

Abstract
To identify sequence domains important for the neurotoxic and neuroprotective activities of the prion protein (PrP), we have engineered transgenic mice that express a form of murine PrP deleted for a conserved block of 21 amino acids (residues 105–125) in the unstructured, N-terminal tail of the protein. These mice spontaneously developed a severe neurodegenerative illness that was lethal within 1 week of birth in the absence of endogenous PrP. This phenotype was reversed in a dose-dependent fashion by coexpression of wild-type PrP, with five-fold overexpression delaying death beyond 1 year. The phenotype of Tg(PrPΔ105–125) mice is reminiscent of, but much more severe than, those described in mice that express PrP harboring larger deletions of the N-terminus, and in mice that ectopically express Doppel, a PrP paralog, in the CNS. The dramatically increased toxicity of PrPΔ105–125 is most consistent with a model in which this protein has greatly enhanced affinity for a hypothetical receptor that serves to transduce the toxic signal. We speculate that altered binding interactions involving the 105–125 region of PrP may also play a role in generating neurotoxic signals during prion infection.
Keywords: lethal, prion, neurodegeneration, transgenic
Introduction
Prion diseases, also known as transmissible spongiform encephalopathies, are fatal neurodegenerative disorders that affect humans and animals. The infectious agent (prion) that causes these diseases is composed primarily of the protein PrPSc (Prusiner, 1998; Aguzzi and Polymenidou, 2004). PrPSc is a conformationally altered isoform of a normal, cell-surface glycoprotein called PrPC. Although a great deal of information is now available about the role of PrPSc in the disease process, relatively little is known about the normal, physiological function of PrPC. Attempts to deduce the function of PrPC from the phenotypes of prion protein (PrP)-null mice have been unrewarding, as lines of these mice in which the adjacent Doppel (Dpl) gene is not artifactually upregulated display no major anatomical or developmental deficits (Büeler et al, 1992; Manson et al, 1994).
Recent evidence raises the intriguing possibility that the normal physiological activity of PrPC is in some way required for manifestation of prion-induced neuropathology. For example, PrPC expression is essential to render neurons in the brain susceptible to the toxic effects of PrPSc emanating from grafted brain tissue (Brandner et al, 1996) or from nearby astrocytes (Mallucci et al, 2003). In addition, scrapie neuropathology is minimal in transgenic mice that express PrPC lacking a C-terminal, glycolipid anchor, implying that membrane attachment of PrPC is essential for transducing a PrPSc-derived neurotoxic signal (Chesebro et al, 2005).
The mechanism by which PrPC contributes to prion-induced neurotoxicity is unclear. One hypothesis is that PrPC normally serves a neuroprotective function that is abolished or subverted by interaction with PrPSc (Harris and True, 2006). In fact, several recent experiments have uncovered a cytoprotective activity of PrPC (Roucou and LeBlanc, 2005). PrP overexpression rescues cultured neurons, some mammalian cell lines, and yeast from several kinds of death-inducing stimuli (Kuwahara et al, 1999; Bounhar et al, 2001; Diarra-Mehrpour et al, 2004; Li and Harris, 2005; Roucou et al, 2005). Moreover, endogenous PrP has been found to protect cultured neurons against oxidative stress, and brain tissue against ischemia, hypoxia, or trauma in vivo (Brown et al, 2002; Hoshino et al, 2003; McLennan et al, 2004; Spudich et al, 2005). Nevertheless, how the putative neuroprotective activity of PrPC might be altered during prion diseases to produce a neurotoxic effect remains unknown.
A compelling demonstration of two contrasting biological activities of PrPC, one neurotoxic and the other neuroprotective, comes from analysis of transgenic mice expressing certain N-terminally truncated forms of PrP (PrPΔ32–134 and PrPΔ32–121, collectively referred to as PrPΔN). These mice suffer from a fatal neurodegenerative illness characterized by massive apoptosis of cerebellar granule neurons or Purkinje cells (depending on where the transgene is expressed) (Shmerling et al, 1998; Flechsig et al, 2003). Importantly, this phenotype is observed only in Prn-p0/0 mice that do not express endogenous PrP. Coexpression of wild-type PrP, either from the endogenous Prn-p allele or from a second transgene, completely prevents neurodegeneration in Tg(PrPΔN) mice. A similar phenomenon has been observed in mice that ectopically express Dpl, a PrP paralog that is structurally similar to PrPΔN. The Dpl gene, which is normally expressed primarily in testis, is expressed ectopically in the brain of certain lines of Prn-p0/0 mice as a result of intergenic splicing events between the adjacent PrP and Dpl genes (Sakaguchi et al, 1996; Moore et al, 1999; Li et al, 2000; Rossi et al, 2001). These lines, as well as transgenic lines expressing elevated levels of Dpl in the brain, display a neurodegenerative phenotype that is stoichiometrically rescued by wild-type PrP (Nishida et al, 1999; Moore et al, 2001; Rossi et al, 2001; Anderson et al, 2004). Taken together with the previously cited evidence for PrP cytoprotection in vitro, the experiments on transgenic mice expressing PrPΔN and Dpl suggest that PrPC possesses neuroprotective properties, but that deletion of specific regions of the molecule can unmask powerful neurotoxic activities.
Several considerations indicate that the central region of the PrP sequence, comprising residues 105–125 in the mouse protein (residues 106–126 in the human protein), constitutes a critical determinant of the neurotoxic and neuroprotective activities of PrP. First, it was reported in the original work by Shmerling et al (1998), that transgenic mice that express PrP molecules carrying N-terminal deletions up through residue 106 were normal, whereas mice expressing PrP molecules with deletions that extended to residue 121 or 134 displayed a neurodegenerative phenotype. Second, a region homologous to PrP residues 105–125 is missing in Dpl, which consists of a three-helix structure similar to that found in the C-terminal half of PrP (Mo et al, 2001; Luhrs et al, 2003). Third, it has been found that the synthetic peptide PrP106–126 is toxic when applied to cultured neurons from Prn-p+/+ but not from Prn-p0/0 mice (Forloni et al, 1993; Brown et al, 1994). Although the mechanism of this toxicity is unknown, its dependence on expression of wild-type PrP suggests some connection with the normal biological activity of PrPC.
To test the role of residues 105–125 in the biological activity of PrP, we created transgenic mice expressing PrP molecules harboring a deletion of this 21 amino-acid region. We found that these mice displayed a dramatic neurodegenerative phenotype that resulted in lethality as early as 1 week after birth. This phenotype was reversed in a dose-dependent fashion by coexpression of wild-type PrP. Our results define a critical functional domain of PrP that determines its neurotoxic and neuroprotective activities. In addition, our data suggest a model for the normal, biological function of PrPC, and how this function may be altered in prion diseases.
Results
Generation of transgenic mice and analysis of protein expression
For convenience, we will refer to PrP carrying a deletion of residues 105–125 as PrPΔCR, as the deleted region lies in the central region of the protein. The deleted segment encompasses a positively charged region along with part of the adjacent hydrophobic domain (Figure 1A). A cDNA encoding murine PrPΔCR was introduced into the moPrP.Xho vector (Borchelt et al, 1996). This vector drives transgene expression under control of a Prn-p promoter in a pattern similar to that of endogenous PrP, with the exception that there is no expression in cerebellar Purkinje cells (Fischer et al, 1996). Founder mice (designated A, B, and E) were obtained by pronuclear injection of fertilized oocytes from C57BL/6J × CBA/J parents. Initially, the founders were bred to Prn-p0/0 mice to produce offspring carrying a single copy of both the PrPΔCR transgene and the endogenous Prn-p gene.
Figure 1.
