996;26:1–7. doi: 10.1002/eji.1830261109. [DOI] [PubMed] [Google Scholar]
- 27.Larsson M, Fonteneau JF, Somersan S, Sanders C, Bickham K, Thomas EK, Mahnke K, Bhardwaj N. Eur J Immunol. 2001;31:3432–3442. doi: 10.1002/1521-4141(200112)31:12<3432::aid-immu3432>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 28.Maranon C, Desoutter JF, Hoeffel G, Cohen W, Hanau D, Hosmalin A. Proc Natl Acad Sci USA. 2004;101:6092–6097. doi: 10.1073/pnas.0304860101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, et al. J Exp Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kita H, Lian Z-X, Van De Water J, He X-S, Matsumura S, Kaplan M, Luketic V, Coppel RL, Ansari AA, Gershwin ME. J Exp Med. 2002;195:113–123. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuo M, Nagata Y, Sato E, Atanackovic D, Valmori D, Chen YT, Ritter G, Mellman I, Old LJ, Gnjatic S. Proc Natl Acad Sci USA. 2004;101:14467–14472. doi: 10.1073/pnas.0405947101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurts C, Heath WR, Carbone FR, Allison J, Miller JFAP, Kosaka H. J Exp Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend ARM, Gotch FM, Davey J. Cell. 1985;42:457–467. doi: 10.1016/0092-8674(85)90103-5. [DOI] [PubMed] [Google Scholar]
- 34.Townsend ARM, Bastin J, Gould K, Brownlee GG. Nature. 1986;423:575. doi: 10.1038/324575a0. [DOI] [PubMed] [Google Scholar]
- 35.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 36.den Haan J, Lehar S, Bevan M. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung S, Unutmaz D, Wong P, Sano G-I, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iyoda T, Shimoyama S, Liu K, Omatsu Y, Maeda Y, Takahara K, Akiyama Y, Steinman RM, Inaba K. J Exp Med. 2002;195:1289–1302. doi: 10.1084/jem.20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, et al. Proc Natl Acad Sci USA. 2006;103:10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz V, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee H-W, Park CG, et al. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 41.Hawiger D, Inaba K, Dorsett Y, Guo K, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. J Exp Med. 2001;194:769–780. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM. J Exp Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boscardin SB, Hafalla JC, Masilamani RF, Kamphorst AO, Zebroski HA, Rai U, Morrot A, Zavala F, Steinman RM, Nussenzweig RS, et al. J Exp Med. 2006;203:599–606. doi: 10.1084/jem.20051639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trumpfheller C, Finke JS, Lopez CB, Moran TM, Moltedo B, Soares H, Huang Y, Schlesinger SJ, Park CG, Nussenzweig MC, et al. J Exp Med. 2006;203:607–617. doi: 10.1084/jem.20052005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo M, Gong S, Maric S, Misulovin Z, Pack M, Mahnke K, Nussenzweig M, Steinman RM. Hum Immunol. 2000;61:729–738. doi: 10.1016/s0198-8859(00)00144-0. [DOI] [PubMed] [Google Scholar]
- 46.Figdor CG, van Kooyk Y, Adema GJ. Nat Rev Immunol. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 47.Sallusto F, Cella M, Danieli C, Lanzavecchia A. J Exp Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geijtenbeek TBH, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, Cornelissen ILMHA, Nottet HSLM, KewalRamani VN, Littman DR, et al. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 49.Granelli-Piperno A, Pritsker A, Pack M, Shimeliovich I, Arrighi J-F, Park CG, Trumpfheller C, Piguet V, Moran TM, Steinman RM. J Immunol. 2005;175:4265–4273. doi: 10.4049/jimmunol.175.7.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yewdell JW, Norbury CC, Bennink JR. Adv Immunol. 1999;73:1–77. doi: 10.1016/s0065-2776(08)60785-3. [DOI] [PubMed] [Google Scholar]
- 51.Thery C, Amigorena S. Curr Opin Immunol. 2001;13:I45–51. doi: 10.1016/s0952-7915(00)00180-1. [DOI] [PubMed] [Google Scholar]
- 52.Romani N, Gruner S, Brang D, Kämpgen E, Lenz A, Trockenbacher B, Konwalinka G, Fritsch PO, Steinman RM, Schuler G. J Exp Med. 1994;180:83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delamarre L, Holcombe H, Mellman I. J Exp Med. 2003;198:111–122. doi: 10.1084/jem.20021542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}