

Abstract
Following entry and reverse transcription, the HIV-1 genome is integrated into the host genome. In contrast to productively infected cells, latently infected cells frequently harbor HIV-1 genomes integrated in heterochromatic structures, allowing persistence of transcriptionally silent proviruses. Microglial cells are the main HIV-1 target cells in the central nervous system and constitute an important reservoir for viral pathogenesis. In the present work, we show that, in microglial cells, the co-repressor COUP-TF interacting protein 2 (CTIP2) recruits a multienzymatic chromatin-modifying complex and establishes a heterochromatic environment at the HIV-1 promoter. We report that CTIP2 recruits histone deacetylase (HDAC)1 and HDAC2 to promote local histone H3 deacetylation at the HIV-1 promoter region. In addition, DNA-bound CTIP2 also associates with the histone methyltransferase SUV39H1, which increases local histone H3 lysine 9 methylation. This allows concomitant recruitment of HP1 proteins to the viral promoter and formation of local heterochromatin, leading to HIV-1 silencing. Altogether, our findings uncover new therapeutic opportunities for purging latent HIV-1 viruses from their cellular reservoirs.
Keywords: CTIP2, HDAC, HIV-1, HP1, SUV39H1
Introduction
After entry into the target cell and reverse transcription, HIV-1 genes are integrated into the host genome. It is now well established that the viral promoter activity is directly governed by its chromatin environment (reviewed in Van Lint, 2000). Nucleosomes are precisely positioned at the HIV-1 promoter (Verdin et al, 1993; Van Lint et al, 1996). Nuc-1, a nucleosome located immediately downstream of the transcriptional initiation site directly impedes LTR activity. Epigenetic modifications and disruption of Nuc-1 are a prerequisite to activation of LTR-driven transcription and viral expression (for review, see Van Lint, 2000). The compaction of chromatin and its permissiveness for transcription are directly dependent on the post-translational modifications of histones such as acetylation, methylation, phosphorylation and ubiquitination (reviewed in Fischle et al, 2003). Euchromatin, a relaxed state of chromatin, is often associated with active transcription. On the other hand, heterochromatin, a compacted and more structured chromatin environment, is repressive for transcription. In contrast to productively infected cells, latently infected cells frequently harbor HIV-1 genomes integrated in heterochromatic structures, which allows viral persistence of silenced integrated proviruses (Jordan et al, 2003). These observations might at least partially explain how the virus can escape the host immune response and current therapeutic tools (Finzi et al, 1997; Pierson et al, 2000). Understanding the molecular mechanisms underlying HIV-1 transcriptional silencing is thus a major challenge in the fight against AIDS.
HP1α is associated with transcriptionally inactive chromatin. At heterochromatic sites, HP1 binds methylated lysine 9 of histone H3 where it promotes gene silencing (Bannister et al, 2001). Transition from an active to inactive transcriptional state implies a series of ordered recruitment of histone-modifying enzymes. This is exemplified for K9/H3 methylation and HP1 recruitment, which requires the stepwise recruitment of histone deacetylase (HDAC) (to first remove the acetyl group) and histone methyltransferase (HMT) activities.
HIV-1 gene transcription has been shown to be activated by trichostatin A (TSA) treatment, and several transcription factors bound to the viral LTR recruit class I or II HDAC (Van Lint et al, 1996; Coull et al, 2000; Williams et al, 2006). However, to date, the mechanisms associated with the establishment of a heterochromatic environment at the HIV-1 promoter remains unclear.
COUP-TF interacting protein 2 (CTIP2) is a recently cloned transcriptional repressor that can associate with members of the COUP-TF family (Avram et al, 2000). This cofactor is expressed in the brain and the immune system (Leid et al, 2004). By regulating both differentiation and survival of thymocytes, CTIP2 is necessary for T-lymphocyte development (Wakabayashi et al, 2003). In the brain, CTIP2 plays a key role in the development of corticospinal motor neuron axonal projections to the spinal cord (Arlotta et al, 2005).
Recently, we reported that CTIP2 inhibits HIV-1 replication in human microglial cells (Rohr et al, 2003; Marban et al, 2005). Microglial cells constitute the central nervous system (CNS)-resident macrophages. They are the main HIV-1 target cells in the brain, and because they are long lived and relatively protected by the blood–brain barrier, they constitute an important reservoir of viruses. Recently, brain macrophages have been described as latently HIV-1-infected cellular reservoirs (Barber et al, 2006). It is now clear that the long-lived reservoirs of HIV-1 can persist for years in the presence of HAART. However, contrary to CD4+ T-lymphocyte reservoirs (reviewed in Marcello, 2006), information on the virus state within macrophages and microglial cells is very limited.
Here, we report that CTIP2 inhibits HIV-1 gene transcription by recruiting a chromatin-modifying complex and by establishing a heterochromatic environment at the HIV-1 promoter in microglial cells. Understanding HIV-1 silencing in the cellular reservoirs is actually the major challenge to viral eradication. Thereby, CTIP2, HDAC and HMT recruitments might uncover new therapeutic opportunities.
Results
CTIP2 associates with TSA-sensitive HDAC activities
Our previous studies (Rohr et al, 2003; Marban et al, 2005) suggested that CTIP2 may repress HIV-1 transcription through association with HDAC activities in microglial cells. To decipher whether TSA-sensitive HDACs participate in CTIP2 repressive function, we examined the effect of TSA, a compound known to inhibit class I and II but not class III HDACs, on CTIP2-mediated repression of HIV-1 promoter in microglial cells (Figure 1A). TSA treatment and CTIP2 knockdown stimulated LTR-driven transcription in microglial cells. Interestingly, CTIP2 knockdown and TSA treatment strongly synergyized in HIV-1 transcriptional activation, suggesting an interaction of CTIP2 and TSA-sensitive HDACs (Figure 1A). Of note, control and HDAC3 shRNAs did not activate LTR-driven transcription, alone or in cooperation with TSA treatment (Supplementary Figure 1A). The specificity of CTIP2 knockdown and TSA cooperation in HIV-1 transcription was assessed by testing other cellular and viral promoters. No significant cooperation was observed in the modulation of the tested promoters (Supplementary Figure 1B). To test whether CTIP2 could associate with HDAC activity, FLAG-tagged CTIP2 was immunoprecipitated from HEK cell extracts and associated HDAC activity was assessed. As shown in Figure 1B, CTIP2 associated with robust HDAC activity. By comparison, the relative amount of HDAC activity associated with CTIP2 was about nine-fold higher than that observed with the control immunoprecipitation. Interestingly, TSA totally abolished CTIP2-associated HDAC activity, demonstrating that this activity was due to class I or II HDACs.
Figure 1.
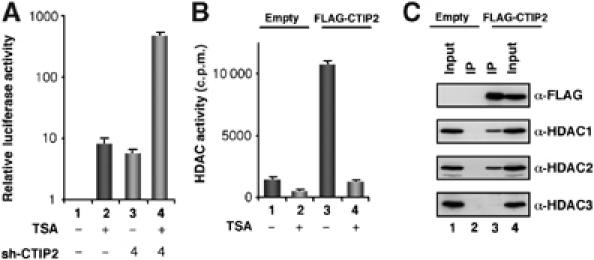
Interactions of TSA-sensitive HDAC1 and HDAC2 with CTIP2. (A) Microglial cells were transfected with the episomal LTR-LUC vector in the presence or absence of 4 μg of pshRNA-CTIP2 vector. Cells were untreated or treated with 450nM TSA for 24 h. Two days post-transfection, LUC activities were measured and expressed relative to the value obtained with the empty vector. The knockdown efficiency of shRNA construction was controlled by Western blot (Supplement Figure 5A). Control shRNAs are also presented (Supplement Figure 1A). (B, C) HEK 293T cells were transfected with the indicated pFLAG-CTIP2 expression vector and the empty vector as control. Immunoprecipitated complexes were tested for HDAC activities (B) and for the presence of HDAC1, HDAC2 and HDAC3 by Western blot (C).
