

Abstract
Cells damaged by environmental insults have to be repaired or eliminated to ensure tissue homeostasis in metazoans. Recent studies suggest that the balance between cell survival signals and pro-apoptotic stimuli controls the decision between cell repair and death. How these competing signals are integrated and interpreted to achieve accurate control over cell fate in vivo is incompletely understood. Here, we show that the Forkhead Box O transcription factor Foxo and the AP-1 transcription factor DFos are required downstream of Jun-N-terminal kinase signaling for the apoptotic response to UV-induced DNA damage in the developing Drosophila retina. Both transcription factors regulate the pro-apoptotic gene hid. Our results indicate that UV-induced apoptosis is repressed by receptor tyrosine kinase-mediated inactivation of Foxo. These data suggest that integrating stress and survival signals through Foxo drives the decision between cell death and repair of damaged cells in vivo.
Keywords: apoptosis, DNA damage, Foxo, JNK, stress signaling
Introduction
Metazoans are able to maintain tissue homeostasis over a long period of time while under constant exposure to environmental challenges that damage macromolecules like DNA and proteins. Tight control of the decision between repair and salvage of damaged cells or elimination of such cells by programmed cell death (apoptosis) is crucial for this ability. It is becoming increasingly recognized that the integration and interpretation of extracellular death and survival cues by complex signal transduction networks ultimately controls such life or death decisions (Janes et al, 2005). However, it remains a challenge to identify and characterize the molecular mechanisms employed by cells to decode and respond to such competing signals in vivo.
The developing Drosophila retina is an ideal system to study mechanisms that control cellular life/death decisions genetically. In the developing eye, tyrosine kinase signaling (mainly via the EGF receptor (EGFR)) is required to provide survival cues during photoreceptor differentiation (Bergmann et al, 1998b, 2002; Baker, 2001; Baker and Yu, 2001; Freeman and Bienz, 2001; Freeman, 2002; Brown and Freeman, 2003). EGFR signaling leads to activation of the Drosophila Ras homologue and protects cells against apoptosis. This effect is in part mediated by the MAPK ERK, which phosphorylates the pro-apoptotic protein Head Involution Defective (Hid) and thus induces its degradation (Bergmann et al, 1998a). A second mechanism to repress apoptosis downstream of EGFR signaling involves transcriptional repression of hid (Kurada and White, 1998).
Apoptosis in the developing retina occurs naturally to shape the adult morphology of this highly ordered tissue (Baker, 2001), but can also be induced by genotoxic stress, for example by UV-induced DNA damage (Jassim et al, 2003). When repair of UV-induced DNA damage by photoreactivation is impaired, excessive apoptosis ensues and results in extensive damage to the fly's compound eye. Overexpression of caspase inhibitors reduces the morphological disruptions observed after UV irradiation, establishing the role of apoptosis in this effect. Furthermore, transcriptional upregulation of hid in response to UV-irradiation in the pupal retina has been reported, suggesting transcriptional induction of pro-apoptotic molecules as an important part of the DNA damage response in the eye (Jassim et al, 2003).
An evolutionarily conserved regulatory system that influences cell survival and death in response to extracellular as well as intracellular cues is the stress-responsive Jun-N-terminal kinase (JNK) signaling pathway. A multitude of studies using cell culture systems and genetic model organisms have demonstrated that JNK plays an important role in apoptosis. However, JNK does not act universally as a pro-apoptotic signaling pathway, as JNK activation can also exert antiapoptotic activity in certain situations (Weston and Davis, 2002; Lamb et al, 2003; Lin, 2003; Liu and Lin, 2005; Ventura et al, 2006). The mechanism(s) by which the cellular response to JNK activation is determined is a subject of intense investigation. In a recent study, signaling interactions in cancer cells have been modeled using a systems biology approach, resulting in the identification and prediction of parameter sets that influence cellular life/death decisions as a function of JNK, EGF and insulin signaling activity (Janes et al, 2005). The models developed in these studies suggest that the strength of survival signals (EGF, insulin) determines the pro or antiapoptotic effect of JNK activation.
Work in Drosophila melanogaster indicates that the pro-apoptotic as well as the prosurvival functions of JNK are evolutionarily conserved (Adachi-Yamada et al, 1999; Igaki et al, 2002; Moreno et al, 2002; Adachi-Yamada and O'Connor, 2004; de la Cova et al, 2004; Ryoo et al, 2004; McEwen and Peifer, 2005; Perez-Garijo et al, 2005; Uhlirova et al, 2005). As compared with mammals, the Drosophila system exhibits reduced genetic redundancy, which makes it easier to derive definitive answers to questions regarding signaling specificity as well as context-dependent responses to JNK activation. The JNK signaling pathway in flies consists, at its core, of the JNK kinase hemipterous (Hep), which is phosphorylated and activated by a variety of upstream JNKK kinases, and in turn activates the JNK Basket (Bsk) by phosphorylation. Bsk is known to phosphorylate transcription factors of the AP-1 family (mainly Djun and Dfos), thus regulating transcription. Drosophila AP-1 induces the transcription of the JNK-specific phosphatase Puckered (Puc), limiting the activity of JNK in a negative feedback loop.
As in mammals, activation of the JNK pathway in flies is not always sufficient to induce apoptosis. JNK activity is, for example, required for morphogenetic processes during development, in which it regulates cellular shape changes (Harden, 2002). Moreover, JNK activation increases stress tolerance and longevity of flies (Wang et al, 2003, 2005). The molecular mechanisms that determine context- or cell type-specific cellular responses to JNK activation remain largely unknown.
Transcription factors of the Forkhead Box O (Foxo) family play an important role in mediating apoptosis in a variety of cellular contexts (e.g. in B lymphocytes and neurons; Dijkers et al, 2000, 2002; Linseman et al, 2002; Accili and Arden, 2004; Greer and Brunet, 2005). Foxo factors are phosphorylated in response to cell survival signals by active Akt or IKK, and are consequently retained in the cytoplasm. When cells are deprived of survival signals, the Akt signal is reduced and Foxo translocates to the nucleus, where it can induce the expression of pro-apoptotic molecules such as the BH3-only Bcl2 family member Bim (Dijkers et al, 2000; Greer and Brunet, 2005). Foxo can, however, also induce gene expression programs with protective functions for the cell (Kops et al, 2002; Murphy et al, 2003). The mechanism that switches the cellular response to Foxo activation from apoptosis to survival remains unclear.
Recently, a function for JNK in the regulation of Foxo activity has been identified in mammalian cells, Caenorhabditis elegans and Drosophila (Essers et al, 2004; Oh et al, 2005; Wang et al, 2005). This activation was shown to enhance oxidative stress resistance and longevity of the animal, but its role in the control of apoptosis has not been addressed. Interestingly, JNK signaling is known to induce Bim expression in neurons (Harris and Johnson, 2001; Whitfield et al, 2001), suggesting that Foxo might act downstream of JNK to promote apoptosis. Foxo is thus a prime candidate for an integrator of survival and stress signals in the control of cell homeostasis.
