

Abstract
The atmospheric budget of methyl bromide (CH3Br), an ozone-depleting gas, is highly uncertain, because it has complex sources and sinks. Although oceans, biomass burning, and industrial production are identified as the major sources, the fraction of CH3Br that is contributed by each source is not well known. A mass-balance approach that exploits differences in the carbon isotopic signature (δ13C) of CH3Br sources and sinks may provide a means of reducing uncertainties in the atmospheric budget. This approach depends on the distinctiveness of industrially produced methyl bromide. Our δ13C measurements of industrial CH3Br from the three largest manufacturers worldwide yield a weighted average of −54.4‰ relative to the Peedee Belemnite standard. This result suggests that industrial CH3Br is isotopically distinct and that the carbon isotopic composition of atmospheric CH3Br may indicate what fraction of atmospheric CH3Br is anthropogenic.
Methyl bromide (CH3Br) is a trace gas with a tropospheric concentration of about 10 parts per trillion by volume and a total lifetime of 0.6 to 0.9 years in the troposphere (1–3). CH3Br has the highest tropospheric concentration of any long-lived organobromide, making it the primary source of bromine to the stratosphere (4). Bromine radicals in the stratosphere have been shown to contribute significantly to stratospheric ozone loss through coupled reactions with ClO, HO2, and NO2 radicals (5–7).
Although the role of bromine in stratospheric ozone loss has been well documented, the CH3Br budget of the troposphere has major uncertainties. Methyl bromide has both industrial and nonindustrial sources to the atmosphere, all of which have highly uncertain magnitudes. Based on our current understanding of the budget, the fraction of tropospheric CH3Br contributed by industrial production could be as low as 20% or as high as 70% (refs. 3, 8, and 9; Table 1). The efficacy of controls or bans on the use of CH3Br to lower the atmospheric burden depend significantly on whether the actual anthropogenic contribution is closer to the higher or lower end of this range.
Table 1.
Summary of estimated CH3Br source fluxes to the atmosphere
| Source | Amount of
CH3Br, ×109 g/year
|
|||
|---|---|---|---|---|
| Flux | Range | Industrial | Nonindustrial | |
| Ocean | 56 | 17–61 | 0 | 56 |
| Biomass burning | 20 | 15–30 | 0 | 20 |
| Fumigation | ||||
| Soils | 32 | 16–47 | 32 | 0 |
| Durables | 6.6 | 4.8–8.4 | 6.6 | 0 |
| Perishables | 5.7 | 5.4–6.0 | 5.7 | 0 |
| Structures | 2 | 2 | 2 | 0 |
| Gasoline Additives | 5 | 0–10 | 0 | 5 |
| Totals | 127 | 60–164 | 46 | 81 |
| (Range) | (28–63) | (32–101) | ||
Stable isotopic signatures have been used profitably for studying the budgets of many trace gases in the atmosphere. The best and most closely related example is that of methane (CH4; refs. 10–14). Like CH3Br, CH4 has industrial sources (primarily natural gas leaks of thermogenic origin), as well as a number of biologically derived sources (e.g., biomass burning, wetlands, and ruminants; ref. 14). Atmospheric CH4 retains the δ13C and δD signatures that it acquires during formation and release (10–12). Because CH3Br has a similar chemical structure, we expect its stable isotopes (C, H, and Br) to be nonexchangeable with other atmospheric species. Thus, isotopic signatures should act as reliable tracers of the sources and sinks of CH3Br to the atmosphere. We present measurements of δ13C for industrial CH3Br and suggest a modeling framework for exploiting this information to further understanding of the CH3Br budget. The isotopic composition of the H and Br in CH3Br may also be useful for fingerprinting purposes.
Methods and Results.
Pure samples of CH3Br were obtained from the three major industrial manufacturers (Albemarle, Baton Rouge, LA; Dead Sea Bromine, Beer Sheva, Israel; and Great Lakes, West Lafayette, IN) for isotopic analysis. These companies manufacture approximately 82% of the world total CH3Br that is used as a fumigant (15). The δ13C values reported here were measured on a micromass isoprime gas chromatograph–isotope ratio mass spectrometer (GC-IRMS) at the Lawrence Berkeley National Laboratory Center for Isotope Geochemistry. GC-IRMS allows separation of CH3Br from other gases, followed by in-line combustion to CO2 and analysis by magnetic sector mass spectrometry; the method is selective, reproducible, and orders of magnitude more sensitive than traditional dual inlet mass spectrometry (DIMS). The δ13C values measured by GC-IRMS for individual tanks typically had standard deviations of less than 0.1‰ and were indistinguishable from DIMS measurements made on CH3Br after off-line closed-tube combustion.