CTIP2 associates with HDAC1 and HDAC2
Based on the above findings, we next tested the presence of HDAC1–3 in the material associated with CTIP2 (Figure 1C). Western blot analysis revealed that CTIP2 specifically associated with HDAC1 and HDAC2 but not with HDAC3. Association of CTIP2 with HDAC1/2 in microglial cells was confirmed by immunofluorescence confocal microscopy (Supplementary Figure 2). Altogether, these results demonstrate that CTIP2 associates with an active HDAC complex.
Several multiproteic complexes containing HDAC1 and HDAC2 have been described, such as Sin3, NuRD or Co-REST. To further investigate the nature of CTIP2-associated HDAC complex, Western blot analysis was performed on affinity-purified CTIP2-associated complex from HEK 293T cell extracts. Although Sin3, NuRD and Co-REST were present in CTIP2-expressing cells, no interaction was detected with CTIP2 (data not shown). This suggests that CTIP2 may not be involved in any of the HDAC1/2-containing complexes identified to date.
The HDAC-interacting domain of CTIP2 is located in its N-terminus
To delineate the CTIP2 region mediating HDAC interaction, we performed HDAC activity assays on affinity-purified materials from cells expressing truncated mutants of CTIP2 (Figure 2A). The N-terminal part of CTIP2 (aa 1–354) was sufficient to recruit 80% of the HDAC activity associated with the full-length CTIP2 (Figure 2A, compare columns 5 and 3). Interestingly, neither the 145–434 nor the 350–813 region is associated with significant HDAC activity, suggesting that aa 1–145 of CTIP2 are responsible for HDAC recruitment.
Figure 2.
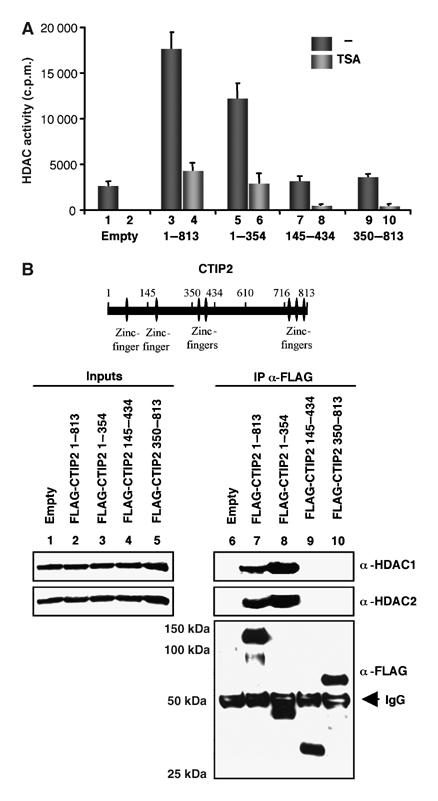
CTIP2 associates with HDAC1 and HDAC2 via its N-terminal domain. (A, B) HEK 293T cells were transfected with the indicated pFLAG-CTIP2 constructs and the empty vector as control. Cells extracts were normalized for the quantities of overexpressed FLAG-CTIP2 proteins and endogenous HDAC1 and HDAC2. Immunoprecipitated complexes were tested for HDAC activities (A) and for the presence of HDAC1, HDAC2 and FLAG-CTIP2 proteins by Western blot (B). (B) Input controls for HDAC1, HDAC2 (columns 1–5) and FLAG-CTIP2 construct expression (α-FLAG panel) are presented. A schematic CTIP2 linear structure is also drawn for a better visualization of CTIP2 domains.
To verify the correlation between CTIP2-associated HDAC activity and the presence of HDAC1/2, we performed Western blot analysis of the material associated with the various CTIP2 mutants (Figure 2B). As expected, the full-length and the N-terminal part of CTIP2 associated with endogenous HDAC1 and HDAC2.
HDAC1 and HDAC2 cooperate with CTIP2 to repress HIV-1 gene transcription and viral replication
To decipher how HDAC1 and HDAC2 participate in CTIP2-repressive function, we examined their effect on CTIP2-mediated repression of chromatin-integrated HIV-1 promoter. TZM-bl cells, which contain a chromatin-integrated LTR, were cotransfected with expression vectors for CTIP2, HDAC1 or HDAC2, as indicated. Overexpression of HDAC1 and HDAC2 alone did not significantly affect LTR-driven transcription (Figure 3A). Moreover, CTIP2-mediated repression was further enhanced by coexpression of HDAC1 or HDAC2. Similar results were obtained in microglial cells transfected with an episomal LTR-LUC reporter (Figure 3B). Of note, a mutant of CTIP2 deficient in HDAC1/2 binding (i.e. 145–434 mutant) did not cooperate with HDAC1/2 (Supplementary Figure 3A). To validate the biological relevance of our finding, we next investigated the effect of knocking down CTIP2, HDAC1 and HDAC2 on HIV-1 LTR-driven transcription. Knockdown of HDAC1 or HDAC2 had very little to no impact on HIV-1 transcription, whereas knockdown of CTIP2 increased LTR transcriptional activity, both on integrated and on episomal HIV-1 LTR (Figure 3C and D). However, simultaneous knockdown of CTIP2 and HDAC1 or HDAC2 led to further increase HIV-1 transcription. To determine the impact of CTIP2-mediated recruitment on HIV-1 replication, microglial cells were transfected with the HIV-1 pNL4-3 vector together with expression vectors for CTIP2, HDAC1 and HDAC2, as indicated (Figure 3E). Forty-eight hours post-transfection, soluble p24 capsid protein was assessed in the culture medium by ELISA. As expected, overexpression of HDAC1 or HDAC2 did not affect HIV-1 replication significantly. In contrast, overexpression of CTIP2 was associated with a dramatic inhibition of HIV-1 replication, which was further enhanced by coexpression of HDAC1 or HDAC2 (Figure 3E). To further evaluate the functional cooperation of endogenous CTIP2 and HDAC1/2 enzymes, we quantified p24 production in HIV-1-infected microglial cells that had been knocked down for CTIP2, HDAC1 or HDAC2, alone or in combination, as indicated (Figure 3F). HDAC1 or HDAC2 knockdown only slightly stimulated HIV-1 replication. In contrast, knockdown of CTIP2 stimulated HIV-1 production up to 60-fold, thus confirming the repressive function of CTIP2 in microglial cells. More importantly, simultaneous knockdown of CTIP2 together with either HDAC1 or HDAC2 enhanced viral production up to 170-fold (Figure 3F). Altogether, these observations demonstrate a functional cooperation between CTIP2 and HDAC1/2 in HIV-1 gene silencing.
Figure 3.