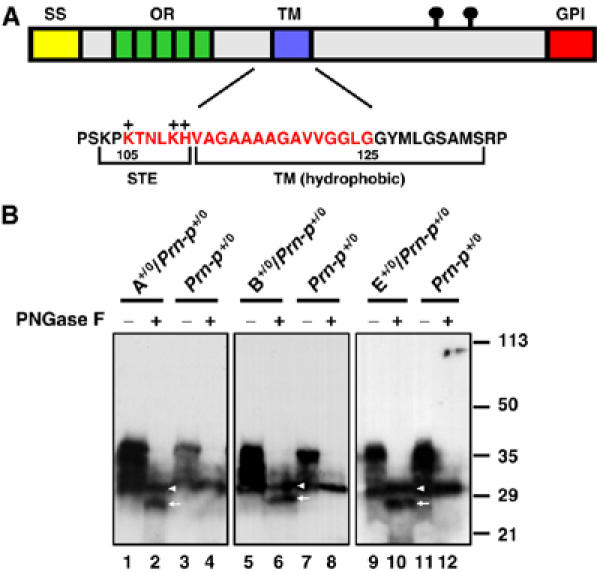
Schematic of PrP structure highlighting the CR region, and analysis of PrP expression in Tg(ΔCR) mice. (A) Structural domains of PrP are indicated by the colored blocks: SS (yellow), signal sequence; OR (green), octapeptide repeats; TM (purple), transmembrane domain; GPI (red), GPI attachment signal. The lollipop symbols indicate positions of N-linked glycosylation. The amino-acid sequence of PrP in the central region is shown below the block diagram, with the region deleted in Tg(ΔCR) mice (residues 105–125) indicated by red letters. STE, stop-transfer effector; TM, transmembrane domain. The + symbols above the sequence indicate positively charged amino acids in the STE region. (B) Brain homogenates from mice of the A, B, or E lines that were hemizygous for the PrPΔCR transgene on the Prn-p+/0 background, or from non-transgenic Prn-p+/0 mice were analyzed by Western blotting using anti-PrP antibody 8H4. Samples in lanes 2, 4, 6, 8, 10, and 12 were enzymatically deglycosylated with PNGase before blotting. The positions of wild-type PrP (arrowhead) and PrPΔCR (arrow) are indicated. Molecular size markers are given in kDa.
Brain homogenates from Tg(PrPΔCR+/0)/Prn-p+/0 mice were subjected to Western blotting using anti-PrP monoclonal antibody 8H4 following enzymatic deglycosylation with PNGase. PrPΔCR could then be distinguished from full-length, endogenous PrP owing to the small size difference (∼2 kDa) between the two polypeptide chains in the absence of N-linked oligosaccharides (Figure 1B). By quantitating the relative amounts of the two bands, we determined that the expression level of PrPΔCR in the A and E lines was similar to that of endogenous PrP in these Prn-p+/0 mice (i.e., 0.5 × with respect to Prn-p+/+ mice), whereas the expression level of PrPΔCR in the B line was ∼2-fold higher (1 ×).
Neurological symptoms of Tg(ΔCR) mice and amelioration by wild-type PrP
F1 offspring from all three founders that were hemizygous for the PrPΔCR transgene and heterozygous for the endogenous Prn-p gene became ill within 2 weeks of birth and died within 1 month (Table I; lines 1, 3, and 6). Symptoms in these neonatal animals included decreased body size and weight, immobility, difficulty righting, myoclonic spasms, and tremor.
Table 1.
Characteristics of Tg(ΔCR) mouse lines
| Genotypea | Onsetb | Deathb | PrPΔCR (fold)c | Wild-type PrP (fold)c | |
|---|---|---|---|---|---|
| 1. | ΔCR-A+/0 Prn-p+/0 | 11±3 (9) | 24±3 (7) | 0.5 | 0.5 |
| 2. | ΔCR-A+/0 Prn-p+/0 Tga20+/0 | 281±31 (8) | >360 (6) | 0.5 | 5.5 |
| 3. | ΔCR-B+/0 Prn-p+/0 | 7 (1) | 16 (1) | 1.0 | 0.5 |
| 4. | ΔCR-B+/0 Prn-p+/0 Tga20+/0 | 43 (1) | 240 (1) | 1.0 | 5.5 |
| 5. | ΔCR-E+/0 Prn-p0/0 | 4±1 (30) | 6±2 (26) | 0.5 | 0 |
| 6. | ΔCR-E+/0 Prn-p+/0 | 12±2 (40) | 25±2 (34) | 0.5 | 0.5 |
| 7. | ΔCR-E+/0 Prn-p+/+ | 17±2 (28) | 48±16 (22) | 0.5 | 1.0 |
| 8. | ΔCR-E+/0 Prn-p0/0 Tga20+/0 | 249±27 (8) | 499±76 (6) | 0.5 | 5.0 |
| 9. | ΔCR-E+/0 Prn-p+/0 Tga20+/0 | 279±36 (16) | 588±57 (10) | 0.5 | 5.5 |
| 10 | ΔCR-E+/0 Prn-p+/+ Tga20+/0 | 298±25 (9) | 491±100 (7) | 0.5 | 6.0 |
| Transgenic lines were designated A, B, and E. | |||||
| Mean age in days±s.e.m., with the number of mice given in parentheses. The > symbol indicates that mice were still alive at the time of writing. | |||||
| Expression relative to the amount of PrP in Prn-p+/+ mice, as determined by Western blotting. | |||||
By analogy to the case of Tg(PrPΔN) mice expressing N-terminally truncated PrP, we hypothesized that coexpression of wild-type PrP would ameliorate the symptoms in Tg(ΔCR) mice. To maintain the lines, we therefore bred the A, B, and E founders to Tga20 mice (Fischer et al, 1996), which overexpress wild-type PrP by five-fold when the transgene array is present in the hemizygous state. Tg(ΔCR-A+/0) and Tg(ΔCR-E+/0) mice on the Tga20+/0/PrP+/0 background did not develop symptoms until ∼280 days and survived more than 360 days (Table I; lines 2 and 9). In contrast, only one Tg(ΔCR-B+/0) mouse was obtained on the Tga20+/0/PrP+/0 background, and this mouse became ill at 43 days of age and did not produce offspring (Table I; line 4). The earlier age of disease onset in Tg(ΔCR-B) mice correlates with the higher expression level of mutant PrP in this line (Figure 1B).
In a previous study, we found that Tg(WT-E1) mice, which express wild-type PrP from the moPrP.Xho vector at a level four-fold higher than endogenous PrP, never develop clinical symptoms (Chiesa et al, 1998). Tga20 mice also do not show spontaneous illness (Fischer et al, 1996). Thus, the neurological illness seen in Tg(ΔCR) mice is specifically related in a dose-dependent fashion to the presence of the PrPΔCR protein.
To investigate quantitatively the relationship between clinical illness and wild-type PrP expression levels, we bred Tg(ΔCR)/Tga20+/0/PrP+/0 mice from the E line to either Prn-p0/0 or Prn-p+/+ mice, to obtain Tg(ΔCR-E+/0) offspring expressing different amounts of wild-type PrP encoded by either the endogenous Prn-p gene or the Tga20 transgene. We found that development of symptoms in Tg(ΔCR) mice was inversely correlated with the expression level of wild-type PrP. Tg(ΔCR-E+/0)/Tga200/0/Prn-p0/0 mice, which completely lack wild-type PrP, appeared runted and displayed righting difficulty and myoclonic spasms by 4 days after birth (Figure 2); these animals died within 1 week (Table I; line 5). Coexpression of wild-type PrP ameliorated the phenotype in a dose-dependent fashion: one Prn-p allele (0.5 × expression level) delayed death until 25 days (Table I; line 6) and two Prn-p alleles (1 × expression level) delayed death until 48 days (Table I; line 7). The presence of one Tga20 allele either with or without a Prn-p allele, (5–6 × expression level), delayed symptom onset to 250–300 days and allowed the mice to survive >1 year (Table I; lines 8–10) (Figure 2). Symptoms in older, clinically ill Tg(ΔCR-E+/0)/Tga20+/0 mice included coarse tremor, staggering gait, hind limb paresis, and difficulty righting.