The measured δ13C values of industrially produced CH3Br are all low, and there is variability both within and between manufacturers (Fig. 1; Table 2). The δ13C measured values vary from −66.4‰ to −43.5‰, and the mean for all measured tanks (shown as the horizontal line in Fig. 1) is −55.7‰ (± 2.0‰ SEM). There are differences in the δ13C of CH3Br from the various manufacturers, and thus, a production-weighted mean should be more accurate. Based on reported CH3Br production (15–17), the weighted mean value is −54.4‰. The industrial CH3Br is isotopically light in comparison to the carbon found in most living organic tissue, and its δ13C is lighter than that of methane. Thermogenic methane is used as a carbon source for methanol, which in turn is the feedstock for manufacturing CH3Br (17).
Figure 1.

Mean measured δ13C values for industrial CH3Br from the major manufacturers and comparison with the expected range of natural sources. The error bars shown are SDs, based on multiple samples from each manufacturer. The horizontal shaded line represents the mean for all tanks, without weighting by manufacturer. The weighted average (Table 2) accounts for the percentage of total production from each manufacturer.
Table 2.
Data used to construct a weighted mean δ13C for the three largest manufacturers of CH3Br
| Manufacturer | No. tanks measured | Mean δ13C* | SD of δ13C | SEM of δ13C | Production† |
|---|---|---|---|---|---|
| Albemarle | 3 | −52.97 | ±1.71 | ±0.96 | 13% |
| Dead Sea Bromine | 6 | −49.48 | ±6.92 | ±2.82 | 41% |
| Great Lakes | 7 | −62.32 | ±4.51 | ±1.70 | 28% |
| Weighted average | — | −54.4 | 82% |
Mean δ13C values for each manufacturer in units of permil relative to the Peedee Belemnite standard.
Approximate fraction of world total production for each manufacturer in 1995 (ref. 15; G. Ter Haar, personal communication; D. McAllister, personal communication).
Nonindustrial Source Signatures.
Biomass burning (Table 1) produces CH3Br through methylation of Br− in the cellular fluids of plants during combustion (18, 19) and, as such, is a distinct and measurable source to the atmosphere. Most combustion-produced hydrocarbons, including CH4, have δ13C values that reflect the biomass source (12, 20). In the case of forests (predominantly C3 plants), this average δ13C is about −27‰. Grasses (predominantly C4 plants) have an average δ13C of about −13‰. Although grassland fires tend to have lower emission ratios of hydrocarbons to CO2 than forest fires (21, 22), the majority of CH3Cl is produced by savanna fires (J. M. Lobert, personal communication). Thus, the same is probably true for CH3Br. The δ13C signature for other combustion hydrocarbons also typically reflects their carbon source; thus, as a starting point, the mean δ13C of CH3Br from biomass burning might be around −20‰. On the other hand, one study shows that CH3Cl produced from forest fires has δ13C of −45‰ (23). Although CH3Cl is a close analogue to CH3Br, it is uncertain whether this large fractionation applies for CH3Br.
CH3Br is produced by marine algae, probably during senescence (24–29), causing coastal upwelling regions of the ocean to be highly supersaturated with CH3Br (1). Until recently, it was assumed that the oceans were supersaturated everywhere with respect to CH3Br and, thus, that they were a net source of the gas to the atmosphere (30–32). Recent measurements, however, show that large regions of the ocean are undersaturated with respect to CH3Br (1, 33). Gross-source estimates range from 17 to 61 Gg/year (8, 9) and do not include the effect of the ocean sink on the net flux. Marine algae typically have a δ13C of −12 to −23‰ (34). CH3Br produced by these algae may be in the same range.
Because current estimates of the sinks for CH3Br exceed the estimated sources (3, 35), it has been suggested that balancing the CH3Br budget requires other large, as yet unidentified, terrestrial sources of CH3Br to the atmosphere. A recent study of Brassica crops has shown that soil Br− can be converted to CH3Br and emitted to the atmosphere in significant quantities (36). Other terrestrial plants and fungi have also been shown to emit methyl halides and thus may be additional sources of CH3Br to the atmosphere (37–39). If these sources were found to be significant, then their δ13C signatures would have to be measured to complete the characterization of natural sources.
Carbon Isotope Fractionation in the CH3Br cycle.