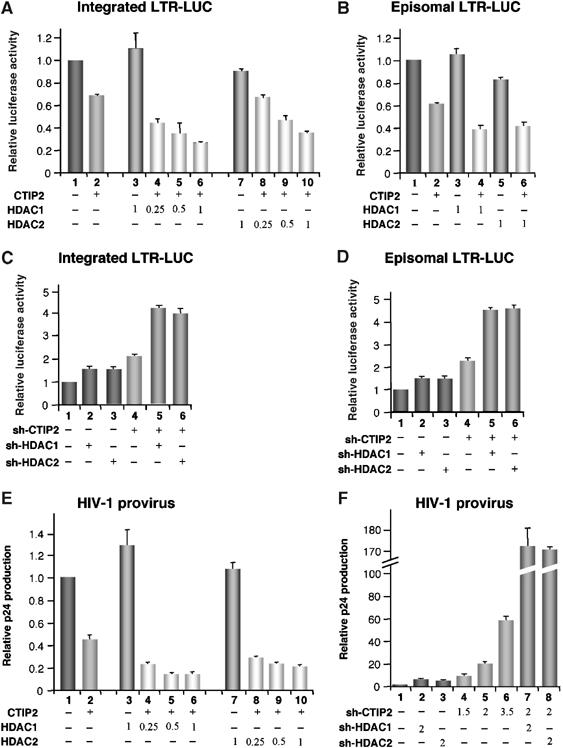
HDAC1 and HDAC2 cooperate with CTIP2 to repress HIV-1 gene transcription and viral replication. (A, C) TZM-bl cells were transfected with the indicated plasmids. Two days post-transfection, LUC activities were measured and expressed relative to the value obtained with the empty vector. (B, D) Microglial cells were transfected with the episomal LTR-LUC and the indicated vectors. LUC activities were measured 2 days post-trasnfection and expressed relative to the value obtained with LTR-LUC alone. DNA quantities were normalized with the corresponding empty vector. (E, F) Microglial cells were transfected with pNL4-3 and the indicated vectors. Culture supernatants were analyzed for p24 Gag contents 48 h post-transfection. The knockdown efficiency of shRNA constructions was controlled by Western blot (Supplement Figure 5A).
Association of CTIP2 with the HIV-1 proximal promoter induces recruitment of HDAC1 and HDAC2 and local histone H3 deacetylation
We previously reported that CTIP2 was recruited to the HIV-1 promoter by the cellular transcription factor Sp1 (Marban et al, 2005). To determine whether endogenous CTIP2, HDAC1 and HDAC2 proteins are associated at the viral promoter, microglial cells were infected with an NL4.3-env− HIV-1 virus and recruitment of CTIP2, Sp1, HDAC1 and HDAC2 was analyzed by chromatin immunoprecipitation (ChIP) (Figure 4A). CTIP2 and Sp1 were found at the proximal region of the HIV-1 promoter (PCR1) (Figure 4A). In addition, both HDAC1 and HDAC2 were detected bound at the same region of the promoter. As a control, no binding of CTIP2, Sp1, HDAC1 or HDAC2 was observed for the control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter (data not shown). We next investigated whether HDAC1 and HDAC2 are recruited to the HIV-1 promoter by CTIP2. As expected, CTIP2 associated specifically with the LTR proximal region, but not with the Nuc-1-binding region (Figure 4B). Overexpression of CTIP2 increased recruitment of HDAC1 and HDAC2 to the Sp1-binding site (Figure 4B, panels α-HDAC1 and α-HDAC2, PCR1) and to the Nuc-1-binding region (Figure 4B, panels α-HDAC1 and α-HDAC2, column PCR2) of the LTR. As a control, overexpression of the N-terminal truncated 145–434 proteins did not increase HDAC1 and HDAC2 binding to the viral promoter (Supplementary Figure 3E).
Figure 4.

Association of CTIP2 with the HIV-1 proximal promoter induces local H3 deacetylation with concomitant recruitment of HDAC1 and HDAC2. (A, D) Microglial cells (A) and CTIP2 knockdown cells (D) were infected with the VSV-pseudotyped pNL4.3-env− virus 24 h before being subjected to ChIP experiments with the indicated antibodies. As a control, immunoprecipitations were performed in the absence of antibody (Ab control). Input (1/1000) and immunoprecipitated DNAs were quantified by real-time PCR using PCR1 LTR-specific oligonucleotides. The amount of immunoprecipitated material was normalized to the input DNA (A) and fold enrichments were normalized to the nonspecific enrichment in the GAPDH DNA (D). (B) ChIP experiments were performed on HEK 293T cells transfected with the HIV-1 LTR-LUC episomal vector in the presence or absence of the FLAG-CTIP2 expression vector as indicated. Cells were subjected to ChIP assays with the indicated antibodies. Specific enrichments in HIV LTR regions were quantitated by real-time PCR with the PCR1, PCR2 and GAPDH oligonucleotides. Results were normalized to enrichment in nonspecific GAPDH DNA. Results are representative of three independent experiments. (C) LTR-LUC-transfected HEK 293T cells were subjected to LUC activity quantification in the presence or absence of overexpressed CTIP2. (E) Initiated and elongated HIV-1 gene transcripts were quantitated by real-time RT–PCR in HIV-1-infected control and CTIP2 knockdown microglial cells. PCR quantifications target the HIV-1 TAR (initiation) and the HIV-1 Tat (elongation) regions. Results are presented relative to the initiated transcripts in control cells and normalized to β-actin copies.
We next assessed the impact of HDAC1/2 recruitment at the HIV-1 promoter by CTIP2 on local acetylation levels. For this purpose, we examined the acetylation of histone H3 at the Nuc-1-binding region and as a control at the proximal promoter region (Figure 4B, PCR2 and PCR1 respectively). No Ac/H3 was detected on the HIV-1 proximal promoter in the absence of CTIP2, whereas acetylated histones H3 were observed at the HIV-1 Nuc-1-binding region and at the constitutively active GAPDH promoter (Supplementary Figure 3C). Interestingly, overexpression of CTIP2 resulted in decreased levels of histone H3 acetylation at the HIV-1 Nuc-1-binding region (Figure 4B, panel α-Ac/H3, PCR2). As a control, no enrichment of Ac/H3 was observed at the proximal promoter (Figure 4B, panel α-Ac/H3, PCR1). These results suggest that CTIP2 targets HDAC1 and HDAC2 to the HIV-1 gene promoter to establish a chromatin environment detrimental to transcription. This conclusion was supported by the CTIP2-mediated repression of LTR-driven transcription in 293T cells (Figure 4C).
To further explore the link between CTIP2 and association of HDAC1 and HDAC2 with the viral promoter, we performed ChIP experiments in a context of HIV-1-infected control and CTIP2 knockdown microglial cells. As expected, reduction of endogenous CTIP2 levels correlated with decreased binding at the HIV-1 proximal promoter region (Figure 4D, panel α-CTIP2). As control, the same amount of Sp1 (Figure 4D, panel α-Sp1) was found at the proximal promoter region in control and CTIP2 knockdown cells. Surprisingly, whereas HDAC2 binding at the viral promoter was reduced in CTIP2 knockdown cells (Figure 4D, panel α-HDAC2), HDAC1 recruitment was enhanced (Figure 4D, panel α-HDAC1). Together with the results from previous studies (Doetzlhofer et al, 1999), this observation suggests that displacement of the CTIP2-repressing complex may allow additional HDAC1 protein to interact via Sp1 with the viral promoter. To further explore the role of CTIP2 in HIV-1 gene transcription, we quantified the initiated and the elongated transcripts in the context of HIV-1-positive microglial cells, knocked down for CTIP2. As shown in Figure 4E, the relative amount of HIV-1-initiated transcripts was eight times increased in CTIP2 knockdown cells compared to control cells. CTIP2 promoted accumulation of initiated transcripts but did not significantly increase elongated transcripts under these experimental conditions (Figure 4E).