Figure 2.
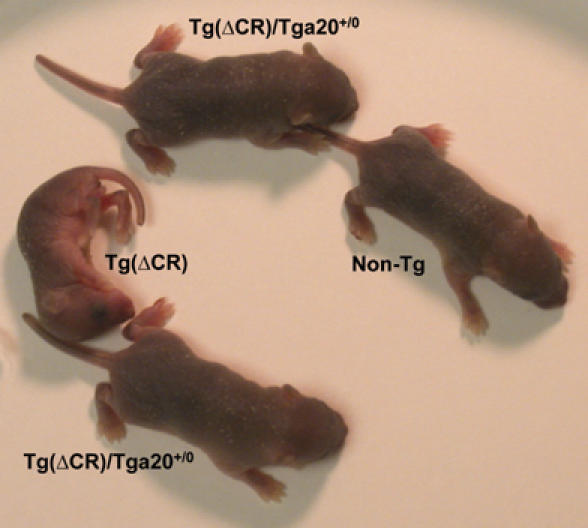
Clinical phenotype of Tg(ΔCR-E+/0) mice at 3 days of age. All mice were on the Prn-p0/0 background. The Tg(ΔCR) mouse, which completely lacks wild-type PrP, is runted and immobile. In contrast, two Tg(ΔCR)/Tga20+/0 mice, which express 5 × the endogenous level of wild-type PrP, are healthy, similar to a non-transgenic Prn-p0/0 mouse (non-Tg).
Neuropathology in Tg(ΔCR) mice
Compared to non-transgenic littermates (Figure 3C), symptomatic mice expressing PrPΔCR showed marked cerebellar atrophy, with reduction in the thickness of the granule cell and molecular layers (Figure 3A). There was a dramatic decrease in the number and density of cerebellar granule cells (Figure 3, compare D to F). In contrast, Purkinje cell number was unaffected (Figure 3, compare G to I). Immunohistochemical staining for glial fibrillary acidic protein (GFAP) demonstrated gliosis and astrocytic hypertrophy, which were most prominent in the granule cell and molecular layers of the cerebellar cortex (Figure 3, compare J to L). Consistent with the clinical observations, overexpression of wild-type PrP from the Tga20 transgene completely rescued cerebellar atrophy, granule cell loss, and astrogliosis in Tg(ΔCR) mice at 25 days of age (Figure 3B, E, and K). Based on hematoxylin and eosin staining, there were no obvious neuropathological abnormalities in areas of the brain outside of the cerebellum (not shown). In a previous study, we did not observe any histological abnormalities in Tg(WT-E1) mice overexpressing wild-type PrP by four-fold (Chiesa et al, 1998).
Figure 3.
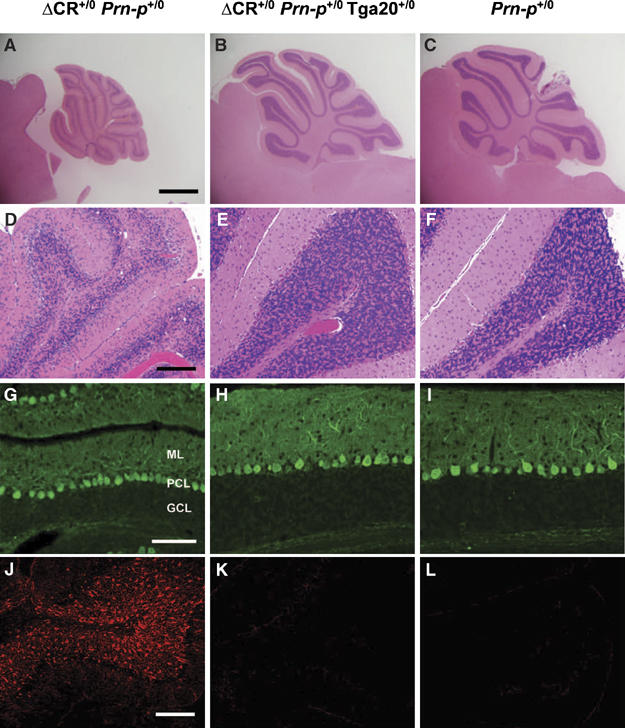
Neuropathological changes in Tg(ΔCR) mice at 25 days of age. Cerebellar sections were prepared from mice of the following genotypes: Tg(ΔCR-E+/0)/Prn-p+/0 (A, D, G, J); Tg(ΔCR-E+/0)/Prn-p+/0/Tga20+/0 (B, E, H, K); and Prn-p+/0 (C, F, I, L). Sections were stained with hematoxylin and eosin (A–F), an antibody to calbindin (G–I), or an antibody to GFAP (J–L). Abbreviations in panel G are as follows: ML, molecular layer; PCL, Purkinje cell layer; GCL, granule cell layer. Scale bars=1 mm (A–C); 50 μm (D–F); 70 μm (G–I); 25 μm (J–L).
To further explore the mechanism underlying neuronal degeneration in Tg(ΔCR) mice, brain sections of symptomatic Tg(ΔCR) mice were analyzed by TUNEL as well as by immunocytochemical staining with antibodies to activated caspase-3. Degenerating cerebellar granule cells were strongly TUNEL-positive (Figure 4A), and some cells also stained positively for activated caspase-3 (Figure 4D). Occasional TUNEL-positive cells were also observed in the hippocampus and neocortex, although loss of neurons in these regions was not obvious in hematoxylin- and eosin-stained sections (not shown). Introduction of the Tga20 transgene abrogated appearance of TUNEL- and caspase-3-positive neurons in the cerebellum (Figure 4B and E). Only very rare cells positive for these markers were observed in age-matched, non-transgenic littermates (Figure 4C and F). Taken together, these results indicate that expression of PrPΔCR causes granule neurons to degenerate via an apoptotic process that is abrogated by overexpression of wild-type PrP.
Figure 4.
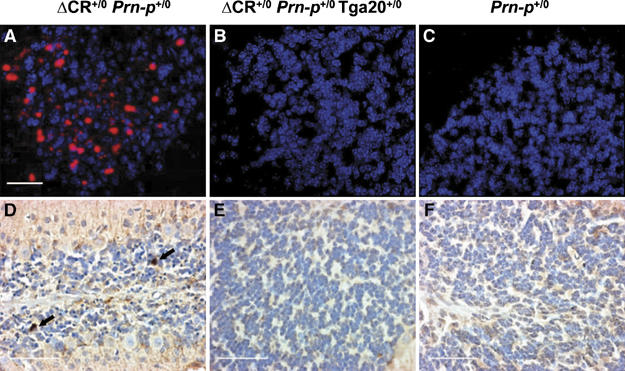
Apoptosis of cerebellar granule neurons in Tg(ΔCR) mice at 25 days of age. Cerebellar sections were prepared from mice of the following genotypes: Tg(ΔCR-E+/0)/Prn-p+/0 (A, D); Tg(ΔCR-E+/0)/Prn-p+/0/Tga20+/0 (B, E); and Prn-p+/0 (C, F). Sections were stained with TUNEL (red) and DAPI (violet) (A–C) or with an antibody to activated caspase-3 (D–F). DAPI stains cell nuclei. The arrows in panel D indicate granule cells positive for activated caspase-3. Counts of caspase-3-immunoreactive cells in five contiguous 100 × fields yielded the following results (mean±s.d.): 7.3±2.6 (Tg(ΔCR-E+/0/Prn-p+/0, three animals); 0.5±0.5 (Tg(ΔCR-E+/0)/Prn-p+/0/Tga20+/0, two animals); 0 (Prn-p+/0, one animal). Scale bars=20 μm (A–C); 30 μm (D–F).
