

Abstract
Animal models of rheumatoid arthritis (RA) have provided substantial insights into basic pathogenic mechanisms of chronic inflammatory arthritis and autoimmune disease in general. Of the variety of models reported, collagen-induced arthritis (CIA) has been the most characterized in terms of both its pathogenesis and its underlying immunological basis. Collagen-induced arthritis has also been the model of choice in terms of testing potential new therapeutic agents for the treatment of human RA. Nevertheless, the complex nature of the balance between T-cell cytokines and the chronic inflammatory processes is only recently becoming clear. This review focuses on these developments, highlighting their implications for our understanding of RA and for the use of CIA as a suitable animal model.
Introduction
Collagen-induced arthritis (CIA) was first reported by Trentham and colleagues who observed the disease in rats following a single intradermal injection of type II collagen (CII) emulsified in Freund's adjuvant.1 Further studies demonstrated that a similar pathology could also be induced in primates2,3 and in susceptible strains of mice.4 CIA can be induced using native autologous or heterologous CII and is specific to CII, since immunization with types I or III collagen fail to induce disease.1,4 While either incomplete (IFA) or complete Freund's adjuvant (CFA) can be used to trigger CIA in rats,3 the induction of disease in mice generally requires the presence of heat-killed Mycobacterium tuberculosis in CFA.4 Immunization with CII/CFA results in a rapid and severe polyarthritis of the peripheral articular joints that first appears around 3–4 weeks after disease challenge and becomes progressively worse for approximately 2–4 weeks before slowly waning. Whilst the pathology is similar when CIA is induced with either autologous or heterologous CII, the nature of the disease differs; autologous CII induces a more chronic disease with a delayed onset and reduced penetrance.5,6 In both cases the histopathology of inflammatory arthritis resembles human rheumatoid arthritis (RA).
Like RA, CIA is characterized by the presence of fibrin deposition, hyperplasia of synovial cells, periosteal bone formation, mononuclear infiltrates, pannus formation and eventual ankylosis of one or more articular joints.1,4 In addition, the presence of rheumatoid factor and systemic manifestations have been reported in animals with CIA.7,8 Moreover, susceptibility to both CIA and RA is strongly associated with the expression of specific major histocompatibility complex (MHC) class II molecules,9,10 with additional roles for non-MHC loci being reported.11–13 In mice, susceptibility to CIA is mediated predominantly by I-Aq, an MHC class II molecule which binds the same immunodominant CII peptide region as the human RA-associated allele HLA-DR4 (DRB1*0401).14 This observation, taken together with the other similarities between the diseases, has led to speculation as to whether CII or a cross-reacting antigen is involved in the initiation of RA itself. Autoreactivity to cartilage CII in human RA patients, although not a defining feature of the disease, has been clearly demonstrated.15,16 In this regard, anti-CII antibody responses have been reported in 30–70% of RA patients depending on the stage of disease.15–17 However, anti-CII reactivity may remain a consequence of the chronic inflammatory processes in RA rather than the cause. Regardless of the involvement of CII in triggering RA, its localization as a major component of diarthroidal joints, the primary site of inflammation in RA, probably means that the underlying processes involved in establishing CIA and RA share similar features. The parallels between these arthritides, combined with the relative ease of inducing a consistent and reproducible experimental arthritis, have led to extensive investigations of autoimmune arthritis using the CIA model. Particular emphasis has been placed on elucidating the mechanisms involved in the initiation and maintenance of the pathogenic anti-CII immune response throughout the course of disease.
The Role of T cells in the initiation of CIA
Collagen-induced arthritis is a multifaceted, immunologically mediated disease involving T cells, B cells and populations of inflammatory cells that infiltrate the joint tissue and induce pathology. While the precise mechanisms by which immunization with heterologous or autologous CII in CFA leads to a chronic arthritis in susceptible mice are not known, there are considerable data to implicate CII-reactive CD4+ T cells as the primary mediators of disease induction, and complement-fixing anti-CII autoantibody production by B cells as the major immune mechanism leading to the localized chronic inflammatory response.7,9,18,19
Since antigen recognition by T cells requires peptide to be presented in association with MHC molecules, an important role for CD4+ T cells in CIA is implied by disease susceptibility being restricted to mice that possess certain MHC class II alleles. This association was first described in 1981 by Wooley and colleagues using strains of congenic B10 mice that had been immunized with chicken CII. Despite the ability of several strains to mount strong anti-CII immune responses, only those mice expressing the I-Aq allele developed arthritis.9,20 This observation suggested that susceptibility to CIA depends on the capability of MHC class II molecules to present specific peptides which leads to the activation of arthritis-promoting CII-reactive T cells. The observations that early administration of monoclonal antibodies to CD4,21 T-cell receptor-αβ (TCR-αβ),22,23 CD2524 and I-A25 can suppress CIA provides further support for the role of T cells in mediating disease.
Type II collagen is a 1018 amino acid, homotrimeric molecule that shares up to 94% amino acid sequence homology between bird and mouse. Due to its large size, attempts to isolate antigenic CII peptides presented by I-Aq molecules necessitated cyanogen bromide (CB) digestion of the protein. A single fragment of chicken CII, CB11 (amino acids 124–402) was found to induce strong serum reactivity, and upon immunization of DBA/1 mice (H-2q), led to an attenuated, but histologically similar arthritis to that induced by native CII.26 Peptide-mapping studies of CB11 have revealed a single immunodominant epitope, CII(260–267), that upon immunization induces a strong T-cell proliferative response and the production of interferon-γ (IFN-γ) in strains susceptible to CIA. Significantly, H-2 congenic strains that are resistant to disease fail to recognize this peptide.27 Furthermore, preimmunization of neonatal mice with CB11,28 and specifically CII(245–270), prior to challenge in adulthood with native chicken CII, dramatically suppressed the incidence of arthritis.29 Taken together, with amino acid substitution experiments,29 these observations indicate that the determinant CII(260–270) plays a central role in regulating the onset of CIA in H-2q mice. In addition to CII(260–270), four subdominant I-Aq-restricted epitopes on CB11 have been characterized and are hypothesized to be involved to varying degrees in the induction and maintenance of the arthritic state.27,30 While H-2q mice are susceptible to CIA upon immunization with chicken, bovine, or human CII, the haplotype H-2r has also been found to confer susceptibility to CIA when porcine or bovine CII is used as immunogen.31 Separate determinants localized on CB8 and CB10 are involved in regulating the arthritic response in these animals.32,33
Extensive research is beginning to define the relative contributions of non-autoreactive CII-specific T cells and autoreactive CII-specific T cells in the induction of CIA. The ability to induce a diminished, but chronic CIA in DBA/1 mice using autologous CII indicates that at least some autoreactive CII-specific T cells escape thymic deletion in healthy animals and either remain ignorant, or are subject to other processes involved in peripheral tolerance induction.7 Nevertheless, studies have shown that despite close sequence homology, the strong proliferative response induced following immunization of DBA/1 mice with rat CII was poorly cross-reactive to mouse CII.34 More recently, elegant experiments by Malmström and co-workers have helped to clarify the development of the autoimmune response to the immunodominant I-Aq-restricted determinant CII(256–270) in CIA.35,36 Within this peptide, a single amino acid distinguishes rat, avian, bovine and human CII [which have a glutamic acid (E) at position (266)] from mouse CII [which has an aspartic acid (D) at this position]. Transgenic mice expressing rat CII(256–270) systemically as part of CI are rendered profoundly tolerant to CII, failing to develop CIA, produce antibodies, or mount lymph node cell responses following challenge with either the peptide itself or whole rat CII. However, immunization of these mice with chicken CII triggers T-cell responses to a subdominant chicken CII-specific epitope, CII(190–200), leading to arthritis. In another transgenic mouse strain bearing a D→E substitution at CII(266), immunization with rat CII fails to trigger significant proliferative responses, but is associated with the production of IFN-γ and normal levels of anti-CII antibodies. These animals develop CIA, and although the incidence of disease was halved, severity remained comparable to non-transgenic littermates. Collectively, these data suggest that following heterologous CII immunization, T-cell recognition is directed primarily against non-self epitopes including CII(256–270). The onset of inflammation triggered by resulting antibodies leads to the subsequent activation of residual self-CII-specific T cells. When autologous CII is used as immunogen, partially tolerized T cells capable of recognizing native CII(256–270), are able to secrete IFN-γ and thus, may provide help for anti-CII antibody production.