CTIP2 associates with histone methyltransferase SUV39H1
We have previously reported that CTIP2 promotes HP1α recruitment to the HIV-1 promoter (Marban et al, 2005). In heterochromatic environments, HP1α associates with the methylated form of histone H3 lysine 9 (Bannister et al, 2001). As methylation of H3/K9 is typically mediated by SUV39H1, we logically tested whether CTIP2 is associated with SUV39H1. As shown in Figure 5, endogenous SUV39H1 was copurified with immunoprecipitated FLAG-tagged CTIP2 (Figure 5A, lane 1). In addition, deletion analysis revealed that the central 145–434 domain (Figure 5A, lane 3) but not the N- and C-terminal domains (Figure 5A, lanes 2 and 4) of CTIP2 was sufficient to mediate interaction with SUV39H1. The association of SUV39H1 with CTIP2 was further tested in vitro (Figure 5B). Pull-down experiments with in vitro-translated CTIP2 showed that 2–5% of CTIP2 proteins specifically interacted with GST-SUV39H1 in vitro (Figure 5B, column 3). Finally, confocal microscopic observations revealed colocalization of CTIP2 and SUV39H1 in the nuclei of microglial cells (Supplementary Figure 4).
Figure 5.
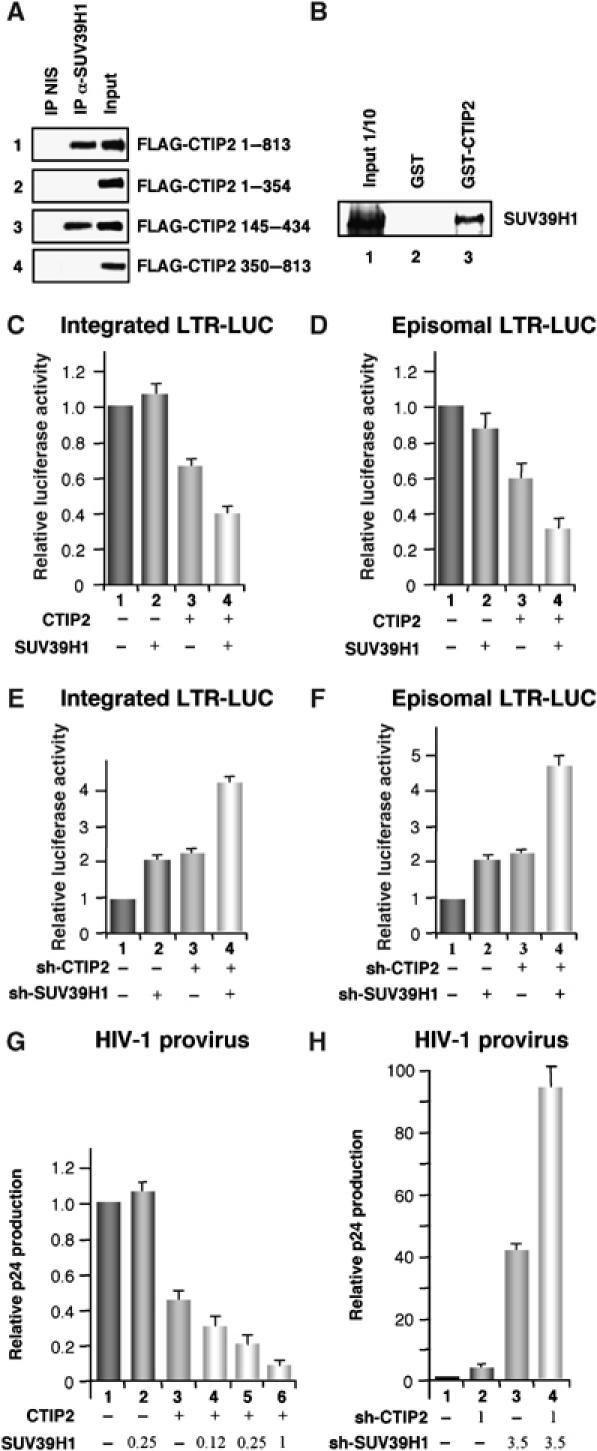
CTIP2 cooperates and associates with HMT SUV39H1 via its central domain. (A) HEK 293T cells were transfected with the indicated pFLAG-CTIP2 constructs. Cell extracts were normalized for the quantities of overexpressed FLAG-CTIP2 proteins and endogenous SUV39H1. Complexes immunoprecipitated with the anti-SUV39H1 antibodies or the control non-immune serum were immunodetected for the presence of FLAG-CTIP2 proteins by Western blot. (B) GST pull-down assays were performed with 35S-labelled CTIP2 protein incubated with GST (column 2) or GST-SUV39H1 fusion proteins (column 3). Approximately 10% of the total 35S-labelled proteins obtained were loaded as input control (column 1). (C, E) TZM-bl cells were transfected with the indicated vectors. Two days post-transfection, LUC activities were measured and expressed relative to the value obtained with the empty vector. (D, F) Microglial cells were transfected with the indicated vectors. LUC activities were measured 2 days post-transfection and expressed relative to value obtained with the LTR-LUC alone taken as 1. DNA quantities were normalized with the corresponding empty vector. (G, H) Microglial cells were transfected with the HIV-1 pNL 4-3 vector and the indicated vectors. Two days post-transfection, culture supernatants were analyzed for p24 Gag contents. The knockdown efficiency of shRNA constructions was controlled by Western-blot (Supplement Figure S3).
CTIP2 cooperates with SUV39H1 to repress HIV-1 gene transcription and viral replication
To further characterize the significance of CTIP2/SUV39H1 interaction, we performed luciferase assays on cellular extracts from TZM-bl cells containing an integrated LTR-LUC construct (Figure 5C) and from microglial cells transfected with an episomal LTR-LUC reporter (Figure 5D). The effects of CTIP2 and SUV39H1 were assessed after overexpression of each protein, independently or in combination. Overexpression of SUV39H1 alone had no effect on LTR-driven transcription (Figure 5C and D, column 2). However, it dramatically increased CTIP2-mediated repression of the HIV-1 promoter (Figure 5C and D, column 4). As a control, SUV39H1 had no effect on the aa 1–354 truncation CTIP2 mutant, which is unable to interact with SUV39H1 (Supplementary Figure 3B). These results suggest a functional cooperation between CTIP2 and SUV39H1 in the context of chromatinized HIV-1 promoter in microglial cells. Next, similar experiments were performed using shRNA constructs targeting SUV39H1 and CTIP2 (Figures 5E and F). As expected knockdown of CTIP2 and SUV39H1 independently or in combination stimulated transcription from genome-integrated as well as episomal HIV-1 LTR. The functional relevance of CTIP2/SUV39H1 to HIV-1 replication was next examined. For this purpose, CTIP2 and increasing amounts of SUV39H1 were expressed in pNL4-3-transfected microglial cells (Figure 5G). Production of p24 HIV-1 capsid was unaffected by overexpression of SUV39H1 (Figure 5G). However, in the presence of CTIP2, SUV39H1 further enhanced CTIP2-mediated repression of viral replication, confirming a functional cooperation between both proteins. As expected, the functional cooperation between CTIP2 and SUV39H1 was also observed in a similar experimental setting using knockdown of CTIP2 and SUV39H1 (Figure 5H).