Radovanovic et al (2005) reported that mice expressing PrPΔN and Dpl display a leukoencephalopathy characterized by vacuolar degeneration of white matter regions of the brain and spinal cord, accompanied by axonal loss and deterioration of myelin sheaths. We observed similar abnormalities in older, symptomatic Tg(ΔCR)/Tga20+/0 mice. Coarse vacuolation was seen in the cerebellar white matter (Figure 5A), as well as in white matter tracts of the spinal cord (Figure 5C). In semi-thin plastic sections of the spinal cord white matter from Tg(ΔCR)/Tga20+/0 mice, we observed extensive loss of myelinated axons, accompanied by the presence of large vacuoles and degeneration of myelin sheaths into condensed spheroid bodies (Figure 5E). Interestingly, these mice did not display significant cerebellar granule cell loss (Figure 5A). This result suggests that leukoencephalopathy and granule cell degeneration are independent processes that both contribute to clinical symptoms in Tg(ΔCR) mice, and that overexpression of wild-type PrP rescues granule cell apoptosis more effectively than white matter degeneration. No white matter pathology was observed in Tga20 mice in the absence of the PrPΔCR transgene (Figure 5B, D, and F).
Figure 5.
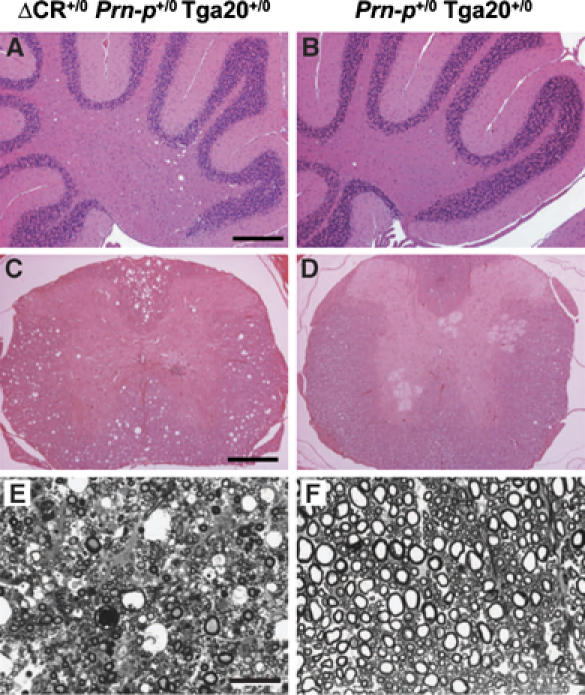
Vacuolar degeneration in the white matter of older, symptomatic Tg(ΔCR)/Tga20+/0 mice. Paraffin sections (A–D) or semi-thin plastic sections (E, F) were prepared from the cerebella (A, B) or spinal cords (C–F) of ill Tg(ΔCR-E+/0)/Prn-p+/0/Tga20+/0 mice at 397 days of age (A, C, E) and healthy Prn-p+/0/Tga20+/0 control mice (B, D, F) at 491 days of age. Paraffin sections were stained with hematoxylin and eosin, and plastic sections with toluidine blue. Scale bars=100 μm (A, B); 120 μm (C, D); 20 μm (E, F).
Biochemical and cell biological properties of PrPΔCR
We performed several experiments to determine whether abnormalities in the biochemical properties or cellular localization of PrPΔCR could contribute to the phenotype of Tg(ΔCR) mice. For these experiments, we utilized Tg(ΔCR) mice that lacked wild-type PrP to allow selective antibody recognition of the mutant protein. Similar to wild-type PrP, PrPΔCR displayed three major bands on Western blots, representing di-, mono-, and unglycosylated isoforms, with the diglycosylated form predominating (Figure 6A, lanes 1 and 3). Following treatment with PNGase F, wild-type PrP appeared as two bands of 30 and 19 kDa, representing unglycosylated versions of full-length PrP and the C1 fragment, respectively (Figure 6A, lane 4). The latter fragment is produced physiologically by cleavage at approximately residue 110 (Harris et al, 1993; Chen et al, 1995). In contrast, PNGase treatment of PrPΔCR produced primarily a single band of 27 kDa, representing an unglycosylated version of the uncleaved protein (Figure 6A, lane 2). PrPΔCR did not produce a fragment equivalent to C1, consistent with the absence of the cleavage site in the deleted protein. These results indicate that PrPΔCR is glycosylated like wild-type PrP, and is therefore processed through the secretory pathway, although it is not subject to cleavage at the C1 site.
Figure 6.
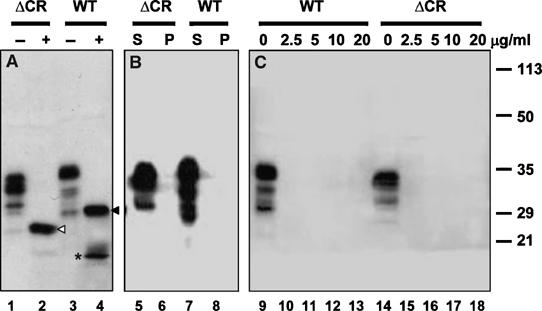
PrPΔCR from the brains of Tg mice is normally glycosylated and is not detergent-insoluble or protease-resistant. Detergent lysates were prepared from the brains of Tg(ΔCR-E+/0)/Prn-p0/0 mice (lanes 1, 2, 5, 6, 14–18; indicated by ΔCR) or Prn-p+/0 mice (lanes 3, 4, 7, 8, 9–13; indicated by WT). (A) Samples were incubated with (lanes 2, 4; indicated by +) or without (lanes 1, 3; indicated by −) PNGase to remove N-linked oligosaccharides and were then Western blotted with anti-PrP antibody 8H4. Uncleaved forms of PrPΔCR and wild-type PrP are indicated, respectively, by the white arrowhead (lane 2) and black arrowhead (lane 4). The asterisk (lane 4) indicates the C1 fragment of wild-type PrP. PrPΔCR does not produce a C1 fragment, although a slightly larger band that may represent the equivalent of the C2 fragment is faintly visible (lane 2). (B) Samples were subjected to ultracentrifugation and PrP present in supernatants (lanes 5, 7; indicated by S) and pellets (lanes 6, 8; indicated by P) was detected by Western blotting. (C) Samples were incubated with the indicated amounts of proteinase K (in μg/ml) for 30 min at 37°C. PrP was then detected by Western blotting.
We tested whether PrPΔCR in the brains of transgenic mice adopted any of the biochemical properties characteristic of PrPSc, including detergent insolubility (assayed by ultracentrifugation) and protease resistance (assayed by treatment with proteinase K). We found that, like wild-type PrP, PrPΔCR remained in the supernatant fraction after ultracentrifugation (Figure 6B), and was completely digested by concentrations of proteinase K as low as 2.5 μg/ml (Figure 6C). Under the same conditions, PrPSc from scrapie-infected brain as well as mutant PrP from the brains of Tg(PG14) mice is pelleted by ultracentrifugation and produces a protease-resistant fragment (PrP27–30) after digestion with proteinase K (Chiesa et al, 1998; data not shown).