The Role of antibody in the establishment of inflammation
Importantly, it has proven difficult to adoptively transfer CIA with CD4+ T cells alone.37,38 These findings support the idea that anti-CII antibodies play a critical role in initiating the disease, a hypothesis for which there is now a great deal of evidence.19,39–41 Immunization of DBA/1 mice with heterologous CII induces the rapid activation of CII-specific B cells;5,42 9 days after challenge with native heterologous CII, large numbers of immunoglobulin (IgG) anti-CII antibody-producing cells can be detected within lymph nodes draining the site of immunization. This reaction is highly cross-reactive against CII from a variety of species in high responder H-2q mice.43 Furthermore, an autoreactive antibody response may be observed in both mice and rats following immunization with autologous CII.5,6
Studies have demonstrated that the total amount of anti-CII immunoglobulin produced by B cells is not a good correlate for the development of arthritis, rather it is qualitative differences in antibody production that determine the outcome of disease.20 Such differences may include the antibody isotype, antibody avidity and the precise epitopes recognized by the autoantibody.44,45 Indeed, many non-susceptible mouse strains can elicit strong anti-CII antibody responses following immunization, some directed to the arthritogenic CB11 region of CII.20,45 Experiments to compare murine CII-specific autoantibodies produced from susceptible and non-susceptible congenic mouse strains immunized with the CB11 fragment of chicken CII, revealed that amongst a pattern of similar autoantibody epitope specificities, a single murine epitope was identified that was recognized only by susceptible I-Aq-bearing mice. Analysis of antibodies binding to this epitope revealed them to be predominantly of the IgG2 subclass.45 These isotypes, particularly complement-fixing IgG2a, dominate the anti-CII response in CIA and are essential for pathogenesis.44 Other isotypes of anti-CII antibodies are also found in CIA; for example, the T helper type 2 (Th2)-associated antibody subclasses, IgG1 and IgE are readily measured in the serum of arthritic mice. A role for IgE-mediated pathology in CIA has been proposed, but remains speculative.46
Binding of complement-activating IgG autoantibodies to CII on articular cartilage has been hypothesized as the primary immune mechanism leading to joint inflammation.47,48 In addition, recent data support a crucial role for the formation of IgG immune complexes and subsequent cross-linking of Fc-receptors to induce inflammation. FcRγ-chain−/− mice (H-2q) do not develop CIA, even though anti-CII antibody production remains similar to that observed in susceptible wild-type mice.49
Is there a requirement for T cells in maintaining established disease?
Immunohistochemical studies of joints taken from DBA/1 mice immunized with CII have provided a basic understanding of the distribution and potential interactions of different cell populations in the joint throughout the inflammatory process. During the initial phase of disease (before clinical onset), cells expressing MHC class II are observed to accumulate in foci along the synovial lining. Interspersed within these foci are CD4+ T cells, many of which are CD25+. In addition, CD11b+ macrophage-like cells are detectable at the synovial membrane, while CD8+ T cells and B cells are scarce.50 Progression of the inflammatory response is associated with a marked infiltration of polymorphonuclear cells into the synovium and the development of oedema. Once this has occurred the synovium becomes inflamed, with a pannus forming at the synovium–cartilage interface. Within the pannus, proliferating synoviocytes, activated macrophages, dendritic cells (DC) and granulocytes predominate, while the proportion of CD4+ T cells is reduced.51,52
Although the importance of CD4+ T cells in the development of CIA is clearly established; as in RA, the role of these cells following disease onset is less apparent. Anti-CD4 or anti-TCR-αβ depleting antibody treatments have failed to attenuate established CIA and in some cases even exacerbate disease.21,53–56 However, treatment of mice transgenic for a CII-specific TCR-Vβ chain with non-depleting anti-CD4 monoclonal antibody was shown to suppress established arthritis57 and blockade of CD28 using a combination of antibodies to CD80 and CD86 or soluble CD152 during established CIA, significantly reduced inflammatory cytokine production and ameliorated disease.58 These apparent discrepancies may result from the fact that once disease is established it becomes a self-perpetuating process driven largely by tissue changes themselves (see below). While T cells may continue to play a role in recruiting more inflammatory cells into the joint, the induction of populations of regulatory T cells as part of the ongoing anti-CII response may also serve to dampen the pathological changes. Such regulatory cells are likely to include recently described populations of T cells such as CD4+CD25+ cells,59 Tr1 cells60 or Th3 cells.61 The role of these lymphocyte populations in CIA has not yet been properly explored.
T-cell-mediated regulation of the inflammatory response in the joint may also involve the activities of populations of synovium-resident T cells. CD4−CD8− γδ T cells, and minor populations of interleukin-2 receptor (IL-2R)-α−β+ and CD4−CD8α+β− T cells are resident within the joints of both naïve and arthritic mice, and have been shown to expand during CIA.62,63 Intraperitoneal administration of a cross-linking anti-TCR-γδ antibody prior to immunization with CII significantly delayed the onset and severity of arthritis. By contrast, if the T cells were stimulated by antibody 40 days after disease induction, CIA was rapidly and severely exacerbated, implying a biphasic regulatory function for γδ T cells in arthritis.62
Unlike many models of autoimmune disease, the potential involvement of CD8+ T cells in CIA has not been thoroughly investigated. Experiments to associate CD8+ T cells with disease onset have demonstrated conflicting data between species. In mice, regular administration of monoclonal antibody to CD8 was shown to dramatically suppress the onset of CIA,63 while in the rat, a similar treatment regime had no significant effect.64 Using CD8+ T-cell-deficient DBA/1 mice, Tada and colleagues demonstrated a requirement for these cells in the induction of arthritis. These same CIA-resistant mice were more susceptible to secondary CIA induced after remission, leading the authors to propose that CD8+ T cells may have an additional regulatory function in the disease process.65 Indirect support for this hypothesis comes from experiments involving CII-specific cytotoxic hybridomas generated from arthritic mice. These cells expressed low levels of CD8, recognized CII by an MHC class I pathway, and following irradiation and adoptive transfer into naïve recipients, prevented the onset of disease. In addition, one of the hybrid clones possessed therapeutic properties; demonstrated by its ability to ameliorate CIA when administered 30 days after CII-challenge.66
Do you really need adaptive immunity to get CIA?
Recent data obtained by immunizing recombination activating gene-1 (RAG-1)-deficient mice (RAG-1−/−) bred onto a susceptible genetic background (DBA/1; H-2q) with CII has provided strong evidence for the involvement of lymphocyte-independent mechanisms in the induction and maintenance of CIA.67 Although disease onset and severity were reduced in these T-cell- and B-cell-deficient mice, the frequency of arthritis and associated histopathology were comparable to hemizygous, immune-competent littermates. The authors suggest that the non-antigenic properties of CII, and specifically its ability to act as a ligand for the discoidin domain receptor tyrosine kinases (DDR)1 and DDR2, may be important for the development of a lymphocyte-deficient arthritis. The DDR molecules are involved in regulating the production of matrix metalloproteinases (MMP); molecules linked to tissue destruction in the arthritides. Thus, CII interactions with DDR may promote the activation of MMP independently of the adaptive immune response. However, a delay in disease onset and a reduced pathology in RAG-1−/−DBA/1 mice maintains support for an important role for the adaptive immune response in CIA.