CTIP2-mediated recruitment of SUV39H1 to the HIV-1 proximal promoter induces K9/H3 methylation and HP1 association
K9/H3 methylation is the ultimate epigenetic modification necessary for HP1 association and polymerization. To determine whether endogenous CTIP2, SUV39H1 and HP1 proteins are associated with the viral promoter in the context of infected microglial cells, cells were infected with a vesicular stomatostatis virus (VSV)-pseudotyped HIV-1 NL4.3-env− virus 24 h before being processed for ChIP experiments (Figure 6A). The presence of CTIP2, HDAC2 and HDAC1 at the viral promoter (Figure 4A) correlated with the recruitment of SUV39H1 and HP1 proteins (Figure 6A). To refine these observations, ChIP experiments using anti-trimethyl-K9/H3, anti-SUV39H1 and anti-HP1 antibodies were performed in the presence or absence of CTIP2 overexpression in LTR-LUC-transfected 293T cells (Figure 6B). The control GAPDH gene promoter, the LTR Nuc-1-binding region (Figure 6B, PCR2 columns) and the LTR proximal region (Figure 6B, PCR1 columns) were analyzed by quantitative real-time PCR.
Figure 6.
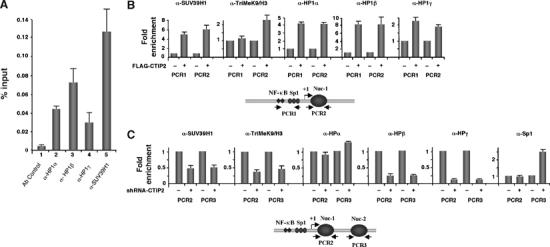
CTIP2-mediated recruitment of SUV39H1 to the viral LTR promotes K9/H3 methylation and HP1 recruitments. (A, C) Microglial (A) and CTIP2 knockdown microglial cells were infected with the VSV-pseudotyped pNL4.3-env− virus 24 h before being subjected to ChIP experiments with the indicated antibodies. As a control, immunoprecipitations were performed in the absence of antibody (Ab control). Input and immunoprecipitated DNA was subjected to real-time PCR using the PCR1 (A) or PCR2 and PCR3 (C) LTR-specific oligonucleotides. The amount of immunoprecipitated material was normalized to the input DNA (A) and the fold enrichments were normalized to enrichment in the nonspecific GAPDH promoter (C). (B) ChIP experiments were performed on HEK 293T cells transfected with the HIV-1 LTR-LUC episomal vector in the presence or absence of the FLAG-CTIP2 expression vector as indicated. Specific enrichments in HIV LTR regions obtained with the indicated antibodies were quantified by real-time PCR with the PCR1, PCR2 and GAPDH oligonucleotides. Results were normalized to enrichment in nonspecific GAPDH DNA. Results are representative of three independent experiments.
CTIP2 overexpression and binding (Figure 4B) correlated with the specific recruitment of HP1α, HP1β and HP1γ to the proximal and to the Nuc-1-binding regions of the HIV-1 LTR (Figure 6B). Binding of CTIP2 was associated with SUV39H1 recruitment and concomitant increase in K9/H3 methylation of the adjacent Nuc-1-binding region (Figure 6B). The increase of SUV39H1 association and H3/K9 methylation was correlated with HIV-1 transcriptional silencing (Figure 4C). ChIP experiments were then performed in a context of HIV-1-infected CTIP2 knockdown microglial cells (Figure 6C). As expected, CTIP2 knockdown correlated with a reduced presence of SUV39H1 at the Nuc-1- and the Nuc-2-binding regions of the HIV-1 promoter (Figure 6C, panel α-SUV39H1). This reduction correlated with a decrease in K9/H3 trimethylation and release of HP1β and HP1γ from Nuc-1 and Nuc-2 nucleosomes (Figure 6C). Surprisingly, in CTIP2 knockdown cells, HP1α binding was only weakly reduced at the Nuc-1 region and even slightly increased at the Nuc-2 region (Figure 6C, panel α-HP1α). CTIP2 knockdown also led to a significant increase in Sp1 binding at the Nuc-2 region, suggesting that CTIP2 knockdown-mediated modifications of the Nuc-2 nucleosome favored Sp1 binding to the adjacent Sp1-binding sites (Van Lint et al, 1997).
Decreased recruitment of CTIP2, SUV39H1 and HP1 to the HIV-1 promoter during transcriptional activation in the latently infected monocytic U1 cell line
To further evaluate the biological relevance of our findings, we performed quantitative ChIP assays in the monocytic HIV-1-infected U1 cell line, a model cell line for post-integration latency. In order to map the molecular flux of all the components of CTIP2-associated complex at the viral promoter during transcriptional activation, quantitative ChIP assays were carried out on mock-treated or PMA-treated U1 cells. As a control for transcriptional activation, these assays revealed that PMA treatment correlated with enhanced recruitment of TBP at the viral proximal promoter (Figure 7A). Remarkably, the viral transcriptional activation by PMA was associated with dislodgment of CTIP2 from the LTR proximal region (Figure 7A). In total agreement with our model, displacement of CTIP2 during transcriptional activation also correlated with a reduction in the level of SUV39H1 associated with the proximal region of the viral LTR. Accordingly, H3/K9 methylation of the adjacent Nuc-1 and the downstream Nuc-2 nucleosomes also decreased during PMA-induced transcriptional stimulation (Figures 7B and C). Moreover, CTIP2 release from the viral promoter and Nuc-1/2 hypomethylation correlated with a general reduction in the levels of HP1β and HP1γ associated with the viral promoter (Figure 7A–C). Except for the Nuc-2 region (Figure 7C), levels of HP1α were also reduced after PMA treatment. In contrast, Sp1 levels, which remained unaltered in the proximal and the Nuc-1 regions, surprisingly increased at the Nuc-2 region (Figure 7C). This latter observation correlates with that made after CTIP2 knockdown in microglial cells (Figure 6C, panel α-Sp1, PCR3)
Figure 7.
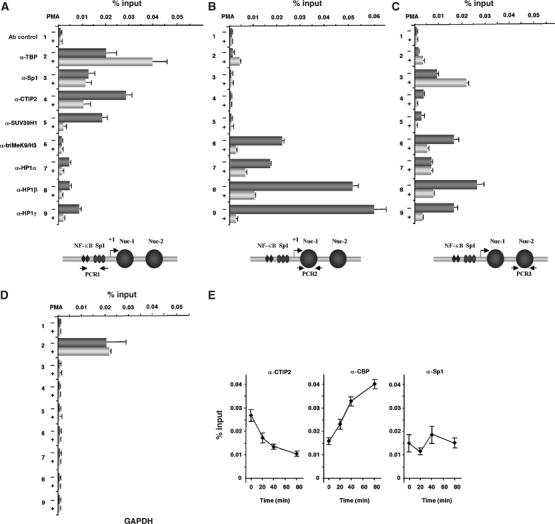
Transcriptional HIV-1 activation is accompanied by a decreased recruitment of CTIP2 and HP1 to the viral proximal promoter in the chromosomal context of integrated proviruses. ChIP assays were used to detect association of TBP, Sp1, CTIP2, HP1α, HP1β, HP1γ, triMeK9/H3 or SUV39H1 with the HIV-1 promoter proximal region (PCR1) (A), the Nuc-1-binding region (PCR2) (B), the Nuc-2-binding region (PCR3) (C) or the GAPDH promoter (D). U1 cells were mock-treated (−) or treated (+) with PMA (100 nM) for 1 h (A–D) and 20–80 min for time-ChIP assays (E). The amount of immunoprecipitated material was normalized to the input DNA. (E) U1 cells were mock-treated or treated with PMA (100 nM) for the indicated times and the proteins were crosslinked with formaldehyde for 10 min and DNA sheared. The crosslinked protein/DNA complexes were immunoprecipitated with the indicated antibodies or with a purified IgG as negative control. The protein/DNA crosslinks were reversed and the purified DNA was amplified and quantified by real-time PCR using the PCR1 primer. The amount of immunoprecipitated material was normalized to the input DNA.