In this study, we use UVC-mediated disruption of retinal morphogenesis of the fly eye as an in vivo model system for DNA damage-induced apoptosis (Jassim et al, 2003). We present genetic evidence that this apoptotic response is mediated by JNK/Foxo signaling. Furthermore, we show that Foxo and the AP-1 transcription factor DFos are required for UV-induced apoptosis, and that both factors control the transcription of hid. Consistent with the notion of Foxo acting as an integration point for competing survival/death signals in Drosophila cells, we find that UV-induced apoptosis is suppressed by cellular survival signaling through the EGFR/Ras and the InR/PI3K/Akt pathways.
Results
Induction of DNA damage by UVC irradiation of the pupal retina results in loss of eye tissue in the adult fly (Jassim et al, 2003). This phenotype is observed only when DNA repair by photoreactivation is impaired, and when the retina is exposed to UV between 23 and 24 h of pupal development. At this time point, the retina consists mainly of post-mitotic, differentiating cells. Studying UV-induced cell death in this system thus allows one to genetically dissect signaling pathways involved in DNA damage-induced cell death of post-mitotic tissues.
Apoptotic response to UV-induced DNA damage in the Drosophila retina is mediated by JNK/Foxo signaling
JNK is activated by UV irradiation, and studies in cell culture suggest that JNK signaling might play an important role in the control of apoptosis after DNA damage (van Dam et al, 1995; Tournier et al, 2000; Jassim et al, 2003; Hamdi et al, 2005). To test this hypothesis genetically, we examined whether JNK signaling is required for apoptosis in the retinal DNA damage response. We exposed the pupal retina of flies carrying the hep loss-of-function alleles hep1 or hepr75 to mild UVC irradiation (5 mJ/cm2 at 254 nm) and prevented photorepair by allowing subsequent pupal development to proceed in the dark.
Comparing the phenotypic effects of this treatment on hep mutants and the corresponding wild-type controls (OreR) showed that reduction of hep function prevents excessive tissue loss (Figure 1A and B; the phenotypic effects of UV were quantified by calculating the ratio between the sizes of irradiated and non-irradiated eyes of the same head (Figure 1E). The resulting values are highly reproducible, and were identical in males and females of the same genotype. See also Materials and methods). This suggests that the JNK signaling pathway is required for apoptosis in the pupal retina in response to UV-induced DNA damage. This interpretation is supported by the observation that mutant animals heterozygous for the JNK phosphatase pucE69 (in which JNK signaling activity is increased) show increased tissue loss upon mild irradiation (Figure 1C and E). The involvement of JNK signaling in the retinal UV response was further confirmed by the induction of puc transcription (detected using RT–PCR, as well as by lacZ staining of pucE69 reporter flies) in response to UV irradiation (Figure 1D). Induction of the JNK target gene puc is a well-described indicator of JNK activation.
Figure 1.
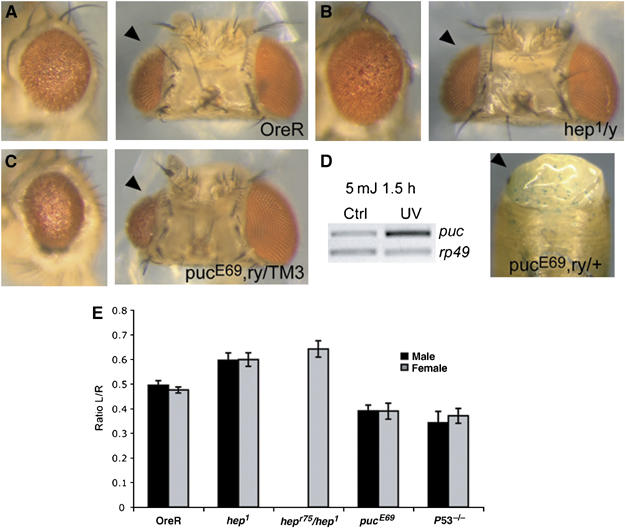
JNK signaling regulates UV-induced apoptosis in the retina. (A–C) JNK signaling regulates the apoptotic response to UV irradiation in the developing retina. Pupal cases were removed from 24-h-old pupae to expose the developing eye. One of the eyes was subjected to UVC irradiation (5 mJ/cm2), whereas the other eye was shielded. After subsequent incubation at 25°C in the dark, morphological defects are observed in the irradiated eye (A, arrowhead). This phenotype is caused by DNA-damage-induced apoptosis (Jassim et al, 2003). (B, C) DNA damage-induced apoptosis requires JNK signaling. Loss of JNKK (Hep) function (in hemizygotes for hep1) protects eyes from UV-induced apoptosis (B), whereas increased JNK activity owing to loss of the JNK-phosphatase puc results in strongly increased defects (C). puc transcription (an indicator of JNK activation in flies) is induced in response to UV irradiation (D). Left panel: RT–PCR demonstrating rapid induction of puc transcripts in the retina within 1.5 h after UV irradiation; right panel: whole-mount X-gal staining showing activation of the puc gene in the irradiated part of the pupal head (arrowhead). The pucE69 allele contains a JNK-responsive lacZ P-element that serves as JNK reporter in vivo. (E) Quantification of tissue loss in irradiated eyes can be used to quantify the extent of apoptosis in a given genotype. The ratio between the area of left (L, irradiated) and right (R, control) eyes for each head was determined for n=10 heads of each genotype (see Materials and methods for details). Means and standard deviations are shown here. Differences between each group are statistically significant (P<0.001, Student's t-test). Quantification is shown for wild-type flies (OreR), hep1 hemizygous mutant males or homozygous mutant females, hepr75/hep1 transheterozygous females, pucE69 heterozygotes and p535A−1−4 (Rong et al, 2002) homozygotes. A requirement for p53 in protection against UV-induced apoptosis was demonstrated by Jassim et al (2003). P53 mutants are included here as control.
UV irradiation induces hid expression in a JNK-dependent manner
To specify the outcome of JNK signaling activity in vivo, additional signaling inputs have to exist that control pro-apoptotic signal transduction downstream of JNK. The mechanism(s) by which JNK induces apoptosis in flies is only beginning to be understood. Although direct activation of the mitochondrial pathway by JNK cannot be ruled out, recent studies in Drosophila support the notion that JNK signaling induces the transcription of pro-apoptotic molecules of the RGH (reaper, grim, hid) family, which inactivate the Drosophila inhibitor of apoptosis (DIAP) and thus induce caspase-mediated cell death (Moreno et al, 2002; McEwen and Peifer, 2005).
We tested whether the requirement for JNK in UV-mediated apoptosis would correlate with transcriptional upregulation of pro-apoptotic molecules. Using a lacZ reporter line (Russell et al, 1998; Cox et al, 2000; Cullen and McCall, 2004; Sen et al, 2004), we found rapid induction of hid transcription in response to genotoxic UV irradiation (Figure 2A). This induction, which we validated using RT–PCR on RNA extracted from irradiated retinae (Figure 2B), was not observed in hep1 mutant animals (Figure 2B). Thus, JNK signaling is required for pro-apoptotic gene expression in response to DNA damage.
Figure 2.