Alternatively, a recent study has suggested that the increased susceptibility of DBA/1 mice for CIA may be partly associated with qualitative differences in the nature of the arthritis it develops. A proportion of DBA/1 mice have been reported to develop spontaneous arthritis with advancing age.68 This condition, which grossly resembles CIA, has been described as a T-cell-independent, enthesopathy of joint tissues, and may develop under conditions similar to those following immunization with CII/CFA.69 It is possible that the susceptibility of DBA/1 mice to this condition contributes to the ease with which CIA can be induced in this mouse strain.
TH1 and TH2 Cytokines in the control of CIA
The production of cytokines by systemic and tissue-specific cell populations is critical for the development (and eventual subsidence) of the autoimmune response to CII and pathology in CIA. There is considerable evidence to suggest that in DBA/1 mice, CIA is a Th1-mediated inflammatory disease. However, a strong humoral component and the production of Th2-associated cytokines and antibodies throughout the course of disease imply that both types of CD4+ Th cell responses are probably involved in modulating arthritis. Two broad experimental strategies have been used extensively to characterize the involvement of cytokines in the induction of CIA: phenotyping CII-specific CD4+ T cells within the periphery, and manipulating the Th1/Th2 cytokine balance at different stages of disease to evaluate the outcome on arthritis. The findings of such studies are summarized in Fig. 1.
Figure 1.
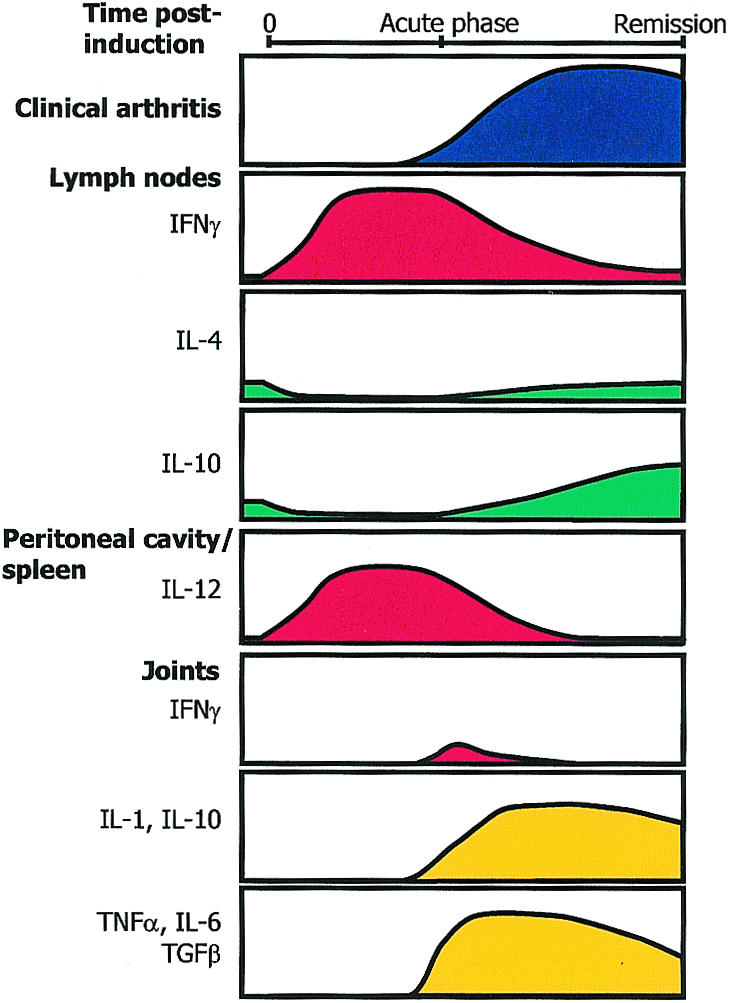
The temporal relationship between cytokine production and disease in CIA in the DBA/1 mouse. Studies of cytokine production and assays for the presence of mRNA have indicated that the initial response following CII/CFA challenge is dominated by a Th1 response, with IFN-γ being readily detectable in the lymph nodes, and basal levels of endogenous IL-4 and IL-10 being markedly reduced.71 Around the time of clinical disease onset, the IFN-γ response begins to decline, concomitant with an increase in the levels of IL-4 and IL-10.71,79 The production of IL-10 increases to above prechallenge levels and is maintained. The Th1 response in the lymph nodes is matched by the presence of high level IL-12 production by cells in the spleen and peritoneal cavity.80 Analysis of cytokine production in the joints has revealed that IFN-γ is only present for a limited period around the time of disease onset, while IL-2, IL-4 and IL-5 are undetectable.89 In contrast, the prolonged presence of the cytokines, IL-1, IL-10, TNFα, TGFβ and IL-6 are detected in the joints.88,89
Numerous reports have documented support for the requirement of a Th1 environment in the induction and pathogenesis of CIA in DBA/1 mice. The Th1 environment is established as a result of the normal use of CFA with the immunizing CII challenge. Thus, restimulation of draining lymph node cells with CII following CII/CFA challenge is associated with high levels of IFN-γ production in the absence of IL-4.70 In contrast, immunization of mice with CII emulsified in IFA gives rise to a strong Th2 response71 and results in only a small proportion of mice developing an atypical CIA.72 Administration of the Th1-associated cytokine IFN-γ early in disease has been shown to exacerbate arthritis,73,74 while treatment with a monoclonal antibody to block IFN-γ at immunization protects mice from disease.75 CIA has also been exacerbated by administration of IL-1272 and/or IL-1876 prior to disease onset, and has been attenuated by transfer of cells engineered to secrete IL-4 or IL-13.77
The dominance of a Th1-associated response in arthritis in DBA/1 mice contrasts with data from arthritic mice of the high antibody responder Biozzi strain (H-2q). In the susceptible substrain of Biozzi mouse, Biozzi HI, induction of CIA is associated with the production of the Th2-associated cytokine IL-5.78 This differs from the CIA-resistant Biozzi HII mouse which secretes predominantly IFN-γ following CII-challenge. Furthermore, in both the DBA/1 and Biozzi models of CIA, the predominant anti-CII antibody isotype secreted has correlates with the cytokine profile associated with disease onset. In DBA/1 mice, production of the Th1-associated, IgG2a subclass is observed, whereas in Biozzi HI mice, antibody production is geared towards a Th2 phenotype, with elevated levels of IgG1 and IgE antibodies being detected. Again, the resistance of Biozzi HII mice to CIA is associated with an increase in Th1-associated IgG2a antibody production. Taken together, these data suggest that in DBA/1 mice, polarization of the immune response towards Th1 is critical for the induction of CIA. However, in other circumstances, polarization towards Th2 can trigger a similar disease process.