To analyze the localization of PrPΔCR, cerebellar granule neurons cultured from postnatal day 3 mouse pups were surface-stained with anti-PrP monoclonal antibody 8H4. PrPΔCR was found to be uniformly distributed on the surface of cell bodies and neurites, similar to wild-type PrP on neurons from Prn-p+/0 mice (Figure 7A and B). The distribution of PrPΔCR was also similar to that of wild-type PrP in immunostained cryostat sections of the cerebellum from postnatal day 6 mice (Figure 7D and E). In both Tg(ΔCR) and Prn-p+/0 mice, PrP was present throughout the internal and external granule cell layers. There was no evidence for the presence of aggregates of PrPΔCR. The specificity of antibody staining was confirmed by the lack of signal on cultured granule neurons and brain sections from Prn-p0/0 mice (Figure 7C and F). The distribution of PrP in other brain regions, including the hippocampus and neocortex, was also similar in Tg(ΔCR) and Prn-p+/0 mice (data not shown).
Figure 7.
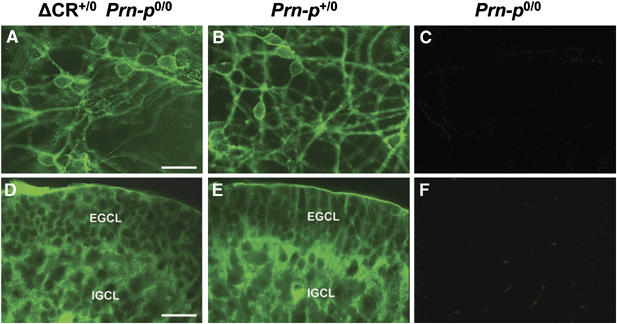
The cellular distribution of PrPΔCR is similar to that of wild-type PrP. Cerebellar granule neurons cultured from postnatal day 3 mice (A–C) and cryostat sections of the cerebella of postnatal day 6 mice (D–F) were stained with anti-PrP antibody 8H4. Neurons in panels A–C were not permeabilized with detergent before staining. Mice had the following genotypes: Tg(ΔCR-E+/0)/Prn-p0/0 (A, D); Prn-p+/0 (B, E); and Prn-p0/0 (C, F). Scale bars=10 μm (A–C); 50 μm (D–F). Abbreviations in panels D and E are as follows: EGCL, external granule cell layer; IGCL, internal granule cell layer.
Taken together, these results indicate that deletion of residues 105–125 did not induce PrP to acquire biochemical properties of PrPSc and did not substantially alter its cellular or anatomical distribution.
Discussion
We have engineered transgenic mice that express a form of PrP deleted for a conserved block of 21 amino acids in the central region of the protein (residues 105–125). These mice spontaneously develop a highly lethal neurodegenerative illness that is reversed in a dose-dependent manner by coexpression of wild-type PrP. This phenotype is reminiscent of, but much more severe than, those described in mice that express PrP harboring larger deletions of the N-terminus (Δ32–121 and Δ32–134), and in mice that ectopically express Dpl in the CNS. Our results define the 105–125 region as a crucial determinant of the neurotoxic and neuroprotective activities of PrP. These data also suggest new models for the normal, biological function of PrPC and how this function may be subverted to generate neurotoxic signals during prion infection.
A common mechanism of neurotoxicity
It was previously reported that mice expressing N-terminally deleted forms of PrP (Δ32–121 and Δ32–134, collectively referred to as PrPΔN) developed a neurodegenerative phenotype that was rescued by coexpression of endogenous, wild-type PrP (Shmerling et al, 1998; Flechsig et al, 2003). A neurodegenerative illness was also produced by ectopic expression in the CNS of Dpl, a PrP paralog that resembles PrPΔN, as it consists of a three-helix structure homologous to the C-terminal half of PrP without the flexible, N-terminal tail (Sakaguchi et al, 1996; Moore et al, 1999, 2001; Nishida et al, 1999; Rossi et al, 2001; Anderson et al, 2004). The Dpl phenotype was also abrogated by coexpression of wild-type PrP.
It seems very likely that the same molecular mechanism underlies the neurotoxicity of PrPΔCR, PrPΔN, and Dpl. All three proteins lack a portion of the flexible, N-terminal tail found in full-length PrP, and the toxicity of each is antagonized by coexpression of wild-type PrP. In addition, it was previously reported that mice expressing PrP molecules deleted from residue 32 through residue 80, 93, or 106 are normal, whereas mice expressing PrP molecules with deletions that extend to residue 121 or 134 display a neurodegenerative phenotype (Shmerling et al, 1998). Thus, it is likely that PrP residues 105–125 constitute a critical functional domain whose absence is responsible for the neurotoxicity of both PrPΔCR and PrPΔN, and that the absence of a homologous domain in Dpl underlies the pathogenicity of this protein as well.
PrPΔCR, PrPΔN, and Dpl also produce similar neuropathological effects in transgenic mice. All three of these proteins cause cerebellar atrophy and apoptosis of granule neurons (this work; Shmerling et al, 1998; Moore et al, 2001). Dpl and PrPΔN also induce degeneration of cerebellar Purkinje cells when expression is directed to these cells (Flechsig et al, 2003; Anderson et al, 2004). Each of the proteins also produces a second kind of pathology: vacuolar degeneration of white matter. In the case of PrPΔN and Dpl, granule cell death is selectively rescued by wild-type PrP expression in neurons and white matter degeneration by wild-type PrP expression in oligodendrocytes (Radovanovic et al, 2005). This result suggests that the two pathologies are likely to represent independent toxic effects of the proteins. This conclusion is consistent with our observation that vacuolation in the white matter of the spinal cord and cerebellum is observed in clinically ill Tg(ΔCR)/Tga20+/0 mice in the absence of cerebellar granule cell loss.
Mutant forms of PrP can be toxic as a result of protein misfolding and aggregation, leading to altered cellular trafficking and deposition of protease-resistant aggregates in the CNS (Harris, 2003). In contrast, PrPΔCR does not become detergent-insoluble or protease-resistant and it appears to undergo normal trafficking to the cell surface. PrPΔN and Dpl also appear to have normal biochemical and cellular properties, to the extent that these have been characterized (Shmerling et al, 1998; Massimino et al, 2004). Thus, it is likely that PrPΔCR, PrPΔN, and Dpl act via a common neurotoxic mechanism that is independent of protein aggregation, and that is more likely to be related to an effect on the normal biological activity of PrPC.
Why is PrPΔCR so toxic?
A striking feature of our results is the greatly enhanced lethality of PrPΔCR compared to PrPΔN and Dpl (at equivalent expression levels) and the requirement for much higher levels of wild-type PrP to rescue the Tg(ΔCR) phenotype. For example, Tg(PrPΔ32–134)/Prn-p0/0 mice, which express the mutant protein at ∼2 × endogenous levels (using the same promoter as in our Tg(ΔCR) mice), become ill at approximately 3–5 weeks of age and die at 2–6 months (Shmerling et al, 1998). A single Prn-p allele (0.5 × expression level) is sufficient to completely rescue the phenotype of these animals. Several lines of Prn-p0/0 mice that ectopically express Dpl in brain at levels likely to be similar to those of endogenous PrP become ill at 6–18 months of age (Sakaguchi et al, 1996; Moore et al, 1999; Rossi et al, 2001). Again, a single Prn-p allele completely abrogates the phenotype in these animals. In contrast, transgenic mice with 0.5 × expression level of PrPΔCR (four-fold less than PrPΔ32-134) become ill at a much younger age (4 days on the Prn-p0/0 background), and supraphysiological levels of wild-type PrP (5 and 6 ×) ameliorate, but are not sufficient to completely rescue, the neurodegenerative phenotype.