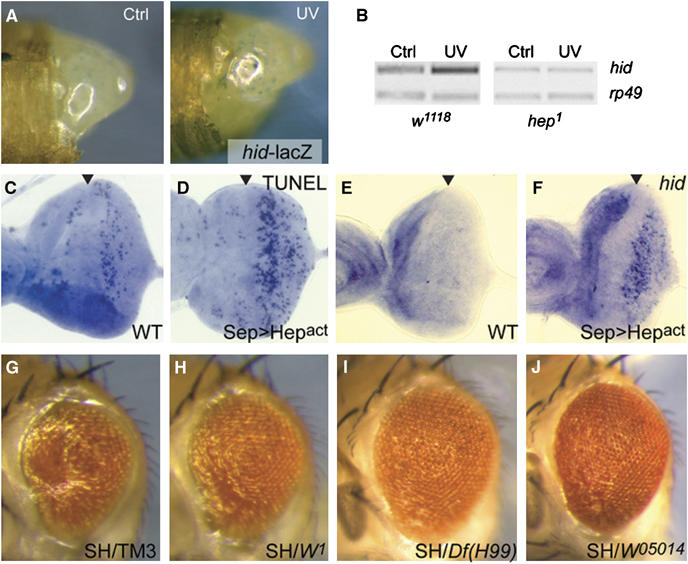
JNK-mediated hid induction induces apoptosis in the retina in response to UV irradiation. (A, B) hid is induced by UV irradiation in the Drosophila retina in a JNK-dependent manner. (A) hid expression 2 h after UV irradiation was detected by monitoring lacZ expression from a hid reporter line (Russell et al, 1998; Cox et al, 2000; Cullen and McCall, 2004; Sen et al, 2004). The irradiated eye is shown on the right (UV) and the shielded eye on the left (Ctrl). (B) Similarly, hid induction in wild-type animals (w1118, left panel) could be observed using RT–PCR on retina dissected from pupae 2.5 h after irradiation (rp49 transcript levels serve as internal controls). Induction of hid was not observed in hep1 hemizygous males (right panel), indicating a requirement for JNK signaling in the transcriptional response to UV. (C, D) Overexpression of constitutively active Hep (Hepact) in developing photoreceptors and cone cells causes apoptosis. Increased TUNEL-positive cells were observed in larval eye imaginal discs expressing Hepact under the control of Sep-Gal4 (D), compared to wild-type discs (C). Sep-Gal4 is active in the developing photoreceptors and cone cells posterior to the morphogenetic furrow (arrowhead; Jasper et al, 2002). (E, F) JNK activation is sufficient to induce hid expression in the developing retina. In situ hybridization demonstrating hid induction in response to overexpression of Hepact under the control of Sep-Gal4 (F). (G–J) The resulting adult eye phenotype (G; sep-Gal4, UAS-Hepact is abbreviated as SH) is reduced in hid mutant backgrounds (in heterozygous conditions for hid (alleles W05014 and W1) as well as all three RGH genes (Df(H99)).
We tested genetically whether hid might act downstream of JNK to induce apoptosis. Excessive JNK signaling during development of the retina, experimentally induced by the expression of constitutively active Hep (Hepact) in differentiating photoreceptors and cone cells (under the control of sepGal4), induces morphological defects that are caused by apoptosis (determined by TUNEL staining; Figure 2C and D). The distribution of apoptotic figures in these eye imaginal discs correlates with the strong upregulation of hid transcription, indicating that JNK activation is sufficient to induce hid (Figure 2E and F). Reducing the gene dose of hid (W05014 and W1), as well as of all three RGH genes (Df(H99)), was sufficient to significantly reduce the extent of damage observed in adult eyes expressing constitutively active Hep (Figure 2G and J). These results confirm a requirement for hid downstream of JNK in the induction of apoptosis.
Transcriptional control of apoptosis by Foxo and Fos downstream of JNK signaling
Direct transcriptional control of hid downstream of JNK is likely to be achieved by the activation of JNK-responsive transcription factors. The AP-1 family members Jun and Fos (in Drosophila encoded by the jra and kayak genes, respectively) are canonical downstream transcription factors that respond to JNK signaling in a variety of physiological situations (Kockel et al, 2001). In addition, recent studies from our and other laboratories have established the forkhead transcription factor Foxo as a downstream effector of JNK signaling in flies, mice and worms (Essers et al, 2004; Oh et al, 2005; Wang et al, 2005). A role for Foxo in inducing apoptosis has been described in mammalian cells, in which Foxo-mediated transcriptional induction of pro-apoptotic molecules, such as Bim, has been found (Accili and Arden, 2004; Greer and Brunet, 2005). In vivo confirmation of such a pro-apoptotic role of Foxo has been elusive.
To test whether AP-1 or Foxo acts downstream of JNK signaling in the control of apoptosis, we assessed whether JNK gain-of-function phenotypes in the eye are modulated when the gene dose of either dfoxo or the Dfos-encoding gene kayak (kay) is reduced. We found that introducing the dfoxo loss-of-function allele dfoxo21 (Junger et al, 2003; Puig et al, 2003) reduced the JNK gain-of-function phenotype (Figure 3A and B). Similar effects were observed when introducing mutations in kay (kay2; Figure 3E) or in both dfoxo and kay (Figure 3G), as well as when expressing a dsRNA molecule directed against the fos transcript (FosRNAi; Hyun et al, 2006; Figure 3F). In contrast, overexpression of Foxo dramatically enhanced the JNK gain-of-function background (Figure 3C; overexpression of Foxo alone under the control of the weak SepGal4 driver had no apparent effect, Figure 3D). These results indicate that Foxo and Fos are both required for JNK-induced apoptosis in the eye. We tested whether this requirement would also be observed in the apoptotic response to UV irradiation, and found that both heterozygotes (not shown) and homozygotes for the dfoxo loss-of-function alleles dfoxo21 (not shown) and dfoxo25, as well as transheterozygotes of dfoxo21 and dfoxo25 have significantly reduced morphological defects after UV irradiation (Figure 3H, I and N). Similarly, decreasing Fos activity resulted in reduced apoptosis (Figure 3J, K, L and N). Increasing Fos expression mildly (using a heat-shock-inducible Fos transgene from which Fos is expressed at low levels at normal temperatures; Zeitlinger and Bohmann, 1999), on the other hand, led to hypersensitivity of the developing retina to UV (Figure 3M and N).
Figure 3.