Temporal studies of cytokine production in DBA/1 mice immunized with CII/CFA have provided a clearer understanding of the involvement of Th1 and Th2 cells in the induction, maintenance and remission of arthritis,71,79,80 Experiments by Mauri and colleagues analysed IFN-γ, IL-4 and IL-10 production in draining lymph nodes at different stages of CIA.71 In naïve mice, moderate levels of IL-4 and IL-10 and low levels of IFN-γ were detected. Six days after immunization with CII/CFA, increased production of IFN-γ, coincident with a dramatic suppression of IL-4 and IL-10 was observed. Production of IFN-γ peaked on the day of clinical disease onset. As disease progressed, the cytokine balance gradually returned to a profile similar to that observed in naïve animals. However, levels of IL-10 remained elevated throughout disease remission. A potential role for IL-10 in the natural suppression of established CIA is supported by experiments in which administration of IL-10, or a combination of IL-4 and IL-10, was shown to rapidly suppress tissue inflammation and cartilage destruction in arthritic mice.81,82 A comparable shift in the Th1/Th2 balance during CIA was observed by Doncarli and co-workers who analysed the production of IFN-γ and IL-4 following restimulation of draining lymph node cells with CB peptide fragments of CII.79 Using similar methods, Okamoto and co-workers recently confirmed the temporal shift in the Th1/Th2 balance in CIA, and extended their findings by demonstrating an increase in the number of splenocytes and peritoneal exudate cells which produce the Th1-promoting cytokine IL-12 prior to the onset of clinical arthritis. After disease onset, the numbers of IL-12-producing cells were seen to decrease concomitant with the observed shift in the CII-specific response from Th1 to Th2.80 Collectively, these data provide strong evidence that in DBA/1 mice, inflammation and the induction of CIA are mediated by a dominant Th1 response characterized by the production of IFN-γ. As chronic disease progresses, down-regulation of the inflammatory response is associated with an increase of Th2 cell-activity and subsequent disease remission.
Although the importance of IFN-γ for the promotion of a Th1 response and the subsequent development of arthritis in DBA/1 mice has been thoroughly demonstrated, recent studies have suggested a more complex role for the production of IFN-γ in the regulation of CIA. This was first defined in experiments by Boissier and colleagues, who demonstrated that IFN-γ had a biphasic effect on arthritis.75 Intraperitoneal administration of anti-IFN-γ antibody to mice, up to 28 days after immunization, reduced the severity of CIA and was associated with a decreased anti-CII antibody response. By contrast, identical treatment given to mice later in disease had no effect or exacerbated arthritis. Further understanding for the role of IFN-γ in CIA was provided by experiments that used mice with an ablation in the IFN-γR gene (IFN-γR−/−, H-2b), back-crossed onto a DBA/1 background.83,84 These mice unexpectedly develop a rapid onset arthritis with increased incidence in comparison to wild-type DBA/1 mice after CII/CFA challenge. T-cell proliferation and Th1-associated cytokine production appeared normal in IFN-γR−/− animals in response to native or CB peptides of CII. However, the humoral response to CII was reduced, with levels of CII-specific IgG2a being most affected. These findings imply that IFN-γ is not an absolute requirement for the induction of arthritis and its production may be an important disease-limiting factor in CIA.
Other models of Th1-mediated autoimmune disease have also demonstrated a paradoxical role for endogenous IFN-γ in the regulation of the autoimmune response.85,86 Induction of both EAE and experimental autoimmune uveitis in mice ablated of IFN-γ indicated a protective, rather than disease-promoting role for IFN-γ. One feature common to these experimental models and CIA is their reliance on CFA to promote a Th1-type environment for developing autoimmune responses to co-administered antigen. Since the production of IFN-γ is an effective host-defence mechanism against mycobacterial infection, Matthys and colleagues hypothesized that the mycobacterial component of CFA may dictate the role of IFN-γ in these models of autoimmunity.87 To test this, groups of IFN-γR−/− mice (H-2q) and wild-type DBA/1 mice were immunized with CII emulsified in either CFA or IFA to induce arthritis. As expected, wild-type and mutant mice given CII/CFA developed CIA, and although the incidence was lower and the onset delayed, wild-type mice challenged with CII/IFA developed some arthritis. Significantly, no IFN-γR−/− mice immunized with CII/IFA developed any clinical or histological signs of arthritis.87 Protection from disease in these mice was associated with decreased humoral and cellular responses to CII. Together, these data indicate that in the absence of mycobacterial products, IFN-γ has a ‘natural’ disease-promoting role in CIA. However, when CFA is used as an adjuvant for CII, the mycobacterial component intensifies the disease process. In doing so, additional processes are invoked that allow endogenous IFN-γ to become protective rather than pathogenic. The rationale for endogenous IFN-γ to act to protect from disease was alluded to following histological analyses of grossly hypertrophic spleens from IFN-γR−/− mice receiving CII/CFA. In these animals, a massive expansion in the numbers of CD11b+ cells was detected, which peaked with disease onset. Although there was also an expansion of CD11b+ cells in wild-type mice that received CII/CFA the increase was much less pronounced and did not lead to splenomegaly. These results strongly suggest that the mycobacterial component of CFA induces myelopoiesis in CD11b+ macrophage-monocyte/granulocyte cell populations which can then act to promote the arthritogenic response at the joint. IFN-γ limits this response, possibly by modulating macrophage activity so that these cells are able to clear the mycobacterial components more rapidly. These findings are important for understanding mechanisms of pathogenesis in CIA, since they extend a role for CFA beyond the induction of arthritis, and demonstrate an additional level of complexity in the Th1/Th2 cytokine balance during disease.
Inflammatory cytokine production in the joints
Systemically, the Th1 environment in which CII-specific T cells first differentiate is directed predominantly by immune responses towards the mycobacterial component of the adjuvant. In contrast, the inflammatory response at the joint is mediated by the Th1-dominant anti-CII immune response, since pro-inflammatory cytokine gene activation within synovial tissue does not occur in mice that are administered CFA alone.88 Studies to characterize and modulate cytokine production within the joint during CIA have identified critical roles for the Th1-associated, pro-inflammatory cytokines, tumour necrosis factor-α (TNF-α) and IL-1 in the regulation of joint inflammation and tissue destruction.88,89 Primarily secreted by mononuclear phagocytes following activation, TNF-α contributes to inflammatory cell infiltration by up-regulating the expression of adhesion molecules on endothelium, while IL-1 promotes neutrophilia and induces the production of prostaglandins and collagenases by joint-resident chondrocytes, fibroblasts and synoviocytes.90,91 Indeed, intra-articular or systemic administration of either TNF-α or IL-1 can exacerbate CIA.90,92–94 Conversely, therapeutic benefits have been observed following systemic administration of anti-TNF-α or anti-IL-1β antibodies.95,96 Recent experiments by Joosten and colleagues have further characterized the relative contributions of TNF-α and IL-1 in the pathogenesis of CIA. Using a combination of radiography, histology and bioassays for collagen degradation and MMP activity, the researchers demonstrated that blockade of IL-1 activity by administration of antibodies to IL-1α and IL-1β at the onset of arthritis was highly effective in preventing cartilage and bone destruction. By contrast, neutralization of TNF-α led to a reduction in both joint inflammation and oedema, but failed to prevent destruction of the joint.97
Other cytokines that have been implicated in the regulation of joint inflammation are IL-6 and transforming growth factor-β (TGF-β). IL-6 is both a growth factor in the latter stage of differentiation for activated B cells and a stimulator of myelopoiesis. It is secreted by mononuclear phagocytes in response to stimulation by TNF-α and IL-1. Sasai and co-workers back-crossed IL-6−/− mice onto a DBA/1 background. Immunization of these mice with CII/CFA resulted in a delayed onset and a reduction in the severity of arthritis. This disease attenuation was associated with decreased cellular and humoral responses to CII and a shift towards Th2 cytokine production.98
Although TGF-β is often associated with immune suppression, its strong chemo-attractant properties for neutrophils and inherent ability to stimulate synovial fibroblasts make it a potent inducer of arthritis if delivered locally. Indeed, intra-articular administration of TGF-β has been shown to induce a mild synovitis in naïve rats and rapidly induces the onset of CIA following CII challenge.93 In contrast, systemic administration of TGF-β around the time of CII-challenge can confer protection from CIA. In this circumstance, disease protection is associated with a reduction in CII-specific antibody production and joint histopathology.94
Immunohistochemical studies of articular joints have provided evidence for the in situ production of TNF-α, IL-6 and TGF-β during CIA.89 These cytokines are detected throughout the synovial lining and pannus at all stages of disease. In addition, Stasiuk and co-workers detected both IL-1β and IL-10 mRNA expression within synovial tissue with maximal levels associated with peak disease severity.88
Contrary to the relative abundance of monokines produced within the arthritic joint, T-cell-associated cytokines are relatively scarce during clinical disease. Müssener and colleagues could only detect IFN-γ in arthritic joints from a panel of four T-cell-associated cytokines that included IL-2, IL-4 and IL-5. Furthermore, IFN-γ production was limited to the period associated with disease onset (between 3 and 12 days after the onset of clinical CIA) and was restricted to areas within the synovium where T cells were concentrated.89 These findings are consistent with the notion that a local Th1 response is important in initiating inflammation through recruiting cells into the joint and inducing mononuclear phagocytes to secrete TNF-α. However, they also add support to the argument that after the inflammatory response is initiated, the production of TNF-α, IL-6 and IL-1 are sustained predominantly by activated populations of CD11b+ cells in the synovium. Maintenance of CD11b+ cell stimulation within chronically inflamed joint tissue may result in part from binding of degraded extracellular matrix components from cartilage to macrophages.99,100 Additionally, coexisting responses to the mycobacterial component of the adjuvant may be sufficient to maintain a steady turnover of activated CD11b+ cells which can then traffic to the joint and promote inflammation.