The marked difference between the specific toxic activities of PrPΔCR on the one hand and PrPΔN/Dpl on the other is most consistent with a model in which these proteins have different affinities for a hypothetical receptor (Tr) that serves to transduce the toxic signal (Figure 8A–C). The strong dose dependence that characterizes wild-type PrP rescue of the neurodegenerative phenotype and the fact that much higher expression levels of wild-type PrP are required to reverse the illness of Tg(ΔCR) mice suggest that wild-type PrP acts by competing with PrPΔCR/PrPΔN/Dpl for binding to this hypothetical receptor, preventing delivery of the toxic signal (or perhaps promoting delivery of a protective or trophic signal; see below). In this scheme, PrPΔCR would have a higher affinity for Tr than PrPΔN or Dpl, thus accounting for the greater specific toxicity of PrPΔCR. In addition, the affinity of wild-type PrP for Tr would be higher than that of PrPΔN/Dpl, but lower than that of PrPΔCR. Thus, endogenous levels of wild-type PrP would be sufficient to completely abrogate neurodegeneration in Tg(PrPΔN) or Tg(Dpl) mice, but supraphysiological levels would be required to significantly delay illness in Tg(ΔCR) mice.
Figure 8.
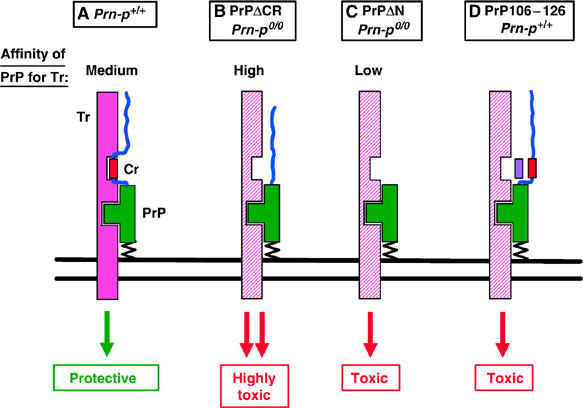
Model for the neurotoxicity of PrPΔCR, PrPΔN, and PrP106–126. The structured, C-terminal half of PrP is shown in green and the flexible, N-terminal tail as a blue line. The CR segment of PrP (residues 105–125) is shown as a red rectangle. Tr, hypothetical signal-transducing protein that normally generates a neuroprotective signal (solid pink), but which can assume an altered conformation (cross-hatched pink) that generates a neurotoxic signal. Two binding sites between PrP and Tr are shown, one involving the C-terminal half of PrP and the other CR segment of PrP. When both binding sites are occupied, Tr elicits a non-essential neuroprotective signal (A). When only the C-terminal site is occupied (as would be the case when the CR segment is absent), the transducer delivers a neurotoxic signal (B, C). The relative binding affinities of PrP for Tr are PrPΔCR>wild-type PrP>PrPΔN. Thus, wild-type PrP can reverse the neurotoxicity of both PrPΔCR and PrPΔN by competing with them for binding to Tr, but PrPΔCR requires supraphysiological levels of wild-type PrP. Dpl presumably binds to Tr with an affinity similar to that of PrPΔN. In (D), the purple rectangle represents the synthetic peptide PrP106–126, which competes for binding of the CR segment of PrP to Tr. This elicits a neurotoxic signal similar to that produced by PrPΔCR and PrPΔN, but only in the presence of PrP.
Functional and structural roles of the 105–125 region
How does deletion of residues 105–125 alter the biological activity of PrP in such a dramatic way? Five of the first six amino acids of this segment are polar, including three positively charged residues, whereas the last 15 amino acids are all hydrophobic (Figure 1A). Strikingly, the 105–125 region of PrP is the most evolutionarily conserved part of the protein, with the positively charged residues and the hydrophobic stretch present in PrP homologs from fish to humans (Rivera-Milla et al, 2006). It is therefore likely that this segment participates in an essential biological function of the protein.
Residues 105–125 lie within a region that plays an important role in determining the membrane topology of PrP. Residues 111–135 constitute a hydrophobic domain that can span the lipid bilayer in transmembrane forms of PrP (CtmPrP and NtmPrP) (Hegde et al, 1998; Stewart et al, 2001), whereas residues 103–111 function as a ‘stop transfer effector' (STE) that regulates membrane insertion of the adjacent hydrophobic segment (Yost et al, 1990) (Figure 1A). Whether the toxicity of PrPΔCR (as well as PrPΔN and Dpl) results from impaired ability of these proteins to adopt a transmembrane topology remains to be determined. The hydrophobic domain of PrP has also been implicated in sorting of the protein in polarized cells (Uelhoff et al, 2005) and in binding to certain ligands (Zanata et al, 2002), functions that might also play a role in the toxicity of PrPΔCR.
Based on NMR analysis of recombinant and brain-derived PrP, residues 23–125 form a relatively unstructured, N-terminal tail, whereas residues 128–230 constitute a folded domain comprised of three α-helices and two short β-strands flanking helix 1 (Zahn et al, 2000; Hornemann et al, 2004). Because residues 105–125 lie within the flexible tail, their deletion would not be expected to dramatically alter the C-terminal, folded domain of PrP (Zahn et al, 2000). These considerations suggest that the toxicity of PrPΔCR results from elimination of a critical binding site encompassing residues 105–125 within the flexible tail (Figure 8), rather than from significant structural alterations induced in the C-terminal half of the molecule. As both PrPΔN and PrPΔCR are missing the 105–125 region, the enhanced lethality of the latter must presumably be due to additional binding interactions between PrPΔCR and Tr involving residues 32–104.
Neuroprotective and neurotoxic effects of PrP
Paradoxically, PrP has been reported to play a role in both neurotoxic and neuroprotective phenomena (Harris and True, 2006). On the one hand, PrPC can protect cells from several kinds of pro-apoptotic stimuli (Kuwahara et al, 1999; Bounhar et al, 2001; Brown et al, 2002; Diarra-Mehrpour et al, 2004; McLennan et al, 2004; Li and Harris, 2005; Roucou et al, 2005; Spudich et al, 2005). Conversely, PrP promotes cell death in some experimental situations (Brown et al, 1994; Solforosi et al, 2004; Sunyach and Checler, 2005).
The phenotype of Tg(PrPΔCR) and Tg(PrPΔN) mice exemplifies the opposing neuroprotective and neurotoxic activities of PrP, as wild-type PrP exhibits a protective effect against the toxic effect of mutant PrP. Our results with Tg(ΔCR) mice suggest that residues 105–125 are essential for eliciting the neuroprotective activity of PrP, and that deletion of this segment converts PrP from a neuroprotective into a neurotoxic molecule. In terms of the model presented above, we hypothesize that PrPC binding to the hypothetical receptor, Tr, normally delivers a neuroprotective signal (Figure 8A). This signal would have to be non-essential, as PrP-null mice display a relatively normal phenotype (Büeler et al, 1992; Manson et al, 1994). We postulate that deletion of residues 105–125 alters or subverts the interaction between PrP and Tr in such a way that a neurotoxic rather than a neuroprotective signal is produced (Figure 8B and C). This subversion of activity might occur because delivery of a neuroprotective signal requires that PrP bind to Tr at two distinct sites: the 105–125 region and the structured, C-terminal domain. Binding to the C-terminal domain alone, in the absence of binding to the 105–125 region, might produce a neurotoxic rather than a neuroprotective signal. The change from neuroprotective to neurotoxic signaling presumably involves a conformational alteration in Tr.
The model outlined here is different from that previously proposed to explain the neurotoxicity of PrPΔN and Dpl (Shmerling et al, 1998). The latter model, which is based on a loss rather than a subversion of function, postulates the existence of two hypothetical molecules, one of which is a ligand that binds to PrP and the other a receptor that binds the ligand when PrP is absent.
A possible mechanism for the toxicity of PrP106–126 and PrPSc
It is striking that a synthetic peptide comprising human PrP residues 106–126 (equivalent to residues 105–125 in murine PrP, the region deleted in PrPΔCR) has been reported to be toxic to neurons cultured from Prn-p+/+ but not Prn-p0/0 mice (Forloni et al, 1993; Brown et al, 1994; Fioriti et al, 2005). This result suggests the hypothesis that the peptide alters interaction between PrP and the hypothetical transducer Tr by competitively blocking binding within the 105–125 region of PrP (Figure 8D). This would then produce a toxic signal equivalent to the one elicited by PrPΔCR, which lacks the 105–125 domain. In the absence of PrPC, no signal would be delivered and the peptide would have no effect.
A similar mechanism could be invoked to explain the toxic effect of PrPSc, which also appears to require expression of PrPC (Brandner et al, 1996; Mallucci et al, 2003). In this case, PrPSc might perturb interaction between PrPC and Tr by blocking binding within the 105–125 domain, thereby producing a toxic signal equivalent to the one elicited PrPΔCR and by PrP106–126. Consistent with this model, PrP106–126 displays certain biochemical properties typical of PrPSc (aggregation, protease resistance), and its mechanism of toxicity has been proposed to be similar to that of PrPSc (Selvaggini et al, 1993). In addition, PrPSc appears to be conformationally altered in the 105–125 region (Peretz et al, 1997).
Future studies
The work presented here identifies residues 105–125 as a critical functional domain of PrP whose deletion endows the protein with powerful neurotoxic properties. To further elucidate the molecular basis of this effect, it will be necessary to identify other proteins, such as the hypothetical receptor Tr, that play a role in transducing the neurotoxic and neuroprotective signals that emanate from PrP. As PrPΔCR appears to engage the signal transducing machinery with very high affinity, it may facilitate discovery of PrP-interacting proteins using biochemical methods. The enhanced toxic potency of PrPΔCR may also allow the development of improved cell culture models to analyze the signaling pathways activated by PrP. Thus far, there has been only limited success in reproducing the toxic effects of PrPΔN and Dpl in cultured cells (Drisaldi et al, 2004). Finally, it will be of great interest to further explore the relationship between the neurotoxic pathways activated by PrPSc and PrPΔCR, and to determine whether common mechanisms are involved.
Materials and methods
Production of transgenic mice
A cDNA encoding murine PrPΔCR (PrPΔ105–125) was generated by preparing the 5′ and 3′ halves of the cDNA separately, and then introducing these into the cloning vector in a three-part ligation reaction. The 5′ half of the cDNA was amplified by PCR using as a template pcDNA3 containing wild-type mouse PrP with a 3F4 epitope tag (Lehmann and Harris, 1995). The upstream primer was Tg51 (5′-GTACAGGACCAAGCTTAGTCTCGAGCCATGG CGAACCTTG GCTACTGGCTGCTG-3′) and the downstream primer was Tg31 (5′-GCTCATGGCGCTCCCCAGCATGTAGCCTGGT TTGCTGGGCTTGTTC CACTGATT-3′). Primer Tg51 contained a HindIII restriction site and primer Tg31 contained the 104–126 junction sequence and an HaeII restriction site. The resulting PCR product was digested with HindIII and HaeII. A fragment encoding the 3′ half of PrPΔCR was generated by digesting wild-type mouse PrP/pcDNA3 with HaeII and BamHI. The 5′ and 3′ halves of the PrPΔCR cDNA were then ligated into pcDNA3.1(+) (Invitrogen, Carlsbad, CA). A fragment encoding the complete PrPΔCR sequence was then released from the resulting plasmid by digestion with HindIII/BamHI, and blunted by treatment with Klenow polymerase. The blunted fragment was then ligated into transgenic expression vector MoPrP.Xho (Borchelt et al, 1996) that had been cleaved with XhoI and then blunted. Recombinant plasmid with the insert in the correct orientation was selected by EcoRI digestion and sequencing. The transgene was excised from the recombinant plasmid by digestion with NotI, purified on GFX PCR DNA purification columns (Amersham Biosciences), and injected into the pronuclei of fertilized eggs from an F2 cross of C57BL/6J × CBA/J F1 parental mice.
Transgenic founders were bred to the following mouse strains: C57BL/6J × CBA/J parental mice; Prn-p0/0 mice obtained from Charles Weissmann that had been produced on a 129 × C57BL/6J background (Büeler et al, 1992); or Tga20 mice (Fischer et al, 1996) to maintain the lines.
Mice were genotyped by PCR analysis of tail DNA prepared using the Puregene DNA Isolation Kit (Gentra Systems, Minneapolis, MN). The primer pairs used were as follows: P1 and P4 (Chiesa et al, 1998) which amplify both the PrPΔCR and Tga20 transgenes; ΔCR (5′-CCTCGAAGCTTAGTCTCGAGCC-3′) and E4 (5′-TCATGGC GCTCCCCAGCATGTA-3′), which amplify only the PrPΔCR transgene; and P2 and P4 (Chiesa et al, 1998) which amplify the Prn-p+ and Prn-p0 alleles.
Histology
Animals were perfusion-fixed and paraffin sections of brain and spinal cord were stained with hematoxylin and eosin or with anti-GFAP antibodies as described previously (Chiesa et al, 1998), except that GFAP antibodies were visualized using AlexaFluor 594-coupled goat anti-rabbit IgG (Invitrogen). Purkinje cells were visualized by staining sections with a rabbit antibody to calbindin (Chemicon, Temecula, CA), followed by visualization with AlexaFluor 488-coupled goat anti-rabbit IgG (Invitrogen).
For TUNEL, paraffin sections prepared as above were treated in permeabilization solution (0.1 M citrate buffer, pH 6.0, 0.05% Tween 20) and labeled with In Situ Cell Death Detection Kit according to the manufacturer's protocol (Roche Diagnostics, Indianapolis, IN). Caspase-3 activation was monitored using an anti-activated caspase-3 antibody (Cell Signaling Technology, Beverly, MA) and visualized using the peroxidase–anti-peroxidase method as described previously (Young et al, 2005). Sections were stained with either DAPI or hematoxylin to visualize cell nuclei.
For PrP immunohistochemistry, brains were immersion-fixed and then 14 μm sagittal sections were cut with a cryostat. Sections were pretreated in PBS containing 0.2% Triton X-100 for 30 min at room temperature. Staining was performed using anti-PrP monoclonal antibody 8H4 (Zanusso et al, 1998), followed by visualization using AlexaFluor 488-coupled goat anti-mouse IgG.
For preparation of semi-thin plastic sections, mice were perfusion-fixed with ice-cold 4% paraformaldehyde/3% glutaraldehyde and spinal cords were embedded in Epon. One micron sections were cut and stained with toluidine blue for viewing by light microscopy.
Biochemical assays
Detergent insolubility and protease resistance of PrP in postnuclear supernatants of brain were assayed as described previously (Chiesa et al, 1998). To deglycosylate PrP, postnuclear supernatants were treated with PNGase F according to the manufacturer's instructions (New England Biolabs, Beverly, MA). Samples were analyzed by Western blotting using anti-PrP antibody 8H4 with the ECL detection system (Amersham Biosciences, Piscataway, NJ).
Cerebellar granule cell cultures
Cultures were prepared from 3-day-old mouse pups as described previously (Miller and Johnson, 1996), and plated at a density of 500 000 cells/cm2 in polylysine-coated eight-well chamber slides. After 4–5 days in culture, cells were stained with anti-PrP antibody 8H4 followed by fixation in 4% paraformaldehyde in PBS and incubation with AlexaFluor 488-coupled goat anti-mouse IgG.
Acknowledgments
We thank Charles Weissmann for Prn-p0/0 and Tga20 mice and Man-Sun Sy for 8H4 antibody. We are grateful to Cheryl Adles and Su Deng for mouse colony maintenance and genotyping and to Robert Schmidt and Karen Green for assistance with semi-thin plastic sections. We thank Mike Green, Heather True, and members of the Harris laboratory for critically reading the manuscript. This work was supported by grants from the NIH to DAH (NS040975) and KAR (NS35107). RC is an Assistant Telethon Scientist (DTI, Fondazione Telethon; S00083). HMC was supported by a pre-doctoral fellowship (NS04691003) from the NIH.
References
- Aguzzi A, Polymenidou M (2004) Mammalian prion biology: one century of evolving concepts. Cell 116: 313–327 [DOI] [PubMed] [Google Scholar]
- Anderson L, Rossi D, Linehan J, Brandner S, Weissmann C (2004) Transgene-driven expression of the Doppel protein in Purkinje cells causes Purkinje cell degeneration and motor impairment. Proc Natl Acad Sci USA 101: 3644–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal Biomol Eng 13: 159–163 [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A (2001) Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 276: 39145–39149 [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A (1996) Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379: 339–343 [DOI] [PubMed] [Google Scholar]
- Brown DR, Herms J, Kretzschmar HA (1994) Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. NeuroReport 5: 2057–2060 [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L (2002) Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 67: 211–224 [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C (1992) Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577–582 [DOI] [PubMed] [Google Scholar]
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L (1995) Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 270: 19173–19180 [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308: 1435–1439 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21: 1339–1351 [DOI] [PubMed] [Google Scholar]
- Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S (2004) Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res 64: 719–727 [DOI] [PubMed] [Google Scholar]
- Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, Chishti MA, Marvi M, Windl O, Ahrens R, Major F, Sy MS, Kretzschmar H, Fraser PE, Mount HT, Westaway D (2004) Genetic mapping of activity determinants within cellular prion proteins: N-terminal modules in PrPC offset pro-apoptotic activity of the Doppel helix B/B′ region. J Biol Chem 279: 55443–55454 [DOI] [PubMed] [Google Scholar]
- Fioriti L, Quaglio E, Massignan T, Colombo L, Stewart RS, Salmona M, Harris DA, Forloni G, Chiesa R (2005) The neurotoxicity of prion protein (PrP) peptide 106-126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol Cell Neurosci 28: 165–176 [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255–1264 [PMC free article] [PubMed] [Google Scholar]
- Flechsig E, Hegyi I, Leimeroth R, Zuniga A, Rossi D, Cozzio A, Schwarz P, Rulicke T, Gotz J, Aguzzi A, Weissmann C (2003) Expression of truncated PrP targeted to Purkinje cells of PrP knockout mice causes Purkinje cell death and ataxia. EMBO J 22: 3095–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F (1993) Neurotoxicity of a prion protein fragment. Nature 362: 543–546 [DOI] [PubMed] [Google Scholar]
- Harris DA (2003) Trafficking, turnover and membrane topology of PrP. Br Med Bull 66: 71–85 [DOI] [PubMed] [Google Scholar]
- Harris DA, Huber MT, van Dijken P, Shyng S-L, Chait BT, Wang R (1993) Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 32: 1009–1016 [DOI] [PubMed] [Google Scholar]
- Harris DA, True HL (2006) New insights into prion structure and toxicity. Neuron 50: 353–357 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, Defea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR (1998) A transmembrane form of the prion protein in neurodegenerative disease. Science 279: 827–834 [DOI] [PubMed] [Google Scholar]
- Hornemann S, Schorn C, Wuthrich K (2004) NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 5: 1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Inoue K, Yokoyama T, Kobayashi S, Asakura T, Teramoto A, Itohara S (2003) Prions prevent brain damage after experimental brain injury: a preliminary report. Acta Neurochir Suppl 86: 297–299 [DOI] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T (1999) Prions prevent neuronal cell-line death. Nature 400: 225–226 [DOI] [PubMed] [Google Scholar]
- Lehmann S, Harris DA (1995) A mutant prion protein displays an aberrant membrane association when expressed in cultured cells. J Biol Chem 270: 24589–24597 [DOI] [PubMed] [Google Scholar]
- Li A, Harris DA (2005) Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem 280: 17430–17434 [DOI] [PubMed] [Google Scholar]
- Li A, Sakaguchi S, Atarashi R, Roy BC, Nakaoke R, Arima K, Okimura N, Kopacek J, Shigematsu K (2000) Identification of a novel gene encoding a PrP-like protein expressed as chimeric transcripts fused to PrP exon 1/2 in ataxic mouse line with a disrupted PrP gene. Cell Mol Neurobiol 20: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T, Riek R, Guntert P, Wuthrich K (2003) NMR structure of the human doppel protein. J Mol Biol 326: 1549–1557 [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302: 871–874 [DOI] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J (1994) 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8: 121–127 [DOI] [PubMed] [Google Scholar]
- Massimino ML, Ballarin C, Bertoli A, Casonato S, Genovesi S, Negro A, Sorgato MC (2004) Human Doppel and prion protein share common membrane microdomains and internalization pathways. Int J Biochem Cell Biol 36: 2016–2031 [DOI] [PubMed] [Google Scholar]
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 165: 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Johnson EM Jr (1996) Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci 16: 7487–7495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo H, Moore RC, Cohen FE, Westaway D, Prusiner SB, Wright PE, Dyson HJ (2001) Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci USA 98: 2352–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D (1999) Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 292: 797–817 [DOI] [PubMed] [Google Scholar]
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P (2001) Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA 98: 15288–15293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S (1999) A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab Invest 79: 689–697 [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR (1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273: 614–622 [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95: 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A (2005) Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci 25: 4879–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R, Zhou Y, Solis GP, Stuermer CA, Malaga-Trillo E (2006) Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J 20: 317–319 [DOI] [PubMed] [Google Scholar]
- Rossi D, Cozzio A, Flechsig E, Klein MA, Rulicke T, Aguzzi A, Weissmann C (2001) Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J 20: 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A (2005) Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ 12: 783–795 [DOI] [PubMed] [Google Scholar]
- Roucou X, LeBlanc AC (2005) Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med 83: 3–11 [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T (1996) Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 380: 528–531 [DOI] [PubMed] [Google Scholar]
- Selvaggini C, De Gioia L, Cantu L, Ghibaudi E, Diomede L, Passerini F, Forloni G, Bugiani O, Tagliavini F, Salmona M (1993) Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. Biochem Biophys Res Commun 194: 1380–1386 [DOI] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C (1998) Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93: 203–214 [DOI] [PubMed] [Google Scholar]
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA (2004) Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303: 1514–1516 [DOI] [PubMed] [Google Scholar]
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM (2005) Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis 20: 442–449 [DOI] [PubMed] [Google Scholar]
- Stewart RS, Drisaldi B, Harris DA (2001) A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic reticulum. Mol Biol Cell 12: 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyach C, Checler F (2005) Combined pharmacological, mutational and cell biology approaches indicate that p53-dependent caspase 3 activation triggered by cellular prion is dependent on its endocytosis. J Neurochem 92: 1399–1407 [DOI] [PubMed] [Google Scholar]
- Uelhoff A, Tatzelt J, Aguzzi A, Winklhofer KF, Haass C (2005) A pathogenic PrP mutation and doppel interfere with polarized sorting of the prion protein. J Biol Chem 280: 5137–5140 [DOI] [PubMed] [Google Scholar]
- Yost CS, Lopez CD, Prusiner SB, Myers RM, Lingappa VR (1990) Non-hydrophobic extracytoplasmic determinant of stop transfer in the prion protein. Nature 343: 669–672 [DOI] [PubMed] [Google Scholar]
- Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J, Qin YQ, Dikranian K, Olney JW (2005) Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis 20: 608–614 [DOI] [PubMed] [Google Scholar]
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wüthrich K (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 97: 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 21: 3307–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS (1998) Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci USA 95: 8812–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]