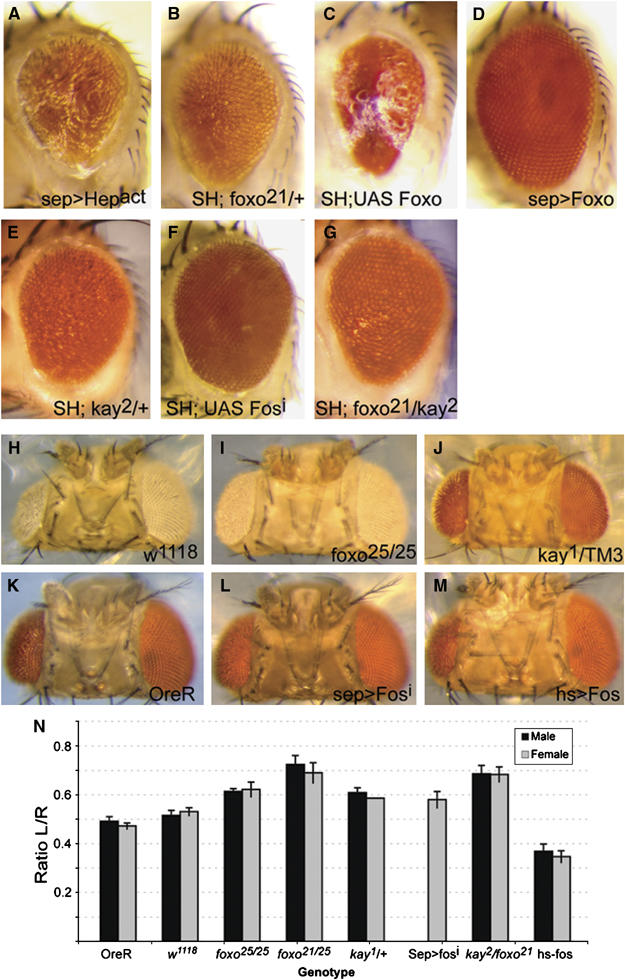
The transcription factors Fos and Foxo are required for UV-induced apoptosis in Drosophila. (A) Constitutive activation of JNKK (Hep) in the fly retina results in a ‘rough' eye phenotype (expressed in photoreceptors and cone cells under the control of sep-Gal4). This eye phenotype (referred to as SH throughout) requires the downstream kinase, JNK (not shown). (B–D) The forkhead transcription factor FOXO genetically interacts with the JNK pathway. Heterozygosity for the loss-of-function allele dfoxo21 suppresses the JNK gain-of-function phenotype (B), whereas co-overexpression of Foxo greatly enhances the eye phenotype (C). Overexpression of Foxo alone with sep-Gal4 does not impact eye morphology (D). (E–G) Similarly, decreasing Fos function by introducing the loss-of-function mutation kay2 (E), or by co-overexpressing dsRNA against Fos (F), rescues the sep>Hepact phenotype. Reducing both dfoxo and kay gene dose (G) results in an almost full recovery of normal eye morphology. (H, I, N) Homozygosity for the dfoxo loss-of-function allele dfoxo25 reduces UV-induced apoptosis. Treatment and quantification were performed as described in Figure 1. (J–N) Increased Fos expression (M, N) enhances, and loss of fos function (J, L, N) reduces UV-induced apoptosis in the eye, as compared to wild-type controls (K, N).
Transcriptional regulation of hid by Foxo and Fos
The requirement for Fos and Foxo in the UV-induced apoptotic response suggested that these transcription factors are involved in the UV-induced transcriptional induction of hid. Supporting such a role of Foxo, we observed activation of the hid gene when wild-type Foxo or Foxo™ (a constitutively nuclear form of Foxo which can not be phosphorylated by Akt; Junger et al, 2003; Puig et al, 2003; Hwangbo et al, 2004) was overexpressed in the larval retina (hid induction was observed using either hid-lacZ (Figure 4A) or by RNA in situ hybridization (Figure 4B). We further found that hid is rapidly induced in response to Foxo activation in imaginal disc cells, mimicking its response to Hepact overexpression (Figure 4C). These experiments were performed using the TARGET system (McGuire et al, 2003). The TARGET system uses a temperature-sensitive Gal80 to inhibit Gal4 at the permissive temperature. Combining this with the Gal4-UAS system in flies allows for temporal control of transgene expression. Temporal control is important for these experiments, as constitutive expression of Hepact or Foxo™ results in cell death, preventing transcriptional analysis of JNK/Foxo target genes. Importantly, our results indicate that the induction of hid by JNK signaling requires Foxo activity, as hid induction in response to Hepact expression in imaginal discs was not observed in a dfoxo21 heterozygous background (Figure 4D).
Figure 4.
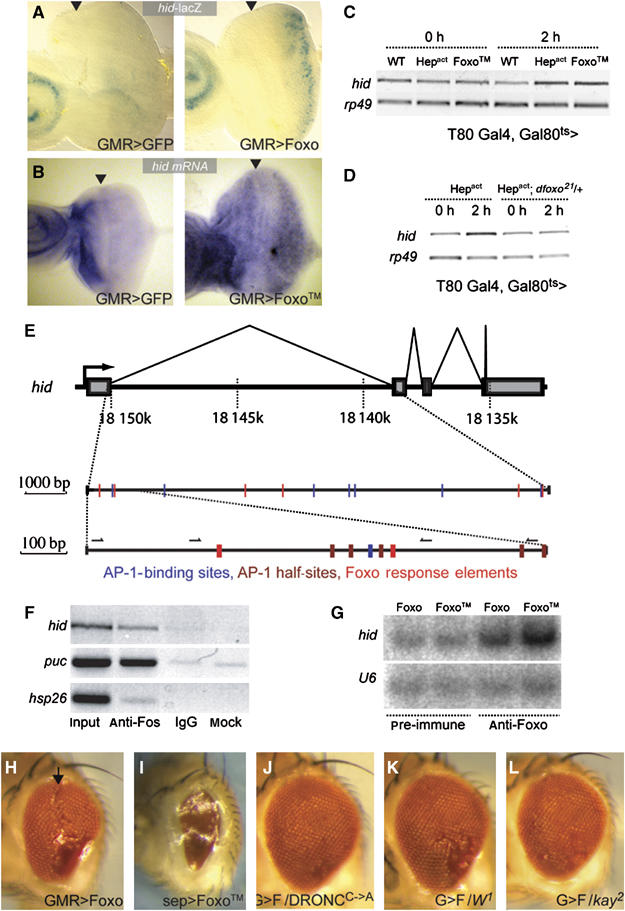
Transcriptional regulation of hid by Fos and Foxo. (A, B) Foxo overexpression is sufficient to induce hid expression in the developing retina. (A) LacZ staining of flies overexpressing GFP (as control, left panel) or Foxo (right panel) posterior to the morphogenetic furrow in a W05014 (hid-lacZ) background. (B) RNA in situ hybridization detecting hid transcript in eye imaginal discs of flies overexpressing GFP (left panel) or constitutively nuclear Foxo (Foxo™; right panel) posterior to the morphogenetic furrow (arrowheads). (C) Foxo and JNK signaling induces hid expression in wing imaginal discs. hid induction was detected using RT–PCR in wing imaginal discs in which Hepact or Foxo expression was induced using the TARGET system (McGuire et al, 2003). In this system, a temperature-sensitive allele of Gal80 inhibits Gal4-mediated transcription until flies are heat-shocked. Heat shock was performed for 30 min at 37°C. At 2 h after induction of Hepact (HA) or of a constitutively nuclear form of Foxo (Foxo™), increased hid expression can be detected. Lanes are as follows: WT, wild-type wing discs (T80Gal4/CyO; Gal80ts/TM3); HA, Hepact-expressing discs (T80Gal4/UASHepact; Gal80ts/TM3); 0 h, dissected immediately after heat shock; 2 h, dissected 2 h after heat shock. (D) hid induction in wing imaginal discs in response to JNK activation is lost in a dfoxo heterozygous mutant background. Experiment was performed as in (B), with flies of the following genotypes: w; T80-Gal4/UASHepact; Gal80ts/+ (left lanes) or w; T80-Gal4/UASHepact; Gal80ts/dfoxo21. (E) Structure of the hid locus. Coordinates for chromosome 3L are listed. The first intron of hid contains multiple AP-1-binding sites (TGANTCA, blue), as well as AP-1 half-sites (TGNNTCA, brown) and Foxo response elements (TTGTTTAC, FREs, red). Arrows indicate binding sites for primers used for ChIPs in panels F and G. (F) Chromatin IP demonstrating binding of Fos to the hid locus in S2 cells. PCR on the same ChIP material against the puc locus is included as positive control and against the hsp26 locus as negative control (Lee et al, 2005). Primers used for PCR flank the 5′ cluster of AP1 and Foxo-binding sites in the first hid intron and are indicated as arrows in panel E. (G) Chromatin IP demonstrating binding of Foxo to the hid locus in S2 cells. PCR on the same ChIP material against the U6 snRNA promoter is included as negative control. Wild-type Foxo (WT) or constitutively nuclear Foxo (TM) was transfected into S2 cells and immunoprecipitated using anti-Foxo antibody or pre-immune serum as control (see also Puig et al, 2003). Note that constitutively nuclear Foxo (TM) binds more efficiently to the hid locus than wild-type Foxo. Primers used for PCR flank the 5′ cluster of AP1 and Foxo-binding sites in the first hid intron and are indicated as arrows in (E). (H, I) Overexpression of wild-type (H) or constitutively nuclear (I) Foxo in the developing retina leads to ommatidia loss in the adult eye. (J) The phenotype caused by Foxo overexpression in the eye can be reduced by co-overexpression of a dominant-negative form of the Drosophila Caspase DRONC (Meier et al, 2000). GMR>Foxo is abbreviated here as G>F for clarity. (K) Similarly, reduction in the gene dose of hid (W1) leads to a partial rescue of the Foxo gain-of-function phenotype. (L) Foxo-induced apoptosis requires Fos function. Reduced apoptotic defects in eyes expressing wild-type Foxo in a kay2 heterozygous background.