It is likely that the δ13C of atmospheric CH3Br is influenced by isotopic fractionation associated with processes that remove it from the atmosphere and with processes that release it from soils and the ocean. We may be able to estimate the size of these fractionations by comparison with methane, for which there are data. For CH4, the primary sink is oxidation by the OH radical in the atmosphere; this sink fractionation has been measured to be about +5‰ (40). The sink fractionation for OH + CH3Br is likely to be of similar magnitude. Sink fractionations associated with degradation in seawater and in soils are also likely and may be larger.
Methyl Bromide Isotopic Budget.
A mass-balance model can be used to understand the effect that various methyl bromide sources and sinks will have on the carbon isotope ratio of atmospheric methyl bromide and, ultimately, to estimate the industrial contribution from the δ13C value of atmospheric CH3Br. The primary known sources are the release from soils of industrially produced CH3Br, biomass burning, and production by algae in the ocean. The primary known sinks are oxidation in the atmosphere by the hydroxyl radical as well as hydrolysis and anion exchange in seawater. The differential equation that describes the time dependence of the 13C/12C ratio (R) of the atmosphere is
 |
1 |
 |
where N is the total number of moles of methyl bromide in the atmosphere, Φ is flux in moles/year, and α is the isotopic fractionation factor (= K12/K13) associated with removal from the atmosphere. This equation can also include other sources, such as terrestrial plants, or sinks, such as soils.
Although differences in concentration persist between the Northern and Southern hemispheres, for each hemisphere, concentrations are essentially stable. Because the lifetime of CH3Br in the troposphere is on the order of 0.7 years (2, 3), it is reasonable to assume that the concentration and isotopic value of CH3Br in the atmosphere will approach steady state on a time scale of 1–2 years. At steady state, Eq. 1 can be solved for the atmospheric 13C/12C ratio of CH3Br and put in the following form:
 |
2 |
where the x variables in the numerator are the fractions of the total flux to the atmosphere by natural and industrial sources and the x′ variables in the denominator are the fractions of the removal attributable to the ocean sink and to oxidation by OH in the atmosphere. In this formulation, the natural sources of CH3Br are combined; this combination assumes that they can be characterized and that a mean value of 13C/12C can be established. Recasting this equation in terms of delta values (δ13C) the result is
 |
3 |
where Δ = 1,000(αi−1 − 1).
The asterisks on the industrial source terms in Eqs. 2 and 3 are added because most industrially produced methyl bromide is applied to soils as a biocide. The CH3Br is partially broken down in the soil by hydrolysis and, to a lesser extent, microbial activity (41), and in the process, it is likely to be fractionated isotopically. The CH3Br released to the atmosphere has a different δ13C, depending on how much of it has been broken down before release and on the conditions under which the microbial action occurred. If it is assumed that the microbial degradation has associated with it a fractionation factor of αsoil (or in terms of delta values: Δsoil) and the process can be described as Rayleigh distillation, then the amount released (x*ind) and the delta value of the released CH3Br (δ13C*ind) is related to the amount applied (xind), and the delta value of industrially produced CH3Br (δ13Cind) is calculated by
 |
4 |
where the parameter fsoil is the fraction of applied CH3Br that is eventually released to the atmosphere. Substituting Eq. 4 into Eq. 3 and solving for the industrial fraction of the total source term x*ind yields
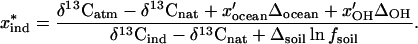 |
5 |
Eq. 5 represents the industrial contribution to the atmospheric budget of CH3Br, based on the δ13C value of atmospheric CH3Br and the isotopic signature of its sources and sinks. Based on our measurements of industrially produced CH3Br and inferences about the natural sources, we expect the difference between the δ13C values of industrial and natural CH3Br to be about −30 to −35‰. The value of the correction factor in the denominator that accounts for microbial degradation in soils is still uncertain, largely because Δsoil depends on the soil microenvironment and therefore must be measured in the field under appropriate conditions. The sensitivity of this expression should ultimately depend on the magnitude of the denominator.
DISCUSSION
The results reported here are significant, because they indicate that the δ13C value of the industrial source of CH3Br, at approximately −54‰, is isotopically light and distinct from the value characteristic of most natural sources of carbon, which is in the range of −15 to −30‰. The low δ13C value of industrial CH3Br is probably the result of the manufacturing process, which uses methane-derived methanol as a carbon source (17). Most methane used in industrial processes is of thermogenic origin and has a δ13C value of −35‰ to −50‰ (11, 12). The CH3Br value may be lower, because additional fractionation occurs during the conversion of methane to methanol or during the production of CH3Br.