Overview
Although the relationship between innate factors and the adaptive immune response in the induction and chronicity of CIA are complex and in many cases require further clarification, a model for the critical events in H-2q mice has emerged from the many recent findings reported here (Fig. 2). Thus, the interactions between the immune responses to CII and CFA in the periphery lead to the development of a Th1-mediated inflammatory response directed towards CII. The consequent production of complement-fixing antibodies against CII leads to the establishment of inflammation in the joint and this serves to recruit macrophages and neutrophils as well as T cells which further fuel the inflammatory response. In addition, the presence of mycobacterial products within the adjuvant promotes the expansion of a population of CD11b+ cells, which can then act as arthritogenic effectors, infiltrating the joint and further amplifying the CII-specific inflammatory response by the release of cytokines and other inflammatory mediators. These cytokines act on local tissue fibroblasts, chondrocytes and synoviocytes to trigger the destruction of cartilage and bone tissue. As disease progresses, the systemic immune response to CII reverts towards Th2, while increased production of IL-10 at the joint acts to down-regulate the local inflammatory response. In combination, these events lead to the eventual remission of arthritis.
Figure 2.

A model for the establishment of CIA in H-2q mice. Under the influence of components of mycobacteria in the CFA, the T-cell response to injected CII develops in the lymph nodes draining the site of challenge. This influence leads to the differentiation of naïve T cells into Th1 cells which then produce IFN-γ; in turn, this acts as an isotype switch factor favouring the production of IgG2a by activated CII-specific B cells. IgG2a antibodies enter the joint and upon binding to CII activate complement which triggers the activation of blood vessel endothelium, facilitating the early entry of activated T cells, monocytes (mφ) and neutrophils (nφ). The activation of macrophages by Th1 cell cytokine production causes the release of TNF-α, further fuelling migration from the blood into the joint space. Inside the joint, the production of IL-1 acts as the primary factor to trigger tissue destruction by infiltrating cells and resident synoviocytes, fibroblasts and chondrocytes. Consequent release of joint antigens and inflammatory mediators triggers a positive feedback circuit which continues to drive the disease process even after the numbers of T cells decline.
References
- 1.Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. J Exp Med. 1977;146:857–68. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoo TJ, Kim SY, Stuart JM, et al. Induction of arthritis in monkeys by immunization with type II collagen. J Exp Med. 1988;168:777–82. doi: 10.1084/jem.168.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cathcart ES, Hayes KC, Gonnerman WA, Lazzari AA, Franzblau C. Experimental arthritis in a nonhuman primate. I. Induction by bovine type II collagen. Lab Invest. 1986;54:26–31. [PubMed] [Google Scholar]
- 4.Courtenay JS, Dallman MJ, Dayan AD, Martin A, Mosedale B. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283:666–8. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 5.Holmdahl R, Jansson L, Gullberg D, Rubin K, Forsberg PO, Klareskog L. Incidence of arthritis and autoreactivity of anti-collagen antibodies after immunization of DBA/1 mice with heterologous and autologous collagen II. Clin Exp Immunol. 1985;62:639. [PMC free article] [PubMed] [Google Scholar]
- 6.Larsson P, Kleinau S, Holmdahl R, Klareskog L. Homologous type II collagen-induced arthritis in rats. Characterization of the disease and demonstration of clinically distinct forms of arthritis in two strains of rats after immunization with the same collagen preparation. Arthritis Rheum. 1990;33:693–701. doi: 10.1002/art.1780330512. [DOI] [PubMed] [Google Scholar]
- 7.Holmdahl R, Andersson M, Goldschmidt TJ, Gustafsson K, Jansson L, Mo JA. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 8.Stuart JM, Townes AS, Kang AH. Type II collagen-induced arthritis. Ann NY Acad Sci. 1985;460:355–62. doi: 10.1111/j.1749-6632.1985.tb51182.x. [DOI] [PubMed] [Google Scholar]
- 9.Wooley PH, Luthra HS, Stewart J, David CS. Collagen arthritis in mice – an MHC linked disease. Fed Proc. 1981;40:971. [Google Scholar]
- 10.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 11.Becker KG, Simon RM, Bailey-Wilson JE, et al. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci USA. 1998;95:9979–84. doi: 10.1073/pnas.95.17.9979. 10.1073/pnas.95.17.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIndoe RA, Bohlman B, Chi E, Schuster E, Lindhardt M, Hood L. Localization of non-Mhc collagen-induced arthritis susceptibility loci in DBA/1j mice. Proc Natl Acad Sci USA. 1999;96:2210–14. doi: 10.1073/pnas.96.5.2210. 10.1073/pnas.96.5.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths MM, Wang J, Joe B, et al. Identification of four new quantitative trait loci regulating arthritis severity and one new quantitative trait locus regulating autoantibody production in rats with collagen-induced arthritis. Arthritis Rheum. 2000;43:1278–89. doi: 10.1002/1529-0131(200006)43:6<1278::AID-ANR10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 14.Andersson EC, Hansen BE, Jacobsen H, et al. Definition of MHC and T cell receptor contacts in the HLA-DR4-restricted immunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. Proc Natl Acad Sci USA. 1998;95:7574–9. doi: 10.1073/pnas.95.13.7574. 10.1073/pnas.95.13.7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook AD, Rowley MJ, Mackay IR, Gough A, Emery P. Antibodies to type II collagen in early rheumatoid arthritis. Correlation with disease progression. Arthritis Rheum. 1996;39:1720–7. doi: 10.1002/art.1780391015. [DOI] [PubMed] [Google Scholar]
- 16.Morgan K, Clague RB, Collins I, Ayad S, Phinn SD, Holt PJL. A longitudinal study of anticollagen antibodies in patients with rheumatoid arthritis. Arthritis Rheum. 1989;32:139–45. doi: 10.1002/anr.1780320205. [DOI] [PubMed] [Google Scholar]
- 17.Ronnelid J, Lysholm J, Engstrom-Laurent A, Klareskog L, Heyman B. Local anti-type II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA-DR4-restricted IgG response. Arthritis Rheum. 1994;37:1023–9. doi: 10.1002/art.1780370707. [DOI] [PubMed] [Google Scholar]
- 18.Seki N, Sudo Y, Yoshioka T, et al. Type II collagen induced murine arthritis. I. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. J Immunol. 1988;140:1477–84. [PubMed] [Google Scholar]
- 19.Holmdahl R, Jansson L, Larsson A, Jonsson R. Arthritis in DBA/1 mice induced with passively transferred type II collagen immune serum. Immunohistopathology and serum levels of anti-type II collagen auto-antibodies. Scand J Immunol. 1990;31:147–57. doi: 10.1111/j.1365-3083.1990.tb02754.x. [DOI] [PubMed] [Google Scholar]
- 20.Wooley PH, Luthra HS, Stuart JM, David CS. Type II collagen induced arthritis in mice. I. Major histocompatibility complex (I-region) linkage and antibody correlates. J Exp Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranges GE, Sriram S, Cooper SM. Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J Exp Med. 1985;162:1105–10. doi: 10.1084/jem.162.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshino S, Cleland LG, Mayrhofer G. Treatment of collagen-induced arthritis in rats with a monoclonal antibody against the alpha/beta T cell antigen receptor. Arthritis Rheum. 1991;34:1039–47. doi: 10.1002/art.1780340814. [DOI] [PubMed] [Google Scholar]
- 23.Chiocchia G, Boissier MC, Fournier C. Therapy against murine collagen-induced arthritis with T cell receptor V beta-specific antibodies. Eur J Immunol. 1991;21:2899–905. doi: 10.1002/eji.1830211202. [DOI] [PubMed] [Google Scholar]
- 24.Banerjee S, Wei BY, Hillman K, Luthra HS, David CS. Immunosuppression of collagen-induced arthritis in mice with an anti-IL-2 receptor antibody. J Immunol. 1988;141:1150–4. [PubMed] [Google Scholar]
- 25.Wooley PH, Luthra HS, Lafuse WP, Huse A, Stuart JM, David CS. Type II collagen-induced arthritis in mice. III. Suppression of arthritis by using monoclonal and polyclonal anti-Ia antisera. J Immunol. 1985;134:2366–74. [PubMed] [Google Scholar]
- 26.Terato K, Hasty KA, Cremer MA, Stuart JM, Townes AS, Kang AH. Collagen-induced arthritis in mice. Localization of an arthritogenic determinant to a fragment of the type II collagen molecule. J Exp Med. 1985;162:637–46. doi: 10.1084/jem.162.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myers LK, Seyer JM, Stuart JM, Terato K, David CS, Kang AH. T cell epitopes of type II collagen that regulate murine collagen-induced arthritis. J Immunol. 1993;151:500–5. [PubMed] [Google Scholar]
- 28.Myers LK, Stuart JM, Seyer JM, Kang AH. Identification of an immunosuppressive epitope of type II collagen that confers protection against collagen-induced arthritis. J Exp Med. 1989;170:1999–2010. doi: 10.1084/jem.170.6.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers LK, Terato K, Seyer JM, Stuart JM, Kang AH. Characterization of a tolerogenic T cell epitope of type II collagen and its relevance to collagen-induced arthritis. J Immunol. 1992;149:1439–43. [PubMed] [Google Scholar]
- 30.Myers LK, Rosloniec EF, Cremer MA, Kang AH. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997;61:1861–78. doi: 10.1016/s0024-3205(97)00480-3. 10.1016/s0024-3205(97)00480-3. [DOI] [PubMed] [Google Scholar]
- 31.Wooley PH, Luthra HS, Griffiths MM, Stuart JM, Huse A, David CS. Type II collagen-induced arthritis in mice. IV. Variations in immunogenetic regulation provide evidence for multiple arthritogenic epitopes on the collagen molecule. J Immunol. 1985;135:2443–51. [PubMed] [Google Scholar]
- 32.Myers LK, Miyahara H, Terato K, Seyer JM, Stuart JM, Kang AH. Collagen-induced arthritis in B10.RIII mice (H-2r): identification of an arthritogenic T-cell determinant. Immunology. 1995;84:509–13. [PMC free article] [PubMed] [Google Scholar]
- 33.Nabozny GH, Griffiths MM, Harper DS, Luthra HS, David CS. Identification of a cyanogen bromide fragment of porcine type II collagen capable of modulating collagen arthritis in B10.RIII (H-2r) mice. Autoimmunity. 1995;20:39–49. doi: 10.3109/08916939508993338. [DOI] [PubMed] [Google Scholar]
- 34.Andersson M, Holmdahl R. Analysis of type II collagen-reactive T cells in the mouse. I. Different regulation of autoreactive vs. non-autoreactive anti-type II collagen T cells in the DBA/1 mouse. Eur J Immunol. 1990;20:1061–6. doi: 10.1002/eji.1830200517. [DOI] [PubMed] [Google Scholar]
- 35.Malmström V, Michaelsson E, Burkhardt H, Mattsson R, Vuorio E, Holmdahl R. Systemic versus cartilage-specific expression of a type II collagen-specific T-cell epitope determines the level of tolerance and susceptibility to arthritis. Proc Natl Acad Sci USA. 1996;93:4480–5. doi: 10.1073/pnas.93.9.4480. 10.1073/pnas.93.9.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malmström V, Kjellen P, Holmdahl R. Type II collagen in cartilage evokes peptide-specific tolerance and skews the immune response. J Autoimmun. 1998;11:213–21. doi: 10.1006/jaut.1998.0198. 10.1006/jaut.1998.0198. [DOI] [PubMed] [Google Scholar]
- 37.Kakimoto K, Katsuki M, Hirofuji T, Iwata H, Koga T. Isolation of T cell line capable of protecting mice against collagen-induced arthritis. J Immunol. 1988;140:78–83. [PubMed] [Google Scholar]
- 38.Holmdahl R, Klareskog L, Rubin K, Larsson E, Wigzell H. T lymphocytes in collagen II-induced arthritis in mice. Characterization of arthritogenic collagen II-specific T-cell lines and clones. Scand J Immunol. 1985;22:295–306. doi: 10.1111/j.1365-3083.1985.tb01884.x. [DOI] [PubMed] [Google Scholar]
- 39.Svensson L, Jirholt R, Holmdahl R, Jansson L. B-cell deficient mice do not develop type II collagen-induced arthritis (CIA) Clin Exp Immunol. 1998;111:521–6. doi: 10.1046/j.1365-2249.1998.00529.x. 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terato K, Hasty KA, Reife RA, Cremer MA, Kang AH, Stuart JM. Induction of arthritis with monoclonal antibodies to collagen. J Immunol. 1992;148:2103–8. [PubMed] [Google Scholar]
- 41.Stuart JM, Dixon FJ. Serum transfer of collagen induced arthritis in mice. J Exp Med. 1983;158:378–92. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmdahl R, Rubin K, Klareskog L, Larsson E, Wigzell H. Characterization of the antibody response in mice with type II collagen-induced arthritis, using monoclonal anti-type II collagen antibodies. Arthritis Rheum. 1986;29:400–10. doi: 10.1002/art.1780290314. [DOI] [PubMed] [Google Scholar]
- 43.Holmdahl R, Andersson ME, Goldschmidt TJ, Jansson L, Karlsson M, Malmström MJ. Collagen induced arthritis as an experimental model for rheumatoid arthritis. Immunogenetics, Pathogenesis Autoimmunity APMIS. 1989;97:575–84. doi: 10.1111/j.1699-0463.1989.tb00446.x. [DOI] [PubMed] [Google Scholar]
- 44.Watson WC, Townes AS. Genetic susceptibility to murine collagen II autoimmune arthritis: proposed relationship to the IgG2 autoantibody sub-class response, complement C5, major histocompatibility complex (MHC) and nonMHC loci. J Exp Med. 1985;162:1878–91. doi: 10.1084/jem.162.6.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brand DD, Marion TN, Myers LK, et al. Autoantibodies to murine type II collagen in collagen-induced arthritis. A comparison of susceptible and nonsusceptible strains. J Immunol. 1996;157:5178–84. [PubMed] [Google Scholar]
- 46.Marcelletti JF, Ohara J, Katz DH. Collagen-induced arthritis in mice. Relationship of collagen-specific and total IgE synthesis to disease. J Immunol. 1991;147:4185–91. [PubMed] [Google Scholar]
- 47.Stuart JM, Tomoda K, Yoo TJ, Townes AS, Kang AH. Serum transfer of collagen-induced arthritis. II. Identification and localization of autoantibody to type II collagen in donor and recipient rats. Arthritis Rheum. 1983;26:1237–44. doi: 10.1002/art.1780261011. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J Immunol. 2000;164:4340–7. doi: 10.4049/jimmunol.164.8.4340. [DOI] [PubMed] [Google Scholar]
- 49.Kleinau S, Martinsson P, Heyman B. Induction and suppression of collagen-induced arthritis is dependent on distinct Fcγ receptors. J Exp Med. 2000;191:1611–16. doi: 10.1084/jem.191.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holmdahl R, Jonsson R, Larsson P, Klareskog L. Early appearance of activated CD4+ T lymphocytes and class II antigen-expressing cells in joints of DBA/1 mice immunized with type II collagen. Lab Invest. 1988;58:53–60. [PubMed] [Google Scholar]
- 51.Holmdahl R, Rubin K, Klareskog L, Dencker L, Gustafson G, Larsson E. Appearance of different lymphoid cells in synovial tissue and in peripheral blood during the course of collagen II-induced arthritis in rats. Scand J Immunol. 1985;21:197–204. doi: 10.1111/j.1365-3083.1985.tb01421.x. [DOI] [PubMed] [Google Scholar]
- 52.Holmdahl R, Tarkowski A, Jonsson R. Involvement of macrophages and dendritic cells in synovial inflammation of collagen induced arthritis in DBA/1 mice and spontaneous arthritis in MRL/lpr mice. Autoimmunity. 1991;8:271–80. doi: 10.3109/08916939109007634. [DOI] [PubMed] [Google Scholar]
- 53.Williams RO, Mason LJ, Feldmann M, Maini RN. Synergy between anti-CD4 and anti-tumor necrosis factor in the amelioration of established collagen-induced arthritis. Proc Natl Acad Sci USA. 1994;91:2762–6. doi: 10.1073/pnas.91.7.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maeda T, Saikawa I, Hotokebuchi T, et al. Exacerbation of established collagen-induced arthritis in mice treated with an anti-T cell receptor antibody. Arthritis Rheum. 1994;37:406–13. doi: 10.1002/art.1780370315. [DOI] [PubMed] [Google Scholar]
- 55.Goldschmidt TJ, Holmdahl R. Therapeutic effects of monoclonal antibodies to alpha beta TCR but not to CD4 on collagen-induced arthritis in the rat. Cell Immunol. 1994;154:240–8. doi: 10.1006/cimm.1994.1072. 10.1006/cimm.1994.1072. [DOI] [PubMed] [Google Scholar]
- 56.Marinova-Mutafchieva L, Williams RO, Mauri C, et al. A comparative study into the mechanisms of action of anti-tumor necrosis factor α, anti-CD4, and combined anti-tumor necrosis factor α/anti-CD4 treatment in early collagen-induced arthritis. Arthritis Rheum. 2000;43:638–44. doi: 10.1002/1529-0131(200003)43:3<638::AID-ANR21>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 57.Mauri C, Chu CQ, Woodrow D, Mori L, Londei M. Treatment of a newly established transgenic model of chronic arthritis with nondepleting anti-CD4 monoclonal antibody. J Immunol. 1997;159:5032–41. [PubMed] [Google Scholar]
- 58.Webb LMC, Walmsley MJ, Feldmann M. Prevention and amelioration of collagen-induced arthritis by blockade of the CD28 co-stimulatory pathway: requirement for both B7-1 and B7-2. Eur J Immunol. 1996;26:2320–8. doi: 10.1002/eji.1830261008. [DOI] [PubMed] [Google Scholar]
- 59.Groux H, Powrie F. Regulatory T cells and inflammatory bowel disease. Immunol Today. 1999;20:442–5. doi: 10.1016/s0167-5699(99)01510-8. 10.1016/s0167-5699(99)01510-8. [DOI] [PubMed] [Google Scholar]
- 60.Groux H, O'Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–42. doi: 10.1038/39614. 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 61.Weiner HL. Oral tolerance: Immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–43. doi: 10.1016/s0167-5699(97)01053-0. 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 62.Peterman GM, Spencer C, Sperling AI, Bluestone JA. Role of gamma delta T cells in murine collagen-induced arthritis. J Immunol. 1993;151:6546–58. [PubMed] [Google Scholar]
- 63.Arai K, Yamamura S, Hanyu T, et al. Extrathymic differentiation of resident T cells in the joints of mice with collagen-induced arthritis. J Immunol. 1996;157:5170–7. [PubMed] [Google Scholar]
- 64.Larsson P, Goldschmidt TJ, Klareskog L, Holmdahl R. Oestrogen-mediated suppression of collagen-induced arthritis in rats. Studies on the role of the thymus and of peripheral CD8+ T lymphocytes. Scand J Immunol. 1989;30:741–7. doi: 10.1111/j.1365-3083.1989.tb02484.x. [DOI] [PubMed] [Google Scholar]
- 65.Tada Y, Ho A, Koh DR, Mak TW. Collagen-induced arthritis in CD4- or CD8-deficient mice: CD8+ T cells play a role in initiation and regulate recovery phase of collagen-induced arthritis. J Immunol. 1996;156:4520–6. [PubMed] [Google Scholar]
- 66.Chiocchia G, Boissier MC, Manoury B, Fournier C. T cell regulation of collagen-induced arthritis in mice. II. Immunomodulation of arthritis by cytotoxic T cell hybridomas specific for type II collagen. Eur J Immunol. 1993;23:327–32. doi: 10.1002/eji.1830230204. [DOI] [PubMed] [Google Scholar]
- 67.Plows D, Kontogeorgos G, Kollias G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J Immunol. 1999;162:1018–23. [PubMed] [Google Scholar]
- 68.Nordling C, Karlsson-Parra A, Jansson L, Holmdahl R, Klareskog L. Characterization of a spontaneously occurring arthritis in male DBA/1 mice. Arthritis Rheum. 1992;35:717–22. doi: 10.1002/art.1780350619. [DOI] [PubMed] [Google Scholar]
- 69.Corthay A, Hansson AS, Holmdahl R. T lymphocytes are not required for the spontaneous development of entheseal ossification leading to marginal ankylosis in the DBA/1 mouse. Arthritis Rheum. 2000;43:844–51. doi: 10.1002/1529-0131(200004)43:4<844::AID-ANR15>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 70.Williams NA, Stasiuk LM, Nashar TO, et al. Prevention of autoimmune disease due to lymphocyte modulation by the B-subunit of Escherichia coli heat-labile enterotoxin. Proc Natl Acad Sci USA. 1997;94:5290. doi: 10.1073/pnas.94.10.5290. 10.1073/pnas.94.10.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mauri C, Williams RO, Walmsley M, Feldmann M. Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur J Immunol. 1996;26:1511–18. doi: 10.1002/eji.1830260716. [DOI] [PubMed] [Google Scholar]
- 72.Germann T, Szeliga J, Hess H, et al. Administration of interleukin 12 in combination with type II collagen induces severe arthritis in DBA/1 mice. Proc Natl Acad Sci USA. 1995;92:4823–7. doi: 10.1073/pnas.92.11.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooper SM, Sriram S, Ranges GE. Suppression of murine collagen-induced arthritis with monoclonal anti-Ia antibodies and augmentation with IFN-gamma. J Immunol. 1988;141:1958–62. [PubMed] [Google Scholar]
- 74.Mauritz NJ, Holmdahl R, Jonsson R, van der Meide PH, Scheynius A, Klareskog L. Treatment with gamma-interferon triggers the onset of collagen arthritis in mice. Arthritis Rheum. 1988;31:1297–304. doi: 10.1002/art.1780311012. [DOI] [PubMed] [Google Scholar]
- 75.Boissier MC, Chiocchia G, Bessis N, et al. Biphasic effect of interferon-gamma in murine collagen-induced arthritis. Eur J Immunol. 1995;25:1184–90. doi: 10.1002/eji.1830250508. [DOI] [PubMed] [Google Scholar]
- 76.Leung BP, McInnes IB, Esfandiari E, Wei XQ, Liew FY. Combined effects of IL-12 and IL-18 on the induction of collagen-induced arthritis. J Immunol. 2000;164:6495–502. doi: 10.4049/jimmunol.164.12.6495. [DOI] [PubMed] [Google Scholar]
- 77.Bessis N, Boissier MC, Ferrara P, Blankenstein T, Fradelizi D, Fournier C. Attenuation of collagen-induced arthritis in mice by treatment with vector cells engineered to secrete interleukin-13. Eur J Immunol. 1996;26:2399–403. doi: 10.1002/eji.1830261020. [DOI] [PubMed] [Google Scholar]
- 78.De Franco M, Gille-Perramant MF, Mevel JC, Couderc J. T helper subset involvement in two high antibody responder lines of mice (Biozzi mice): HI (susceptible) and HII (resistant) to collagen-induced arthritis. Eur J Immunol. 1995;25:132–6. doi: 10.1002/eji.1830250123. [DOI] [PubMed] [Google Scholar]
- 79.Doncarli A, Stasiuk LM, Fournier C, Abehsira-Amar O. Conversion in vivo from an early dominant Th0/Th1 response to a Th2 phenotype during the development of collagen-induced arthritis. Eur J Immunol. 1997;27:1451–8. doi: 10.1002/eji.1830270623. [DOI] [PubMed] [Google Scholar]
- 80.Okamoto Y, Gotoh Y, Tokui H, Mizuno A, Kobayashi Y, Nishida M. Characterization of the cytokine network at a single cell level in mice with collagen-induced arthritis using a dual color ELISPOT assay. J Interferon Cytokine Res. 2000;20:55–61. doi: 10.1089/107999000312739. 10.1089/107999000312739. [DOI] [PubMed] [Google Scholar]
- 81.Walmsley M, Katsikis PD, Abney E, et al. Interleukin-10 inhibition of the progression of established collagen-induced arthritis. Arthritis Rheum. 1996;39:495–503. doi: 10.1002/art.1780390318. [DOI] [PubMed] [Google Scholar]
- 82.Joosten LAB, Lubberts E, Durez P, et al. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Arthritis Rheum. 1997;40:249–60. doi: 10.1002/art.1780400209. [DOI] [PubMed] [Google Scholar]
- 83.Manoury-Schwartz B, Chiocchia G, Bessis N, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J Immunol. 1997;158:5501–6. [PubMed] [Google Scholar]
- 84.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J Immunol. 1997;158:5507–13. [PubMed] [Google Scholar]
- 85.Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–6. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 86.Caspi RR, Chan CC, Grubbs BG, et al. Endogenous systemic IFN-gamma has a protective role against ocular autoimmunity in mice. J Immunol. 1994;152:890–9. [PubMed] [Google Scholar]
- 87.Matthys P, Vermeire K, Mitera T, et al. Enhanced autoimmune arthritis in IFN-γ receptor-deficient mice is conditioned by mycobacteria in Freund's adjuvant and by increased expansion of Mac-1+ myeloid cells. J Immunol. 1999;163:3503–10. [PubMed] [Google Scholar]
- 88.Stasiuk LM, Abehsira-Amar O, Fournier C. Collagen-induced arthritis in DBA/1 mice: Cytokine gene activation following immunization with type II collagen. Cell Immunol. 1996;173:269–75. doi: 10.1006/cimm.1996.0277. 10.1006/cimm.1996.0277. [DOI] [PubMed] [Google Scholar]
- 89.Müssener Å, Litton MJ, Lindroos E, Klareskog L. Cytokine production in synovial tissue of mice with collagen-induced arthritis (CIA) Clin Exp Immunol. 1997;107:485–93. doi: 10.1046/j.1365-2249.1997.3181214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hom JT, Bendele AM, Carlson DG. In vivo administration with IL-1 accelerates the development of collagen-induced arthritis in mice. J Immunol. 1988;141:834–41. [PubMed] [Google Scholar]
- 91.Klareskog L, McDevitt HO. Rheumatoid arthritis and its animal models: the role of TNF-α and the possible absence of specific immune responses. Curr Opin Immunol. 1999;11:657–62. doi: 10.1016/s0952-7915(99)00033-3. 10.1016/s0952-7915(99)00033-3. [DOI] [PubMed] [Google Scholar]
- 92.Killar LM, Dunn CJ. Interleukin-1 potentiates the development of collagen-induced arthritis in mice. Clin Sci. 1989;76:535–8. doi: 10.1042/cs0760535. [DOI] [PubMed] [Google Scholar]
- 93.Cooper WO, Fava RA, Gates CA, Cremer MA, Townes AS. Acceleration of onset of collagen-induced arthritis by intra-articular injection of tumour necrosis factor or transforming growth factor-beta. Clin Exp Immunol. 1992;89:244–50. doi: 10.1111/j.1365-2249.1992.tb06939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thorbecke GJ, Shah R, Leu CH, Kuruvilla AP, Hardison AM, Palladino MA. Involvement of endogenous tumor necrosis factor alpha and transforming growth factor beta during induction of collagen type II arthritis in mice. Proc Natl Acad Sci USA. 1992;89:7375–9. doi: 10.1073/pnas.89.16.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Geiger T, Towbin H, Cosenti-Vargas A, et al. Neutralization of interleukin-1 beta activity in vivo with a monoclonal antibody alleviates collagen-induced arthritis in DBA/1 mice and prevents the associated acute-phase response. Clin Exp Rheumatol. 1993;11:515–22. [PubMed] [Google Scholar]
- 97.Joosten LA, Helsen MM, Saxne T, van De L, Heinega van Den B. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol. 1999;163:5049–55. [PubMed] [Google Scholar]
- 98.Sasai M, Saeki Y, Ohshima S, et al. Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-deficient mice. Arthritis Rheum. 1999;42:1635–43. doi: 10.1002/1529-0131(199908)42:8<1635::AID-ANR11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 99.Hershkoviz R, Cahalon L, Gilat D, Miron S, Miller A, Lider O. Physically damaged extracellular matrix induces TNF-alpha secretion by interacting resting CD4+ T cells and macrophages. Scand J Immunol. 1993;37:111–15. doi: 10.1111/j.1365-3083.1993.tb01672.x. [DOI] [PubMed] [Google Scholar]
- 100.Cahalon L, Hershkoviz R, Gilat D, et al. Functional interactions of fibronectin and TNF alpha: a paradigm of physiological linkage between cytokines and extracellular matrix moieties. Cell Adhes Commun. 1994;2:269–73. [PubMed] [Google Scholar]