As clusters of Foxo and AP-1 response elements are present in the first intron of the hid gene (Figure 4E), we tested whether Fos and Foxo would bind to the hid locus. Using chromatin-immunoprecipitation (ChIP) against endogenous Fos in S2 cells, we found that Fos specifically and selectively binds to sites in the first hid intron (Figure 4F). Similarly, immunoprecipitation of transfected wild-type or constitutively active Foxo demonstrated binding of Foxo to the hid intron. Binding was more efficient for constitutively nuclear Foxo™, supporting a role for Akt-mediated regulation of Foxo in the control of hid expression (Figure 4G, and see below). Control ChIPs targeting the second intron of hid yielded negative results (not shown), supporting the notion that Foxo and Fos selectively bind to the first intron of the hid locus.
Increasing Foxo expression in the fly retina (using the retinal driver GMR-Gal4) is sufficient to induce a weak phenotype that includes loss of ommatidia in the ventral areas as well as in the central midline of the eye (Figure 4H, arrow; see also Junger et al, 2003; Puig et al, 2003). When constitutively active Foxo (Foxo™) is overexpressed in the retina, complete ablation of the ommatidia is observed (Figure 4I; sep-Gal4 is used here, as overexpression of Foxo™ with GMRGal4 leads to ablation of most head structures and pupal lethality), indicating that the pro-apoptotic function of wild-type Foxo is repressed under normal conditions, most likely by survival signals (see below). The loss of ommatidia observed in GMR>Foxo eyes was reduced when a dominant-negative form of the Drosophila caspase 9 homologue DRONC (Meier et al, 2000) was co-overexpressed, further supporting a pro-apoptotic role for Foxo in the fly (Figure 4J). Similarly, heterozygosity for W1 reduces this phenotype, indicating that hid is required downstream of Foxo to induce apoptosis (Figure 4K).
The genetic and biochemical results presented here suggest that binding by both Foxo and Fos to the first hid intron is required for JNK-induced apoptosis. Supporting this notion, the Foxo gain-of-function phenotype in the eye is reduced in a kay2 mutant background (Figure 4L).
Modulation of the apoptotic response by survival signaling
Our data suggest that JNK-induced activation of Fos and Foxo and subsequent transcriptional induction of hid are required for DNA damage-induced apoptosis. At the same time, JNK-mediated activation of Foxo has beneficial effects for the organism, promoting stress tolerance and extending lifespan (Oh et al, 2005; Wang et al, 2005). These protective and pro-apoptotic roles of JNK and Foxo seem contradictory. Two possible explanations could reconcile these observations: (i) stress-induced apoptosis by JNK/Foxo signaling might be beneficial owing to the elimination of damaged cells that would otherwise contribute to deregulated overgrowth and consequent senescence of the organism. (ii) The effects of JNK/Foxo signaling might be context-dependent. Thus, activation of JNK and Foxo might elicit apoptotic responses only in conditions of severe cellular damage, whereas when cells are stressed, but able to adapt to the environment, the JNK/Foxo signaling axis might promote protective gene expression. This latter model has gained credence in recent years with the observation that JNK- as well as Foxo-induced apoptosis can be modulated by survival signals (Brunet et al, 2004; Janes et al, 2005).
Foxo is negatively regulated by a number of survival signaling pathways, most notably signaling through Akt. Cells that receive abundant survival signals are thus expected to be resistant to JNK-mediated apoptosis. We tested this by analyzing whether the activity of EGFR or insulin receptor (InR) signaling would modulate JNK/Foxo-mediated apoptosis in the retina. EGFR signaling plays a crucial role in cell division and photoreceptor determination in the developing fly retina, but is also an important source for survival stimuli in differentiating neurons (Freeman and Bienz, 2001; Brown and Freeman, 2003). Similarly, InR signaling has been proposed to provide cell survival signals in the fly (Scanga et al, 2000). EGFR signals through Ras, which in turn activates a number of downstream signaling pathways, including phosphatidyl inositol 3 kinase (PI3K)—Akt signaling. Supporting a role for EGFR in preventing excessive apoptosis after JNK and Foxo activation, we found that a reduction in the EGFR or Ras gene dose dominantly enhanced JNK and Foxo gain-of-function phenotypes in the eye (Figure 5A–C and E–G). A similar enhancement was observed when the gene dose of chico, which encodes the Drosophila IRS-1 homologue, was reduced (Figure 5D). Chico relays signals from the Drosophila insulin receptor to PI3K, ultimately activating Akt. Both EGFR and InR signaling might therefore have survival functions in the eye that are mediated by Akt-induced Foxo inactivation. Accordingly, Foxo gain-of-function phenotypes were rescued when Akt or activated PI3K were overexpressed (Figure 5H and I). The role of Akt phosphorylation in inhibiting Foxo-induced apoptosis is supported by the complete ablation of normal ommatidial structures when constitutively nuclear Foxo (Foxo™, in which the Akt phosphorylation sites are mutated) was overexpressed in the retina (Figure 4I).
Figure 5.