The δ13C value of atmospheric CH3Br could provide an indication of the relative strengths of the natural and industrial sources (Eq. 5). The isotopic fractionation associated with microbial degradation of methyl bromide in soils before release to the atmosphere must be accounted for and could significantly impact the interpretation of the δ13C value of atmospheric CH3Br. The other major uncertainties include the isotopic fractionation factor associated with removal of CH3Br to soils and the oceans as well as the δ13C value of the nonindustrial sources of CH3Br. There is evidence that significant fractionations could be associated with the production of natural CH3Br, as observed for CH3Cl from biomass burning (24), but the algal source of CH3Br may be shifted in a compensatory fashion toward higher δ13C values by in situ degradation processes that have kinetic isotope effects greater than 1.0.
Variations in the δ13C of atmospheric CH3Br with time and latitude should provide additional information about the factors controlling the atmospheric CH3Br budget. In addition to the interhemispheric gradient in CH3Br concentration, there is probably a related δ13C gradient. Differences in the δ13C of atmospheric CH3Br would indicate differences in the source and sink fluxes to each hemisphere providing new constraints on the global budget. Temporal variations in the δ13C of atmospheric CH3Br could provide information on seasonality of the various sources and sinks.
CONCLUSIONS
The measurements presented here indicate that industrially manufactured CH3Br has a carbon isotopic signature that is light (δ13C = −54‰) and potentially distinguishable from the major nonindustrial sources. Isotopic measurements of CH3Br sources and sinks as well as the atmospheric reservoir could therefore greatly improve our understanding of the biogeochemical processes controlling the atmospheric CH3Br budget. This approach could also provide an independent tool for assessing the effectiveness of recent amendments to the Montreal Protocol that call for the complete phaseout of CH3Br by 2005 in industrialized nations (42).
Acknowledgments
We thank Mark Conrad of the Lawrence Berkeley National Laboratory for his assistance and helpful comments and Tom Duafala, David McAllister, David Sichermann, Gary Ter Haar, and Gene Brown for providing valuable information on industrial CH3Br production and for helping us obtain samples of industrial CH3Br. This work was supported by National Aeronautics and Space Administration Upper Atmosphere Research Program Award NAG5-7173, by National Science Foundation Atmospheric Chemistry Program Award ATM-9729110, and by the Laboratory-Directed Research Development Program of the E.O. Lawrence Berkeley National Laboratory.
References
- 1.Lobert J M, Butler J H, Montzka S A, Geller L S, Myers R C, Elkins J W. Science. 1995;267:1002–1005. doi: 10.1126/science.267.5200.1002. [DOI] [PubMed] [Google Scholar]
- 2.Yvon S A, Butler J H. Geophys Res Lett. 1996;23:53–56. [Google Scholar]
- 3.Yvon S A, Butler J H. Geophys Res Lett. 1997;24:1227–1230. [Google Scholar]
- 4.Schauffler S M, Heidt L E, Pollock W H, Gilpin T M, Vedder J F, Solomon S, Lueb R A, Atlas E L. Geophys Res Lett. 1993;20:2567–2570. [Google Scholar]
- 5.Wofsy S C, McElroy M B, Yung Y L. Geophy Res Lett. 1975;2:215–218. [Google Scholar]
- 6.Yung Y L, Pinto J P, Watson R T, Sander S P. J Atmos Sci. 1980;37:339–353. [Google Scholar]
- 7.McElroy M B, Salawitch R J, Wofsy S C, Logan J A. Nature (London) 1986;321:759–762. [Google Scholar]
- 8.Penkett S A, editor. World Meteorological Organization. Scientific Assessment of Ozone Depletion: 1994. Geneva: World Meteorol. Organ.; 1995. pp. 10.1–10.26. [Google Scholar]