Altogether, these results demonstrate that CTIP2, SUV39H1 and HP1 proteins are associated with the HIV-1 promoter proximal region in the context of latent integrated HIV-1 proviruses in vivo. In contrast, transcriptional activation of the HIV-1 promoter is associated with the release of CTIP2, SUV39H1 and HP1 proteins from the viral LTR.
Very recently, CBP/p300 has been shown to be recruited by Sp1 following PMA treatment (Hung et al, 2006). To further explore the mechanism of CTIP2 displacement upon PMA stimulation, we performed time-ChIP experiments targeting CTIP2, CBP/p300 and Sp1 proteins at the proximal Sp1-binding sites of the viral promoter. As shown in Figure 7E, levels of LTR-bound Sp1 remained unchanged after PMA treatments. Interestingly, CTIP2 displacement was correlated with concomitant recruitment of CBP. In addition, the relative amount of lost CTIP2 was very close to the amount of the newly recruited CBP. This observation thus suggests that displacement of CTIP2 from Sp1 upon PMA treatment may allow the recruitment of CBP coactivator. These data further support the notion that CTIP2 plays an important role in HIV-1 transcriptional repression by dictating a repressive chromatin structure in the viral LTR.
Discussion
Microglial cells, the CNS-resident macrophages, are the main HIV-1 target cells in the brain. Protected by the blood–brain barrier, microglial cells constitute a major viral reservoir insensitive to HAART. In a recent study, we reported that the nuclear cofactor CTIP2 inhibits HIV-1 replication in microglial cells by affecting HIV-1 gene transcription by two different mechanisms. CTIP2 represses the initial phase of HIV-1 transcription via a direct association with the LTR-bound transcription factor Sp1 (Marban et al, 2005) and the late phase via viral Tat protein relocation to transcriptionally inactive regions of chromatin (Rohr et al, 2003). By forcing a local heterochromatic environment at the viral promoter, CTIP2 inhibits HIV-1 replication in human microglial cells, the main HIV-1 target and reservoir cells of the CNS.
Here, we address the intimate molecular mechanisms whereby CTIP2 promotes a local heterochromatic environment to repress HIV-1 gene transcription in microglial cells. Biochemical analysis revealed that CTIP2 associates with HDAC1 and HDAC2. HDAC1 and HDAC2 are often found in association, as illustrated for the HDAC complexes identified to date (for review, see Ahringer, 2000). Interestingly, no component of known HDAC1/2 complexes could be detected in association with CTIP2. This suggests that CTIP2 could interact directly with HDAC1/2 or as part of the new HDAC1/2-containing complex. Biochemical purification and complete identification of CTIP2-associated proteins should allow one to discriminate between both hypotheses. A previous study has reported that CTIP2 physically and functionally interacted with SirT1, a human class III TSA-insensitive HDAC (Senawong et al, 2003). However, enzymatic assays or Western blot analysis did not allow us to detect class III members in association with CTIP2 (data not shown).
Expression of truncated forms of CTIP2 and the analysis of associated HDAC activity revealed that interaction with HDAC enzymes was mediated by the N-terminus of CTIP2 (aa 1–145). In previous studies, we reported that the central domain of CTIP2 (aa 145–434) was involved in interactions with the cellular Sp1, COUP-TF and the viral Tat proteins, whereas its C-terminus (aa 717–813) associated with HP1α (Rohr et al, 2003; Marban et al, 2005). CTIP2 does not directly bind to DNA but is recruited to the HIV-1 promoter via its association with Sp1. Therefore, association of HDACs with the N-terminal region of CTIP2 is fully compatible with the recruitment of their activity to the HIV-1 promoter. ChIP experiments confirmed the concomitant recruitment of CTIP2, HDAC1 and HDAC2 to the viral promoter in infected microglial cells. Overexpression of CTIP2 promoted HDAC1 and HDAC2 recruitment to the viral promoter. Moreover, CTIP2-mediated recruitment of HDAC1/2 to the viral promoter was correlated with local deacetylation of H3. These observations indicate that HDAC1 and HDAC2 interact with CTIP2 anchored to the HIV-1 LTR and suggest that HDAC enzymes collaborate with CTIP2 for HIV-1 gene silencing. We confirmed in HIV-1-infected CTIP2 knockdown cells the need of CTIP2 recruitment for HDAC2 association with the viral promoter. However, as HDAC1 recruitment was increased after CTIP2 knockdown, it should be further recruited by other molecular interactions favored by CTIP2 displacement, such as direct association with Sp1 (Doetzlhofer et al, 1999). Here, we show that both HDAC enzymes cooperate with CTIP2 to repress HIV-1 transcription in integrated or episomal contexts. Remarkably, the best cooperations were observed in the provirus context, suggesting the need of the whole HIV-1 genome to have the best-chromatinized structure and the more physiological view of these phenomena. These results confirm the biological relevance of the association between CTIP2 and HDACs that we unravel in this study. In addition, they imply that both HDAC1 and HDAC2 are necessary for CTIP2 repressive function and contribute to the establishment of a repressive state of HIV-1 gene transcription in microglial cells. Quantification of the HIV-1 gene transcripts revealed that CTIP2 repressed their initiation but not elongation. Very recently, the same observation was made for the NF-κB p50 factor that also recruits HDAC1 to the HIV-1 promoter (Williams et al, 2006).
In contrast to histone acetyltransferases (HATs), there have been few articles to report HDAC recruitment to the HIV-1 promoter. The HIV-1 transcriptional repressors YY1 and LSF have been shown to recruit HDAC1 (Coull et al, 2000). Blocking LSF-mediated recruitment of HDAC1 to the HIV-1 promoter produced a rebound of HIV-1 replication in resting CD4+ T cells from HIV-infected patients whose viremia had been suppressed by therapy (Ylisastigui et al, 2004). These results are in favor of a role for TSA-sensitive HDACs in the establishment of persistent and quiescent reservoir of HIV-1 infection. Thus, understanding HDAC recruitment to HIV-1 promoter is crucial for developing strategies to purge the quiescent reservoir and eradicate the virus.