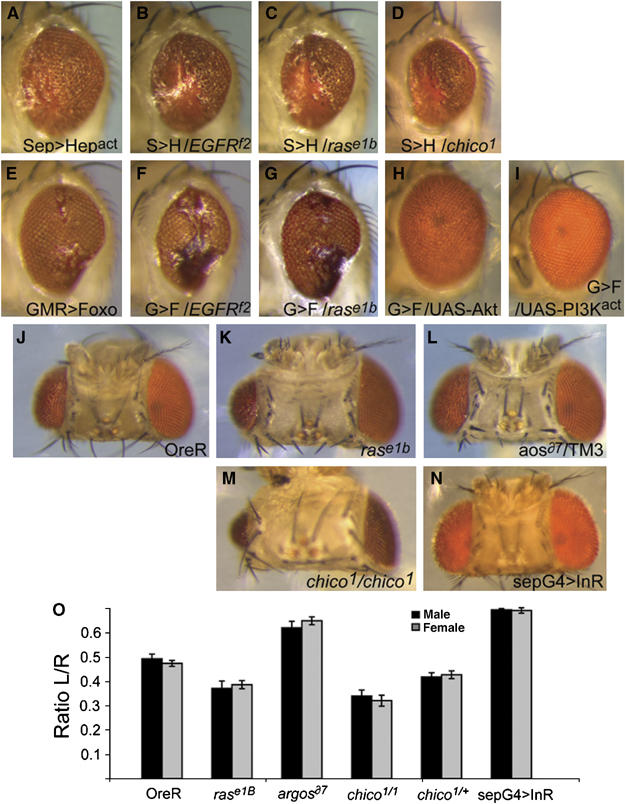
EGFR and Insulin signaling suppress JNK-mediated apoptosis. Decreased level of EGFR activity by mutations in either EGFR (B) or ras (C) and reduced insulin signaling owing to loss of chico (D) substantially enhances the apoptotic phenotype induced by overexpression of Hepact (A). Similarly, the eye phenotype induced by Foxo overexpression (E) is enhanced by reduced EGFR (F) or ras gene dose (G). Conversely, blocking Foxo activity by co-overexpressing Akt or active PI3K blocks the Foxo gain-of-function phenotype (H, I). (J–O) UV-induced apoptosis is reduced by EGF/insulin-mediated survival signaling. Reduced activity of ras (K, O) or chico (M, O) results in strongly increased UV-induced defects in the eye. Conversely, loss of aos, a negative regulator of EGFR, or increased expression of InR, decreases UV-induced apoptotic defects (L, N, O). Genotypes are as follows: A, w; sep-Gal4, UAS-Hepact/+; B, w; sep-Gal4, UAS-Hepact/egfrf2; C, w; sep-Gal4, UAS-Hepact/+; rase1B/+; D, w; sep-Gal4, UAS-Hepact/chico1; E: w; GMR-Gal4, UAS-Foxo/+; F, w; GMR-Gal4, UAS-Foxo/egfrf2; G, w; GMR-Gal4, UAS-Foxo/+; rase1B/+; H, w; GMR-Gal4, UAS-Foxo/UAS-Akt; I, w; GMR-Gal4, UAS-Foxo/UAS-PI3Kact; J, OreR. K: rase1B/TM3; L, aosδ7/TM3; M, chico1/chico1; N, w1118; sep-Gal4/UAS-InR.
To test whether survival signals initiated by receptor tyrosine kinases would influence the apoptotic response to DNA damage, we assessed the extent of UV-induced morphological defects in the retina of EGFR and InR pathway mutants. We observed a marked increase in UV-induced apoptosis in flies heterozygous for ras or homozygous mutant for chico (Figure 5J, K and M–O). Furthermore, reduction in the gene dose of the EGF antagonist argos (thus increasing EGFR activity in the eye), or increased expression of InR, was sufficient to reduce the extent of UV-induced apoptosis in the retina (Figure 5L, N and O).
Discussion
Our results indicate that transcriptional regulation of hid is a crucial component to the DNA damage-induced apoptotic response in flies. The JNK-responsive transcription factors Foxo and Fos are both required for this response, suggesting that binding of both factors to their binding sites in the hid locus is required for hid induction. Thus, negative regulation of either of these factors would lead to repression of JNK-induced apoptosis (Figure 6). Accordingly, we show that RTK-induced Akt signaling, which inhibits Foxo, desensitizes cells towards DNA damage-induced apoptosis.
Figure 6.
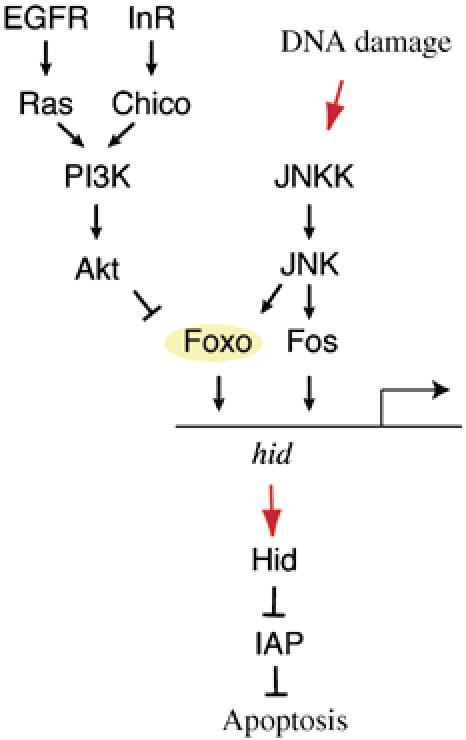
Model for the regulation of UV-induced apoptosis in the Drosophila retina. JNK activation results in Foxo- and Fos-dependent transcriptional upregulation of hid. Hid inhibits IAP and induces caspase-dependent apoptosis. Induction of hid can be blocked by InR or EGFR-initiated survival signals that inhibit Foxo activity. The relative balance between stress (JNK) and survival (RTK) signaling determines the cellular response to UV-induced DNA damage.
The requirement for Foxo in JNK-mediated apoptosis allows fine tuning of the decision between life and death of a cell, as the balance between Akt-mediated survival stimuli that inhibit Foxo activity and JNK-induced pro-apoptotic activation of Foxo would ultimately decide the cellular response to DNA damage. This interpretation is supported by our observation that UV-induced apoptosis in the pupal retina is influenced by the activities of the EGFR and insulin-signaling pathways and by the fact that overexpression of a mutant of Foxo that cannot be phosphorylated by Akt (and is therefore constitutively active) results in complete ablation of ommatidial structures. Mathematical models of signaling networks in cancer cells have predicted the importance of balancing survival signals and JNK activity for accurate control of the cellular decision between death and survival (Janes et al, 2005). Our work presented here validates these predictions in vivo and identifies the action of Foxo and Fos on the hid promoter as a crucial sensor and readout of this balance.
How is the cellular response to JNK/Foxo signaling regulated?
Our model proposes that JNK-mediated activation of Foxo leads to its nuclear translocation, where it activates pro-apoptotic gene expression. Interestingly, JNK signaling and Foxo activity have also been found to induce protective stress response molecules and damage repair proteins in flies as well as in C. elegans and mammalian cells, suggesting a role for Foxo in promoting cell repair and survival in response to stress (Kops et al, 2002; Murphy et al, 2003; Brunet et al, 2004; Wang et al, 2005). Evidently, the cellular context determines how a JNK/Foxo signal is interpreted. A variety of mechanisms governing the decision between Foxo-induced protective gene expression and Foxo-induced apoptosis can be envisioned. One candidate determinant of Foxo function is its acetylation status, which is influenced by the protein deacetylase Sir2 and which is believed to affect its promoter specificity (Brunet et al, 2004; Motta et al, 2004).
Specific responses to JNK/Foxo activity could equally be achieved by the availability of transcriptional cofactors of Foxo that are required to selectively activate the expression of certain genes. Our results suggest that Fos is a pro-apoptotic transcriptional cofactor of Foxo in the fly. Among other recently identified cofactors of Foxo in mammalian cells and C. elegans are β catenin and SMK-1, both of which appear to be required for selected functions of Foxo and are thus prime candidates for additional specificity-providing inputs (Essers et al, 2005; Wolff et al, 2006).
An alternative mechanism by which the decision between death and survival downstream of JNK could be controlled in vivo has been proposed (McEwen and Peifer, 2005). JNK-induced cell death might be governed by a timing mechanism, in which short-term activation of JNK (which is normally inhibited by a negative feedback loop involving Puc) would allow cell repair, whereas long-term activation would lead to cell death. Such a time-dependent cellular response to JNK activation has been observed in mammalian cells (Karin and Gallagher, 2005; Ventura et al, 2006). It could explain why apoptosis in the eye is observed only when photorepair is deficient (and thus JNK is active for a long period of time), but not when JNK is activated transiently by UV, but DNA is repaired. In this model, the activity of EGFR/Akt signaling might change the threshold that distinguishes between pro-apoptotic and prorepair functions of JNK/Foxo signaling.
Accurate understanding of the above-mentioned mechanisms and their importance for the regulation of cell fate in vivo is required to gain insight into how organisms fend off environmental insults and is imperative for understanding disease processes like cellular senescence during aging or cancer.
Materials and methods
Fly strains and handling
The following fly stocks were obtained from the Bloomington Stock Center: OreR, w1118, W05014, Df(H99), rase1B, chico1 and argosdelta-7. hep1/FM6 was a gift from S Noselli; pucE69 was a gift from E Martín-Blanco; UASHepact, GMRGal4 and sepGal4 were gifts from M Mlodzik; Dfoxo21/TM3 and Dfoxo25/TM3 were gifts from E Hafen; UASpro.DroncCtoA was a gift from G Evan; UASDfoxo is described in Puig et al (2003); UASDFoxo™ was a gift from Marc Tatar (Hwangbo et al, 2004). Other fly strains are as described: UASfosRNAi (Hyun et al, in preparation); hs-fos and kay2/TM3 (Zeitlinger and Bohmann, 1999). In the transient overexpression experiments, flies were raised at room temperature (21°C). Wandering third instar larva were collected and heat-shocked at 37°C for 1 h and subsequently kept at room temperature for another 2 h.
Pupal UV irradiation and phenotype quantification
Mid-aged pupae (24 h after puparium formation) were collected and subjected to surgical removal of the pupal shell surrounding the head area. UV irradiation was carried out on larvae that were immobilized on the side, so that only one retina was exposed to UV. A UV crosslinker (Stratalinker, 1800) was used with energy set at 5 mJ/cm2. After irradiation, pupae were kept in the dark until being processed.
The images of UV-damaged adult eyes were taken from the top so that both eyes were visible. The boundary of each eye was outlined using Photoshop. The eye size was determined by measuring the number of pixels contained within this area. Ratios between the area of irradiated and non-irradiated eyes were then determined.
TUNEL staining
TUNEL stainings were performed using the ApopTag kit (Chemicon International) following the instructions.
RT–PCR
Twenty wing or eye imaginal discs were collected for each RNA extraction. Total RNA was isolated using TRIzol (Invitrogen), and RT–PCR was performed as previously described (Jasper et al, 2001; Wang et al, 2003). Primers include hid (sense, 5′-TGCGAAATA CACGGGTTCA-3′ and antisense 5′-CCAATATCACCCAGTCCCG-3′) puc (sense, 5′-CGAGGATGGGTTTGATTACGA-3′, and antisense, 5′-TCAGTCCCTCGTCAAATTGCT-3′) and rp49 (sense, 5′-TCCTAC CAGCTTCAAGATGAC-3′ and antisense, 5′-CACGTTGTGCACCAG GAACT-3′)
Beta-gal staining
Samples were fixed in 1% glutaraldehyde at room temperature and washed with PBS+2 mM MgCl2. The staining solution contained 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6 and 0.1% X-gal. Stainings were performed at 30 or 37°C for 48 or 24 h, respectively.
Chromatin immunoprecipitation
Chromatin from continuously dividing S2 cells was collected according to the protocol of a commercial ChIP assay kit (Upstate Biotechnologies). Rabbit anti-D-Fos antibody (Ciapponi et al, 2001) was used for immunoprecipitation. Promoter regions with or without AP-1 sites of puc, and hsp26 were detected in the precipitated material by PCR using the following primer sets: hid (5′-ATTGTGTGGGTTAATCAGGA-3′ and 5′-TTGTAAGATTCCCAC TTTGG-3′); hsp26 (5′-TTAATAAAGAGGAAAACCAG-3′ and 5′-AA AAATAAAACTAACTAACCTT-3′); and puc (5′-GGTTTGAGCCCGA GATAA-3′ and 5′-ACTGAAGACTTTGCGGTTGAA-3′); see also Lee et al (2005).
ChIP assays for Foxo were performed as described (Puig et al, 2003), using an anti-DFoxo antibody and the following primers: hid (5′-TTTGCTGATAAGCTGACAAAGTGCTG-3′ and 5′-CAAGAAGGA TTTTCATAGGATCTCCTTG-3′) and U6 snRNA (5′-GCAGAGGGTTCTTAAGACCATTTGCC-3′ and 5′-GCTTCACGATTTTGCGTGTCATCC-3′).
Acknowledgments
We thank Benoît Biteau and Mette D Nielsen for critical reading of the manuscript, and Willis X Li and members of his lab for discussion. We are grateful to E Hafen, M Mlodzik, E Martín-Blanco, S Noselli, G Evan and the Bloomington Stock Center for sharing fly lines. This work was supported by a grant from the Upstate Coalition for Aging Research—American Federation for Aging Research to HJ.