- 9.Butler J H, Rodriguez J M. In: The Methyl Bromide Issue. Bell C H, Price N, Chakrabarti B, editors. Vol. 1. Chichester, NH: Wiley; 1996. pp. 27–90. [Google Scholar]
- 10.Schoell M. Geochim Cosmochim Acta. 1980;44:649–661. [Google Scholar]
- 11.Whiticar M J, Faber E, Schoell M. Geochim Cosmochim Acta. 1986;50:693–709. [Google Scholar]
- 12.Whiticar M J. Adv Org Geochem. 1989;16:531–547. [Google Scholar]
- 13.Quay P D, King S L, Stutsman J, Wilbur D O, Steele L P, Fung I, Gammon R H, Brown T A, Farwell G W, Grootes P M, et al. Global Biogeochem Cycles. 1991;5:25–47. [Google Scholar]
- 14.Wahlen M. In: Annual Review of Earth and Planetary Sciences. Wetherill G W, Albee A L, Burke K C, editors. Vol. 21. Palo Alto, CA: Annu. Rev.; 1993. pp. 407–426. [Google Scholar]
- 15.United Nations Environment Programme. Production and Consumption of Ozone-Depleting Substances, 1986–1995. Geneva: United Nations Environment Programme; 1997. [Google Scholar]
- 16.Ter Haar G. Health and Environment. Baton Rouge, LA: Albemarle; 1997. [Google Scholar]
- 17.McAllister D. Agricultural Products Business. West Lafayette, IN: Great Lakes; 1997. [Google Scholar]
- 18.Manö S, Andreae M O. Science. 1994;263:1255–1257. doi: 10.1126/science.263.5151.1255. [DOI] [PubMed] [Google Scholar]
- 19.Andreae M O, Atlas E, Harris G W, Helas G, de Kock A, Koppmann R, Maenhaut W, Manö S, Pollock W H, Rudolph J, et al. J Geophys Res. 1996;101:23603–23613. [Google Scholar]
- 20.Wahlen M. In: Stable Isotopes in Ecology and Environmental Science. Lathja K, Michener R H, editors. Oxford: Blackwell; 1994. pp. 93–113. [Google Scholar]
- 21.Hao W-M, Ward D E. J Geophys Res. 1993;98:20657–20661. [Google Scholar]
- 22.Lobert J M, Scharffe D H, Hao W M, Crutzen P J. Nature (London) 1990;346:552–554. [Google Scholar]
- 23.Rudolph J, Lowe D C, Martin R J, Clarkson T S. Geophys Res Lett. 1997;24:659–662. [Google Scholar]
- 24.Manley S L, Dastoor M N. Limnol Oceanogr. 1987;32:709–715. [Google Scholar]
- 25.Manley S L, Dastoor M N. Mar Biol. 1988;98:477–482. [Google Scholar]
- 26.Wuosmaa A M, Hager L P. Science. 1990;249:160–162. doi: 10.1126/science.2371563. [DOI] [PubMed] [Google Scholar]
- 27.Tokarczyk R, Moore R M. Geophys Res Lett. 1994;21:285–288. [Google Scholar]
- 28.Tait V K, Moore R M. Limnol Oceanogr. 1995;40:189–195. [Google Scholar]
- 29.Scarratt M G, Moore R M. Mar Chem. 1996;54:263–272. [Google Scholar]
- 30.Singh H B, Salas L J, Stiles R E. J Geophys Res. 1983;88:3684–3690. [Google Scholar]
- 31.Singh H B, Kanakidou M. Geophys Res Lett. 1993;20:133–136. [Google Scholar]
- 32.Khalil M A K, Rasmussen R A, Gunawardena R. J Geophys Res. 1993;98:2887–2896. [Google Scholar]
- 33.Lobert J M, Yvon-Lewis S A, Butler J H, Montzka S A, Myers R C. Geophys Res Lett. 1997;24:171–172. [Google Scholar]
- 34.Faure G. Principles of Isotope Geology. New York: Wiley; 1986. [Google Scholar]
- 35.Lee-Taylor J M, Doney S C, Brasseur G P. J Geophys Res. 1998;103:16039–16057. [Google Scholar]
- 36.Gan J, Yates S R, Ohr H D, Sims J J. Geophys Res Lett. 1998;25:3595–3598. [Google Scholar]
- 37.Harper D B. Nature (London) 1985;315:55–57. [Google Scholar]
- 38.Harper D B. J Gen Microbiol. 1986;132:1231–1246. [Google Scholar]
- 39.Harper D B. Phytochemistry. 1988;27:3147–3153. [Google Scholar]
- 40.Cantrell C A, Shetter R E, McDaniel A H, Calvert J G, Davidson J A, Lowe D C, Tyler S C, Cicerone R J, Greenberg J P. J Geophys Res. 1990;95:22455–22462. [Google Scholar]
- 41.Miller L G, Connell T L, Guidetti J R, Oremland R S. Appl Environ Microbiol. 1997;63:4346–4354. doi: 10.1128/aem.63.11.4346-4354.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.United Nations Environment Programme. September 1997 Amendments. Geneva; 1997. [Google Scholar]