We previously reported that CTIP2 recruited HP1α to the HIV-1 promoter. In heterochromatic environment, HP1 proteins specifically interact with the methylated forms of histone H3 lysine 9 (Bannister et al, 2001). As CTIP2-mediated recruitment of HDAC1 and HDAC2 induced K9/H3 deacetylation, we further investigated the ability of CTIP2 to associate with HMT activity. Co-immunoprecipitation experiments revealed that CTIP2 associates with the HMT SUV39H1. ChIP experiments confirmed the concomitant association of CTIP2 and SUV39H1 on the HIV-1 promoter in infected microglial cells. Moreover, we observed CTIP2-mediated recruitment of SUV39H1 to the viral LTR and the concomitant methylation of K9/H3 to the HIV-1 Nuc-1-binding region. In eukaryotic cells, lysine 9 of histone H3 is specifically methylated by SUV39H1 (Rea et al, 2000) creating a binding site for the chromodomain of HP1 (Bannister et al, 2001; Lachner et al, 2001). Targeting HP1 to chromatin also requires direct physical interaction with SUV39H1 (Stewart et al, 2005). Here, we reveal that CTIP2 can recruit SUV39H1 and HP1α, HP1β and HP1γ proteins to the viral promoter. Interestingly, CTIP2 knockdown induces a decreased recruitment of HP1β and HP1γ but not HP1α to the Nuc-1 and Nuc-2 regions. Stimulation of the latently infected U1 cells by PMA did not release Hp1α from the Nuc-2 region. These results suggest that the establishment of transcriptional latency induced by CTIP2 and the transcriptional reactivation are not strikingly linked by reversal molecular mechanisms. Moreover, it also suggests that distinct roles could be attributed to the different isoforms of HP1 proteins. Interestingly, SUV39H1 has been shown to associate with many other proteins, including HDACs. HDAC1, HDAC2 and HDAC3 were shown to associate with SUV39H1, suggesting that transcriptional repression by SUV39H1 could be the consequence of HDAC recruitment (Vaute et al, 2002). Moreover, methylation of K9/H3 can suppress transcription independently of HP1 through a mechanism involving histone deacetylation (Stewart et al, 2005). Altogether, HDAC1, HDAC2, SUV39H1 and HP1 proteins are deeply involved in the formation of heterochromatic environments and gene silencing. Moreover, in the context of latently infected cells, transcriptional activation of the HIV-1 promoter is accompanied by a decreased recruitment of CTIP2, SUV39H1, HP1 proteins and K9/H3 methylation. Moreover, as CTIP2-induced modifications appeared propagated to the downstream Nuc-2 region, it suggests that this heterochromatic structure can spread along the viral genome. In addition, displacement of CTIP2 from Sp1 upon PMA stimulation and the subsequent recruitment of the CBP coactivator further confirm the notion that CTIP2 plays an important role in HIV-1 transcriptional repression by dictating a repressive chromatin structure at the viral LTR.
The present work strongly suggests the concomitant recruitment of HDAC1, HDAC2 and SUV39H1 enzymes to the viral promoter by CTIP2. The ordered histone modifications would allow HP1 binding, heterochromatin formation and HIV-1 silencing (Figure 8). As heterochromatin formation at the HIV-1 promoter has been linked to post-integration latency, it suggests a possible involvement of transcriptional repressors such as CTIP2 in the establishment of viral persistence and post-integration latency in the brain. Therefore, our findings uncover new therapeutic opportunities for purging latent HIV-1 viruses from their cellular sanctuaries in infected patients.
Figure 8.
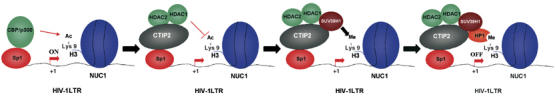
Model for CTIP2-mediated establishment of heterochromatin environment to HIV-1 gene promoter and viral silencing. CTIP2 first recruits HDAC1 and HDAC2 enzymes to deacetylate H3 histones located at the viral promoter and particularly Nuc-1 H3 histones. After H3 deacetylation, CTIP2 recruits SUV39H1 to methylate Nuc-1 histone H3 lysine 9. This last H3 modification allows HP1 binding and polymerization, heterochromatin formation and HIV-1 silencing.
Materials and methods
Plasmids
Most of the constructs used in our assays have been described previously: pcDNA3, GST-CTIP2 and full-length construct pFLAG-CTIP2 1–813 (Avram et al, 2000); pNL4-3, pNL4.3-env−, pVSV.G and deletions constructs pFLAG-CTIP2 (Rohr et al, 2003); pRFP-CTIP2 (Marban et al, 2005); pcDNA3-HDAC1, pcDNA3-HDAC2 and pcDNA3-HDAC3 expression vectors (Fischle et al, 1999). pshRNA-HDACs were provided by MA Lazar (Ishizuka and Lazar, 2003) and pMyc-SUV39H1, pshRNA-SUV39H1 and pGST-SUV39H1 by T Jenuwein (Schotta et al, 2004). The episomal LTR-LUC vector was constructed by cloning the luciferase gene under the control of the HIV-1 LTR into a modified pREP10 episomal vector. pshRNA-CTIP2 and pSirenZsGreen-shRNA-CTIP2 were constructed by inserting the sequence directed against CTIP2 in the pSuper plasmid (Oligoengine Inc.) or the pSirenZsGreen RetroQ vector (Clontech Lab. Inc.) as recommended by the manufacturer.
Cell culture
The human microglial cell line (provided by M Tardieu, Paris, France) (Janabi et al, 1995), TZM-bl (Derdeyn et al, 2000; Wei et al, 2002), U1 and HEK 293T cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum and 100 U/ml penicillin–streptomycin. CTIP2 knockdown microglial cells expressing shRNA-CTIP2 were stably established by infection of microglial cells with a pSirenZsGreen-ShRNA-CTIP2-based retrovirus as described by the manufacturer (Clontech Lab. Inc.). CTIP2 shRNA-expressing cells were sorted by flow cytometry for the concomitant expression of the ZsGreen protein and cultured in DMEM. The CTIP2 knockdown efficiency was controlled by quantitative RT–PCR and ChIP experiments.
GST pull-down assays
Production of GST fusion proteins was described previously (Rohr et al, 1997). The 35S-labelled SUV39H1 was prepared by in vitro transcription–translation using the TNT® T7 Coupled Wheat Germ Extract System (Promega). GST pull-down assays were performed essentially as described previously (Rohr et al, 2003).
Co-immunoprecipitation assays
HEK 293T cells cultured in 100-mm-diameter dishes were transfected using the calcium phosphate co-precipitation method with the indicated pFLAG-CTIP2 deletion constructs (30 μg) or pcDNA3 (30 μg) vectors. Two days post-transfection, immunoprecipitations were performed using the standard technique with M2 anti-FLAG (Sigma) or the anti-SUV39H1 (Santa Cruz) antibodies overnight at 4°C. Finally, the immunoprecipitated complexes were processed for SDS–PAGE and Western blot analysis or HDAC assays.
Histone deacetylase assays
The immunoprecipitated complexes were washed twice with HDAC buffer (10 mM Tris–HCl, pH 8.0, 10 mM NaCl, 10% glycerol). For inhibition studies, they were preincubated with TSA (450 nM) in HDAC buffer for 45 min at 4°C. Beads were resuspended in 30 μl of HDAC buffer containing 20 000 c.p.m. of an acetylated H4 peptide. HDAC activity was determined after incubation for 2 h at 37°C. The reaction was stopped by adding 0.04 M acetic acid and 250 mM HCl. The mixture was extracted with ethyl acetate and the released [3H]acetic acid was quantified by scintillation counting.
SDS–PAGE and Western blot analysis
SDS–PAGE were performed using standard techniques. Proteins were detected using antibodies directed against the FLAG epitope (M2 mouse monoclonal from Sigma), HDAC1, HDAC2 and HDAC3 proteins (Santa Cruz), CTIP2 (abcam), Myc epitope (Santa Cruz) and β-actin (Sigma). Proteins were visualized by chemiluminescence using the Super Signal Chemiluminescence Detection System (Pierce).