References
- Accili D, Arden KC (2004) FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117: 421–426 [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, O'Connor MB (2004) Mechanisms for removal of developmentally abnormal cells: cell competition and morphogenetic apoptosis. J Biochem (Tokyo) 136: 13–17 [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K (1999) Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400: 166–169 [DOI] [PubMed] [Google Scholar]
- Baker NE (2001) Cell proliferation, survival, and death in the Drosophila eye. Semin Cell Dev Biol 12: 499–507 [DOI] [PubMed] [Google Scholar]
- Baker NE, Yu SY (2001) The EGF receptor defines domains of cell cycle progression and survival to regulate cell number in the developing Drosophila eye. Cell 104: 699–708 [DOI] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, Steller H (1998b) Mechanisms and control of programmed cell death in invertebrates. Oncogene 17: 3215–3223 [DOI] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, McCall K, Steller H (1998a) The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95: 331–341 [DOI] [PubMed] [Google Scholar]
- Bergmann A, Tugentman M, Shilo BZ, Steller H (2002) Regulation of cell number by MAPK-dependent control of apoptosis: a mechanism for trophic survival signaling. Dev Cell 2: 159–170 [DOI] [PubMed] [Google Scholar]
- Brown KE, Freeman M (2003) Egfr signalling defines a protective function for ommatidial orientation in the Drosophila eye. Development 130: 5401–5412 [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015 [DOI] [PubMed] [Google Scholar]
- Ciapponi L, Jackson DB, Mlodzik M, Bohmann D (2001) Drosophila Fos mediates ERK and JNK signals via distinct phosphorylation sites. Genes Dev 15: 1540–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RT, McEwen DG, Myster DL, Duronio RJ, Loureiro J, Peifer M (2000) A screen for mutations that suppress the phenotype of Drosophila armadillo, the beta-catenin homolog. Genetics 155: 1725–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen K, McCall K (2004) Role of programmed cell death in patterning the Drosophila antennal arista. Dev Biol 275: 82–92 [DOI] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA (2004) Drosophila myc regulates organ size by inducing cell competition. Cell 117: 107–116 [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ (2002) FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol 156: 531–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ (2000) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 10: 1201–1204 [DOI] [PubMed] [Google Scholar]
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC (2005) Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308: 1181–1184 [DOI] [PubMed] [Google Scholar]
- Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 23: 4802–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M (2002) A fly's eye view of EGF receptor signalling. EMBO J 21: 6635–6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M, Bienz M (2001) EGF receptor/Rolled MAP kinase signalling protects cells against activated Armadillo in the Drosophila eye. EMBO Rep 2: 157–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24: 7410–7425 [DOI] [PubMed] [Google Scholar]
- Hamdi M, Kool J, Cornelissen-Steijger P, Carlotti F, Popeijus HE, van der Burgt C, Janssen JM, Yasui A, Hoeben RC, Terleth C, Mullenders LH, van Dam H (2005) DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene 24: 7135–7144 [DOI] [PubMed] [Google Scholar]
- Harden N (2002) Signaling pathways directing the movement and fusion of epithelial sheets: lessons from dorsal closure in Drosophila. Differentiation 70: 181–203 [DOI] [PubMed] [Google Scholar]
- Harris CA, Johnson EM Jr (2001) BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem 276: 37754–37760 [DOI] [PubMed] [Google Scholar]
- Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M (2004) Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429: 562–566 [DOI] [PubMed] [Google Scholar]
- Hyun J, Becam I, Yanicostas C, Bohmann D (2006) Control of g2/m transition by Drosophila fos. Mol Cell Biol 26: 8293–8302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M (2002) Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 21: 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB (2005) A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 310: 1646–1653 [DOI] [PubMed] [Google Scholar]
- Jasper H, Benes V, Atzberger A, Sauer S, Ansorge W, Bohmann D (2002) A genomic switch at the transition from cell proliferation to terminal differentiation in the Drosophila eye. Dev Cell 3: 511–521 [DOI] [PubMed] [Google Scholar]
- Jasper H, Benes V, Schwager C, Sauer S, Clauder-Munster S, Ansorge W, Bohmann D (2001) The genomic response of the Drosophila embryo to JNK signaling. Dev Cell 1: 579–586 [DOI] [PubMed] [Google Scholar]
- Jassim OW, Fink JL, Cagan RL (2003) Dmp53 protects the Drosophila retina during a developmentally regulated DNA damage response. EMBO J 22: 5622–5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junger MA, Rintelen F, Stocker H, Wasserman JD, Vegh M, Radimerski T, Greenberg ME, Hafen E (2003) The Drosophila Forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol 2: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Gallagher E (2005) From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 57: 283–295 [DOI] [PubMed] [Google Scholar]
- Kockel L, Homsy JG, Bohmann D (2001) Drosophila AP-1: lessons from an invertebrate. Oncogene 20: 2347–2364 [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321 [DOI] [PubMed] [Google Scholar]
- Kurada P, White K (1998) Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95: 319–329 [DOI] [PubMed] [Google Scholar]
- Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ (2003) JunD mediates survival signaling by the JNK signal transduction pathway. Mol Cell 11: 1479–1489 [DOI] [PubMed] [Google Scholar]
- Lee N, Maurange C, Ringrose L, Paro R (2005) Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature 438: 234–237 [DOI] [PubMed] [Google Scholar]
- Lin A (2003) Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25: 17–24 [DOI] [PubMed] [Google Scholar]
- Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA (2002) Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci 22: 9287–9297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lin A (2005) Role of JNK activation in apoptosis: a double-edged sword. Cell Res 15: 36–42 [DOI] [PubMed] [Google Scholar]
- McEwen DG, Peifer M (2005) Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132: 3935–3946 [DOI] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL (2003) Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302: 1765–1768 [DOI] [PubMed] [Google Scholar]
- Meier P, Silke J, Leevers SJ, Evan GI (2000) The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 19: 598–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12: 1263–1268 [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563 [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283 [DOI] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA (2005) JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci USA 102: 4494–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garijo A, Martin FA, Struhl G, Morata G (2005) Dpp signaling and the induction of neoplastic tumors by caspase-inhibited apoptotic cells in Drosophila. Proc Natl Acad Sci USA 102: 17664–17669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Marr MT, Ruhf ML, Tjian R (2003) Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev 17: 2006–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YS, Titen SW, Xie HB, Golic MM, Bastiani M, Bandyopadhyay P, Olivera BM, Brodsky M, Rubin GM, Golic KG (2002) Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev 16: 1568–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MA, Ostafichuk L, Scanga S (1998) Lethal P-lacZ insertion lines expressed during pattern respecification in the imaginal discs of Drosophila. Genome 41: 7–13 [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 7: 491–501 [DOI] [PubMed] [Google Scholar]
- Scanga SE, Ruel L, Binari RC, Snow B, Stambolic V, Bouchard D, Peters M, Calvieri B, Mak TW, Woodgett JR, Manoukian AS (2000) The conserved PI3′K/PTEN/Akt signaling pathway regulates both cell size and survival in Drosophila. Oncogene 19: 3971–3977 [DOI] [PubMed] [Google Scholar]
- Sen A, Kuruvilla D, Pinto L, Sarin A, Rodrigues V (2004) Programmed cell death and context dependent activation of the EGF pathway regulate gliogenesis in the Drosophila olfactory system. Mech Dev 121: 65–78 [DOI] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870–874 [DOI] [PubMed] [Google Scholar]
- Uhlirova M, Jasper H, Bohmann D (2005) Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci USA 102: 13123–13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P (1995) ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J 14: 1798–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ (2006) Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell 21: 701–710 [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H (2003) JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell 5: 811–816 [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H (2005) JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 121: 115–125 [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ (2002) The JNK signal transduction pathway. Curr Opin Genet Dev 12: 14–21 [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29: 629–643 [DOI] [PubMed] [Google Scholar]
- Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A (2006) SMK-1, an essential regulator of DAF-16-mediated longevity. Cell 124: 1039–1053 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Bohmann D (1999) Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126: 3947–3956 [DOI] [PubMed] [Google Scholar]