Luciferase assays
Microglial and TZM-bl cells cultured in 48-well plates were transfected with the indicated vectors using the calcium phosphate co-precipitation method. Two days later, cells were collected and luciferase activity was determined using the Dual-Glo™ Luciferase Assay System (Promega). Values correspond to an average of at least three independent experiments performed in duplicate.
Viral replication
Microglial cells cultured in 12-well plates were transfected using the calcium phosphate co-precipitation method with HIV-1 pNL4-3 and the expression plasmids as indicated. Total amounts of DNA were normalized with the corresponding empty vector. HIV-1 replication was monitored as described previously (Rohr et al, 2003). Values correspond to an average of at least three independent experiments carried out in duplicate.
Pseudotyped virion production and single-round infection
The plasmid pNL4.3-env− was cotransfected with the envelope plasmid encoding the pVSV.G envelope protein into HEK 293T cells. The virions were collected 48 h post-transfection. For single-round infection, microglial cells were incubated with the VSV-pseudotyped HIV-1 NL4.3-env− virus for 24 h at 37°C.
Chromatin immunoprecipitation assays
Microglial and CTIP2 knockdown microglial cells cultured in 150-mm-diameter dishes were subjected to single-round infection by the VSV-pseudotyped viruses 24 h before being processed for ChIP experiments. HEK 293T cells cultured in 100-mm-diameter dishes were transfected using the calcium phosphate co-precipitation method with the indicated vectors. ChIP assays were performed using the ChIP assay kit (Upstate) 48 h post-transfection. To reach the desired resolution, we have optimized our sonication procedure to obtain 100–150 bp DNA fragments. U1 cells were mock-treated (−) or treated (+) with PMA (100 nM) for 1 h (20–80 min for the time-ChIP experiments) before ChIP assays. The primary antibodies used for ChIP were as follows: M2 anti-FLAG (Sigma), anti-CTIP2 (Abcam), anti-Sp1 (Upstate), anti-TBP (Upstate), anti-HDAC1, anti-HDAC2, anti-Ac/H3, anti-triMeK9/H3, anti-HP1α (Upstate) anti-HP1β (Euromedex), anti-HP1γ (Euromedex), anti-SUV39H1 (Abcam) and anti-CBP (Santa Cruz Biotech.). Immunoprecipitated DNA was subjected to real-time PCR quantification. Three LTR-HIV-1 regions were selected for amplification (PCR1, PCR2 and PCR3) as well as the human constitutively expressed cellular gene for GAPDH. Primers are presented in Supplementary Figure 6.
Initiation and elongation quantification
Quantification of the initiated and the elongated HIV-1 gene transcripts was performed in infected control and shRNA-CTIP2-expressing microglial cells by real-time RT–PCR quantification, as described previously (Williams et al, 2006). Results were normalized to the β-actin copies.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank T Jenuwein, MA Lazar and E Seto for providing vectors. We also thank Sylvette Chasserot-Golaz, C Moog, V Holl and F Cammas for technical support. This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), by grants from the Agence Nationale de Recherches sur le SIDA (ANRS) to OR and from the French Ministry of Research (‘ACI JC 5364' to OR and doctoral grant to SS) and by a doctoral INSERM/Conseil Régional d'Alsace grant to CM. FD is ‘chercheur qualifié' at the FNRS. The work in CVL's laboratory was supported by grants from the ‘Fonds National de la Recherche Scientifique' (FNRS, Belgium), the Télévie-Program of the FNRS, the ‘Action de Recherche concertée du Ministère de la Communauté Française' (ULB, ARC program no. 04/09-309), the Internationale Brachet Stiftung (IBS), the Région Wallonne-Commission Européenne FEDER (Intergenes Project, Interreg III program), the Theyskens-Mineur Foundation and the ‘Agence Nationale de Recherches sur le SIDA (ANRS, France)'. CVL is ‘Directeur de Recherches' of the FNRS. SdW is supported by a postdoctoral fellowship from the ‘Région Wallonne' (Program WALEO2 convention 616295).
References
- Ahringer J (2000) NuRD and SIN3 histone deacetylase complexes in development. Trends Genet 16: 351–356 [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005) Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45: 207–221 [DOI] [PubMed] [Google Scholar]
- Avram D, Fields A, Pretty On Top K, Nevrivy DJ, Ishmael JE, Leid M (2000) Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J Biol Chem 275: 10315–10322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE (2006) Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus–Macaque model. J Infect Dis 193: 963–970 [DOI] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM (2000) The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74: 6790–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E (2000) Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74: 8358–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Wintersberger E, Seiser C (1999) Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol 19: 5504–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF (1997) Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278: 1295–1300 [DOI] [PubMed] [Google Scholar]
- Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, Verdin E (1999) A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J Biol Chem 274: 11713–11720 [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172–183 [DOI] [PubMed] [Google Scholar]
- Hung JJ, Wang YT, Chang WC (2006) Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol Cell Biol 26: 1770–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Lazar MA (2003) The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol Cell Biol 23: 5122–5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janabi N, Peudenier S, Heron B, Ng KH, Tardieu M (1995) Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci Lett 195: 105–108 [DOI] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22: 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- Leid M, Ishmael JE, Avram D, Shepherd D, Fraulob V, Dolle P (2004) CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr Patterns 4: 733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, Leid M, Aunis D, Schaeffer E, Rohr O (2005) COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res 33: 2318–2331 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Marcello A (2006) Latency: the hidden HIV-1 challenge. Retrovirology 3: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson T, McArthur J, Siliciano RF (2000) Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol 18: 665–708 [DOI] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Rohr O, Aunis D, Schaeffer E (1997) COUP-TF and Sp1 interact and cooperate in the transcriptional activation of the human immunodeficiency virus type 1 long terminal repeat in human microglial cells. J Biol Chem 272: 31149–31155 [DOI] [PubMed] [Google Scholar]
- Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, Leid M, Schaeffer E (2003) Recruitment of Tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol 77: 5415–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senawong T, Peterson VJ, Avram D, Shepherd DM, Frye RA, Minucci S, Leid M (2003) Involvement of the histone deacetylase SIRT1 in chicken ovalbumin upstream promoter transcription factor (COUP-TF)-interacting protein 2-mediated transcriptional repression. J Biol Chem 278: 43041–43050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MD, Li J, Wong J (2005) Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol 25: 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C (2000) Role of chromatin in HIV-1 transcriptional regulation. Adv Pharmacol 48: 121–160 [DOI] [PubMed] [Google Scholar]
- Van Lint C, Amella CA, Emiliani S, John M, Jie T, Verdin E (1997) Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J Virol 71: 6113–6127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E (1996) Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15: 1112–1120 [PMC free article] [PubMed] [Google Scholar]
- Vaute O, Nicolas E, Vandel L, Trouche D (2002) Functional and physical interaction between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res 30: 475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Paras P Jr, Van Lint C (1993) Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J 12: 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, Aizawa S, Kominami R (2003) Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol 4: 533–539 [DOI] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC (2002) Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46: 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC (2006) NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylisastigui L, Coull JJ, Rucker VC, Melander C, Bosch RJ, Brodie SJ, Corey L, Sodora DL, Dervan PB, Margolis DM (2004) Polyamides reveal a role for repression in latency within resting T cells of HIV-infected donors. J Infect Dis 190: 1429–1437 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures