

Abstract
Alterations in the signaling pathways of bone morphogenetic proteins (BMPs) and activation of the ERK/MAP kinase (MAPK) pathway by growth factors have been implicated in the development and progression of prostate cancer. Smad1 acts as a substrate for MAPKs and also performs a central role in transmitting signals from BMPs. We found that BMPs/Smad1 signaling inhibits the growth of androgen-sensitive prostate cancer cells. Upon the incorporation of ERK/MAPK signals at its linker region, Smad1 physically interacts with androgen-activated androgen receptor (AR) and suppresses its functions. BMPs induce the function of Smad1 as an AR transcriptional corepressor. We demonstrated in vivo that Smad1 signaling is low in androgen-regulated growth of prostate cancer, is activated after castration, and also is decreased in hormone-independent tumors. The activation status of ERK/MAPK parallels Smad1 in the progression of prostate cancer; thus, our findings indicate a molecular basis for the integration of signals of MAPK and Smad1 in the progression and androgen regulation of prostate cancer.
Keywords: androgen receptor, BMP, MAPK, prostate cancer, Smad1
Introduction
Adenocarcinoma of the prostate is the most common malignancy of males in the US. The normal prostate gland as well as nontreated adenocarcinomas of the prostate require androgens for proliferation and maintenance of cellular viability and the production of prostate-specific androgen (PSA). Castration or anti-androgens are used to inhibit prostate cancer growth; however, either chemical or surgical castration only inhibits the progression of prostate cancer as prostate tumors eventually develop hormone-independent phenotypes and resume growth so that prostatic adenocarcinomas become life-threatening. Androgen receptor (AR), a transcription factor, mediates the action of androgen. Ligand binding results in dimerization and translocation of the AR to the nucleus where it regulates the expression of a series of prostate-specific proteins, including PSA as well as facilitates the maintenance and growth of prostate epithelial cells. Following medical or surgical castration, the activities of AR are still maintained by adrenal androgens, residual testosterone, and ligand-independent activators. Aberrant regulation of AR activity by local factors including members of the transforming growth factor-β superfamily (TGF-β) (Danielpour, 2005) and peptide growth factors that activate MAP kinase (MAPK) signaling pathways (Weber and Gioeli, 2004) may play important roles in the progression of prostate cancer.
The more than 20 types of bone morphogenetic proteins (BMPs), including growth and differentiation factors (GDFs), are the largest group of proteins in the TGF-β family. The R-Smads, including Smad1/5/8, are the major transducers of BMP receptor signals. Phosphorylation of the C-terminal of R-Smads via ligand-bound BMP type I receptor (BMPR-I) causes R-Smads to interact with Smad4 and translocate into the nucleus where they regulate transcription of specific genes (Wan and Cao, 2005). Many members in the BMP subfamily are expressed in the prostate including BMP-2, -3, -4, -6, -7 (Bentley et al, 1992), and GDF-7 (Settle et al, 2001). Prostate-derived factor (PDF) (Paralkar et al, 1998), also called placental BMP (Hromas et al, 1997) or GDF-15/MIC-1 (Bottner et al, 1999), is expressed strongly and specifically in the prostate and placenta. The BMPs play critical roles in maintenance of prostate function and male reproductive track development. For example, BMP-4 restricts ductal budding and branching morphogenesis in the developing prostate (Lamm et al, 2001), and GDF-7 is required for development of the seminal vesicle (Settle et al, 2001). In addition, the mutant Smad1 that lacks MAPK phosphorylation sites at the linker region causes male reproductive defects in transgenic mice (Aubin et al, 2004).
Alterations in the BMP signaling pathway are frequently found in human prostate cancers. BMP-6 and BMP-7 are upregulated during progression of prostate cancer (Barnes et al, 1995; Hamdy et al, 1997; Yang et al, 2005). Specifically, PDF expression is significantly higher in prostate cancer than in normal prostatic tissues (Nakamura et al, 2003). However, the components of the BMP signaling, such as the BMP receptors, BMPR-II, BMPR-1A, and BMPR-1B; and Smad proteins, Smad8 and Smad4, are frequently lost in high-grade prostate cancer (Kim et al, 2000, 2004) or decreased during the progression to the androgen-independent phenotype (Horvath et al, 2004). Although the functions of BMPs and PDF in prostate cancer, especially the role of Smad signaling, have not been defined, a limited amount of information is available on the effects of specific BMPs on prostate cell lines. For example, although BMP-2 inhibited the growth of androgen-sensitive (AR positive) LNCaP cells, it did not have any effect on the proliferation of androgen-insensitive (AR deficient) PC3 and DU145 cells (Ide et al, 1997). Similarly, BMP-4 inhibited the growth of LNCaP but not PC3 cells (Brubaker et al, 2004). It is important to note that LNCaP cells expressed BMP receptors and have activated Smad signaling in response to BMP stimulation (Brubaker et al, 2004; Yang et al, 2005). The obvious question is whether or not the results of signaling of BMPs/Smad require the incorporation of AR signaling pathways in prostate cancer cells? Given that the expression of BMP-7 and PDF are androgen inducible (Paralkar et al, 1998; Thomas et al, 1998), it is of interest to understand the inter-relationship between BMP/Smad signaling and the AR axis in prostate cancer.
Smad1 both performs a central role in transmitting signals from BMPs and acts as a substrate for MAPKs. MAPK phosphorylation of Smad1 linker has been shown to antagonize the effects of C-terminal phosphorylation of Smad1 by BMPs (Kretzschmar et al, 1997a; Pera et al, 2003; Kuroda et al, 2005); however, both Smad1 linker and C-terminal phosphorylation sites are required by the germ-cell lineage, suggesting that MAPK-dependent Smad1 phosphorylation may not only serve to inhibit BMP signaling but also may be incorporated in BMP/Smad1 function (Aubin et al, 2004). The interdependent functions of MAPK have been observed with some other Smad proteins (Adachi-Yamada et al, 1999; Engel et al, 1999). We report that Smad1 mediates BMP signaling, including the PDF signal in prostate cancer cells, and inhibits growth of these cells through BMP/androgen-induced interaction of Smad1 with AR. Importantly, MAPK phosphorylation of Smad1 linker is necessary for the inhibitory effects of Smad1. We demonstrate in vivo that the activation status of MAPK–ERK parallels with Smad1 in the clinical course of prostate cancer progression as demonstrated by the CWR22 human prostate cancer xenograft. Thus, our results suggest a molecular basis for the incorporation of MAPK signals within Smad1 signaling in the progression and androgen regulation of prostate cancer.
Results
Smad1 signaling inhibits growth of prostate cancer cells and androgen receptor is required for the inhibition
To examine the possible role of BMPs in prostatic adenocarcinoma, we examined if PDF could induce Smad1 phosphorylation like BMP-2. LNCaP cells, stably expressing Smad1, either were treated with PDF, BMP-2, or vehicle for 30 min or were transfected with a constitutively active BMP type 1 receptor (caALK6). Phosphorylated Smad1 was measured by Western blotting with antibodies against either Smad1 or phosphorylated Smad1. As shown in Figure 1A, PDF induced levels of phosphorylation of Smad1 comparable to the levels induced by BMP-2 and caALK6. Noggin, a BMP antagonist, blocked both PDF- and BMP-induced phosphorylation of Smad1 (Figure 1A, lanes 5 and 6). The effect of PDF on the functional complex formation of endogenous phosphorylated Smad1 with Smad4 also was examined. The cell lysates of LNCaP cells that were either treated with PDF or BMP-2 or transfected to overexpress caALK6 were immunoprecipitated with an antibody against Smad1 and blotted with Smad4. The results were similar to BMP-2 treatment and caALK6 transfection; PDF induced a Smad1/Smad4 complex formation (Figure 1B). These results suggest that PDF may act as a ligand to BMP receptors in LNCaP cells (Paralkar et al, 1998).
Figure 1.
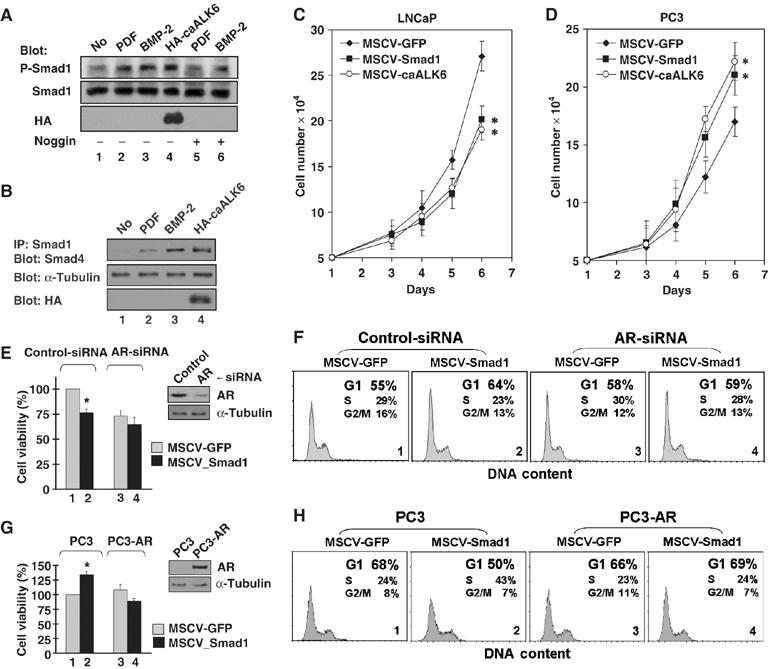
Smad1 inhibits growth of prostate cells and AR is required for the inhibition. (A) PDF induced phosphorylation of Smad1. LNCaP-Smad1 cells were transduced with MSCV-HA-caALK6 retrovirus or control vector for 48 h, and then treated for 20 min with PDF (300 ng/ml) or BMP-2 (100 ng/ml) with or without Noggin (200 ng/ml). Total cell lysates were immunoblotted with antibodies as indicated. (B) PDF induced interaction of endogenous Smad1 and Smad4. LNCaP cells were transduced with caALK6 retrovirus or control vector for 48 h, then treated for 1 h with PDF (300 ng/ml) or BMP-2 (100 ng/ml). The cellular extracts were immunoprecipitated with anti-Smad1 antibody and associated Smad4 was detected by immunoblotting with anti-Smad4 antibody. The amounts of caALK6 and α-tubulin were determined by immunoblotting cell lysates. (C, D) CaALK6/Smad1 inhibits AR-positive LNCaP cellular growth, but promote AR-deficient PC3 cellular growth. Cells were transduced with caALK6, Smad1, or GFP retrovirus. *P<0.01 (Smad1 versus GFP; caALK6 versus GFP). (E, F) Knockdown of endogenous AR in LNCaP cells abrogated the statistical significance of Smad1-induced growth inhibition and cell cycle arrest in the G1 phase. The cells transfected with either control-siRNA or AR-siRNA and transduced with control MSCV-GFP or MSCV-Smad1 retroviruses, and then subjected to a viability assay (E) or flow cytometry analysis (F). The expression level of AR was determined by immunoblotting. (E: inset). (G, H) Smad1 retained the potential to inhibit cellular growth and induce G1 arrest in PC3 cells induced to express AR. PC3 and PC3-AR cells transduced with MSCV-GFP or MSCV-Smad1 were treated with 10 nM DHT, and then subjected to a viability assay (G) or an analysis by flow cyto-metry (H). Expression level of AR was determined by immunoblotting (G: inset). *P<0.01 (Smad1 versus GFP). Representative data are shown in (F) and (H).
To understand the mechanism of BMP-induced growth inhibition, we examined the effect of Smad1 signaling on growth of AR-positive LNCaP or AR-deficient PC3 cells. The cells were transduced for retroviral MSCV-mediated expression of either Smad1 or caALK6, and GFP as a control. In a medium containing 10% fetal bovine serum (FBS), expression of either Smad1 or caALK6 inhibited growth of LNCaP cells in comparison with the GFP control (Figure 1C). In contrast, expression of either Smad1 or caALK6 promoted growth of PC3 cells (Figure 1D). Given that BMPs inhibit the growth of LNCaP cells, but do not inhibit the growth of PC3 cells (Ide et al, 1997; Brubaker et al, 2004), our results indicate that the inhibitory effect of BMP is mediated by Smad1 signaling and that the BMP/Smad1-induced inhibition may be AR dependent.
To test the effect of AR, endogenous AR expression in LNCaP cells was eliminated with AR-siRNA, and the influence on cell proliferation was determined by the analysis of cell viability (Figure 1E). In comparison with the control (Figure 1E, lanes 1 and 2), knockdown of AR abrogated the Smad1-induced growth inhibition (Figure 1E, lanes 3 and 4). FACS analysis revealed that Smad1 induced cell cycle arrest in the G1 phase (G1/GFP=55.12±0.86%; G1/Smad1=63.89±0.63%) (Figure 1F, lanes 1 and 2). When expression of AR was inhibited, the effect of Smad1 (G1/Smad1=59.31±0.67%) was close to the control (G1/GFP=58.27±0.48%) (Figure 1F, 3 and 4), demonstrating that Smad1 induced growth inhibition by arresting cells in G1 and that AR was required for this inhibition. A similar experiment was conducted in AR-deficient PC3 cells after the cells were induced to express AR. In this particular state, when AR is expressed and the cells are treated with 10 nM DHT, Smad1 retained the potential to inhibit the growth of PC3 cells (Figure 1G, lanes 1 and 4). FACS analysis demonstrated that Smad1 decreased G1 arrest (G1/GFP=67.88±0.55%; G1/Smad1=50.09±0.78%) in PC3 cells, but in PC3-AR cells slightly increased G1 arrest (G1/GFP=65.83±0.77%; G1/Smad1=68.75±0.83%) (Figure 1H, 1 and 4). In this particular case, forced activation of AR signaling apparently contributed to the inhibitory effect of Smad1 in PC3 cells. Taken together, these results demonstrate that AR signaling is crucial for the inhibitory effect of BMP/Smad1 in prostate cancer cells.
Both androgen and BMP induce the interaction between Smad1 and AR
The simplest explanation for the interactions between the BMP/Smad1 and androgen/AR signaling pathways might be a direct interaction between pathway-specific proteins. We therefore examined the possible interaction between Smad1 and AR by cotransfection of HEK293 cells with AR and flag-tagged plasmids for Smad1, TGF-β-specific Smad2, or the antagonists, Smad6 or Smad7. In the presence of DHT, the cellular lysate was immunoprecipitated to isolate flagged Smads and the associated AR was immunoblotted. Only Smad1, not other Smads, specifically co-precipitated with AR (Figure 2A and B). Conversely, AR was immunoprecipitated first and the flagged Smad1 subsequently was demonstrated by immunoblotting (Figure 2C). The interaction was further examined with endogenous Smad1 and AR. LNCaP cells were treated with BMP-2 or DHT, or transfected with HA-caALK6. Endogenous Smad1 was immunoprecipitated and associated AR was immunoblotted (Figure 2D). A Smad1 interaction with AR was not detected, but DHT, BMP-2, and caALK6 each separately induced the interaction. Pretreatment with Noggin consistently attenuated the BMP-2-induced interaction. In addition, increased expression of Smad6 led to loss of the Smad1 interaction with AR in a dose-dependent manner (Figure 2E). As Smad6 inhibits nuclear translocation of Smad1 by directly binding to the C-terminal-phosphorylated Smad1 in the cytoplasm (Hata et al, 1998), this result indicates that BMP-activated Smad1 enhances interaction with AR. In summary, both androgen and BMP induce the interaction between Smad1 and AR.
Figure 2.
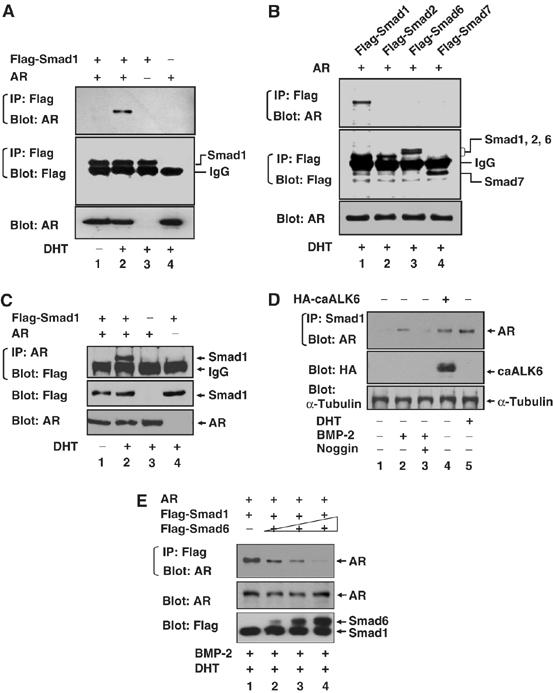
Smad1 physically interacts with the AR in response to both androgen and BMP. (A, B) Smad1 specifically interacts with the AR in response to DHT. HEK293 cells were cotransfected with plasmids as indicated, and then treated with 10 nM DHT for 2 h or left untreated (ethanol as control). Cell extracts were immunoprecipitated by anti-flag antibody and associated AR was immunoblotted with anti-AR antibody. The amount of Smads in the immune complexes and AR in total lysates was determined by immunoblotting. (C) Inverted experiment of (A, B) in which cell extracts were immunoprecipitated with anti-AR antibody and immunoblotted with anti-flag antibody. (D) Smad1 endogenously interacts with AR in response to both androgen and BMP. LNCaP cells were transduced with caALK6 (lane 4) retrovirus or control vector for 48 h, then treated with 10 nM DHT (lane 5), 100 ng/ml BMP-2 (lanes 2 and 3), 200 ng/ml Noggin (lane 3), or left untreated before immunoprecipitation of Smad1. The associated AR was detected by anti-AR antibody. (E) Smad6 disrupts Smad1 interaction with AR. HEK293 cells were cotransfected with AR and Flag-Smad1, or in combination of increasing amount of Flag-Smad6 (lanes 2, 3, and 4). Cells were treated with 100 ng/ml BMP-2 for 2 h, followed by addition of 10 nM DHT. Cell lysates were immunoprecipitated by flag antibody and associated AR was detected by anti-AR antibody.
Phosphorylation of Smad1 linker region is necessary for Smad1/AR interaction
We examined the in vitro effects of BMP and androgen signaling on Smad1 interaction with AR. A yellow fluorescent protein (YFP)-based protein-fragment complementation assay (PCA) was employed (Remy et al, 2004) in which an YFP1 fragment was fused to the N-terminal of Smad1 and an YFP2 fragment was fused to the C-terminal of AR. The PCA strategy facilitates visualization of the AR–Smad1 interaction in living cells when the interaction brings the complementary fragments of the YFP into close proximity (Figure 3F). When both YFP1-Smad1 and AR-YFP2 were expressed, YFP fluorescence was induced in cells treated with BMP-2 or DHT (Figure 3A, panels 1–3), or transfected with caALK6 (Figure 3A, panels 6 and 7). Noteworthy, DHT had a much stronger effect than BMP-2 or caALK6, but the effect was suppressed by MAPK inhibitors PD98059 or SB203580 (Figure 3A, panels 4 and 5). MAPK is known to phosphorylate four serine residues at the Smad1 linker region (Kretzschmar et al, 1997a); thus, the four serine residues were mutated in the YFP1-Smad1 construct (LM-Smad1) (Figure 3F). In addition, we mutated three serine residues at the C-terminal of Smad1 (CM-Smad1) so that Smad1 cannot be phosphorylated by BMPs (Kretzschmar et al, 1997b) (Figure 3F). As shown in Figure 3C, LM-Smad1 disrupted the DHT-induced AR–Smad1 interaction as determined by the PCA method, but CM-Smad1 still produced fluorescence similar to wild-type Smad1. The observations were quantitated (Figure 3B and D) and the immunoprecipitation assay demonstrated that LM-Smad1 lost its interaction with AR (Figure 3E, lanes 3 and 6) in the presence of DHT and BMP-2, whereas CM-Smad1 still interacted with AR (Figure 3E, lanes 4 and 7). These results demonstrate that phosphorylation of the linker region by MAPK is necessary for the AR–Smad1 interaction.
Figure 3.

Phosphorylation of Smad1 linker region is necessary for Smad1/AR interaction. (A) Both androgen and BMP induce the AR–Smad1 interaction in the YFP PCA assay. HEK293 cells were transfected with YFP1-WT-Smad1 and AR-YFP2 plasmids illustrated in (F) or cotransfected with HA-caALK6 for 48 h, then treated with 100 ng/ml BMP-2 or 10 nM DHT for 2 h, or in combination with 10 μM PD98059 or 50 μM SB203580. Ethanol was used as a negative control. Upper: fully confluent cells. Lower: reconstituted YFP fluorescence. Protein expression was determined by immunobotting. (C) LM-Smad1 disrupted the DHT-induced AR–Smad1 interaction in the YFP PCA assay, but CM-Smad1 still produced fluorescence similar to WT-Smad1. (B, D) The relative amount of reconstituted YFP in intact cells was measured by fluorometry. (E) LM-Smad1 does not interact with AR in the immunoprecipitation assay. LNCaP cells were transfected with plasmids as indicated for 48 h, and then treated with 100 ng/ml BMP-2 or 10 nM DHT for 2 h. Cell extracts were immunoprecipitated by anti-flag antibody and associated AR was immunoblotted with anti-AR antibody. (F) Diagram of Smad1 phosphorylation mutant proteins and a cartoon demonstrating the principle of the YFP PCA assay.
Smad1 inhibits androgen-induced transcription
The effect of Smad1 on AR-mediated transactivation was examined using luciferase reporters driven by androgen response elements (ARE) or native PSA promoter-bearing AREs which were transfected in normal prostate-epithelial cells, RWPE-1, or prostate epithelial cancer cells, LNCaP or PC3-AR. Both Smad1 or caALK6 independently repressed androgen-induced transactivation, and expression of both enhanced the repression (Figure 4A). Because Smad6 blocks the interaction between Smad1 and AR, Smad6 was coexpressed with ARE reporter to confirm the Smad1-mediated repression of AR transactivation; as expected, Smad6 blocked the repression (Figure 4B). The effect of Smad1 phosphorylation mutants on AR transactivation activity was evaluated with a PSA luciferase reporter (Figure 4C). Smad1 and CM-Smad1, but not LM-Smad1, inhibited DHT-induced transactivation, indicating Smad1 interaction with AR contributes to the inhibition.
Figure 4.
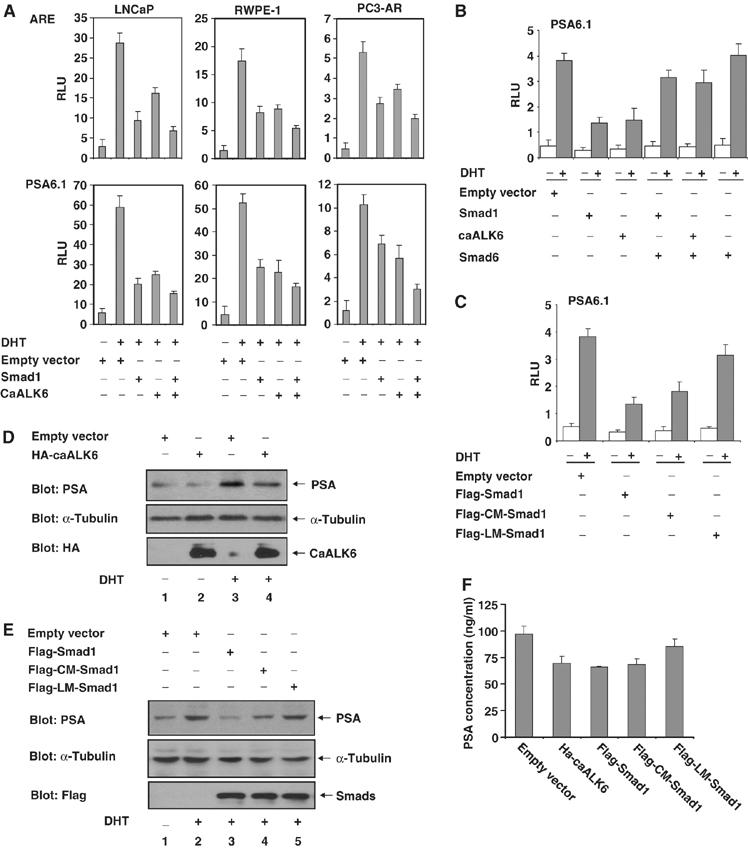
Smad1 inhibits androgen-induced transcription. (A) caALK6/Smad1 represses androgen-induced AR transactivation activity. RWPE-1, LNCaP, and PC-AR cells were transfected with the indicated plasmids for 24 h, and then treated with 10 nM DHT for 24 h. Ethanol was used as the negative control for DHT. Luciferase activities were determined by ARE (upper) or PAS6.1 (lower) reporters. RLU: relative luciferase units. (B, C) Effects of Smad6 (B) or Smad1 phosphorylation mutants (C) to AR transactivation activity were determined by PSA6.1 luciferase reporter in LNCaP cells. (D, E) CaALK6/Smad1 inhibits PSA expression. LNCaP cells transfected with the indicated plasmids were treated with 10 nM DHT for 48 h or left untreated. PSA in cell lysates were determined by immunoblotting with anti-PSA antibody. (F) CaALK6/Smad1 inhibits PSA secretion. Cell culture mediums of (D, E) were collected and PSA concentration was assayed by ELISA.
The effect of Smad1 on the endogenous expression of PSA in LNCaP cells was evaluated using Western blotting (Figure 4D and E) and ELISA analysis (Figure 4F) of PSA in both cell lysates and culture mediums. CaALK6 (Figure 4D), Smad1, and CM-Smad1 (Figure 4E) inhibited PSA expression induced by DHT, whereas LM-Smad1 did not inhibit expression of PSA. In summary, Smad1 inhibits AR-mediated transcription and phosphorylation of the Smad1 linker region is required for the inhibition. Significantly, the inhibitory effect of CM-Smad1 is apparently weaker than wild-type Smad1 (Figure 4C, E and F), indicating that phosphorylation at the C-terminal also contributes to the inhibition.
Smad1 functions as an AR transcription corepressor
To understand the mechanism of the inhibitory effect of Smad1, we attempted to identify the cellular localization of the AR–Smad1 interaction. YFP1-WT-Smad1 and YFP2-AR were coexpressed with and without DHT, or with transfected caALK6 in HEK293 cells. The YFP fluorescence generated by the DHT-induced Smad1/AR interaction was visualized using confocal microscopy (Figure 5A). Smad1 primarily associated with AR in the cytoplasm when cells were treated with DHT; however, coexpression of caALK6 resulted in the interaction moving into the nucleus. Intriguingly, the cytoplasmic YFP fluorescence is mainly concentrated on the membrane-like structures at the perinuclear area where compartmentalized Ras/MAPK signaling is activated (Mor and Philips, 2006); however, in the cells transfected with caALK6, the nuclear fluorescence was restricted to the DAP1-stained DNA sites. These results support the concept that C-terminal phosphorylation by caALK6 induces Smad1 translocation into the nucleus and the association of Smad1 with AR on DNA. Because AR is a nuclear receptor and its active form mainly is localized to the nucleus, the result accounted for the BMP/caALK6-induced Smad1/AR interaction (Figures 2D, 3A, and E). These data indicate that DHT induces Smad1 interaction with AR in the cytoplasm, whereas BMP causes the interaction to move into the nucleus.
Figure 5.
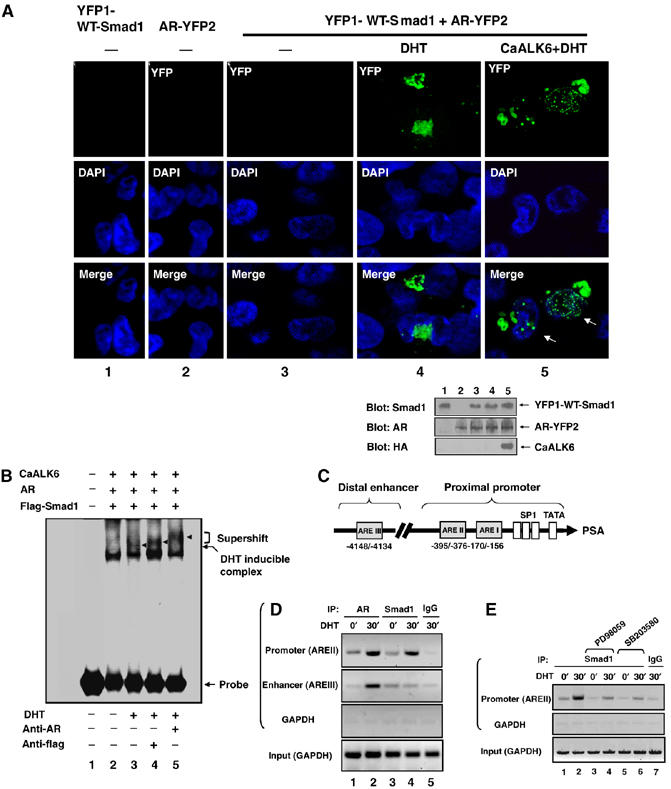
Smad1 functions as an AR transcription corepressor. (A) Expression of caALK6 enables the Smad1–AR complex to move into the nucleus. HEK293 cells were cotransfected with plasmids as indicated, and then treated with 10 nM DHT for 1 h. Ethanol was used as the negative control. The cells were stained with DAPI and visualized through reconstituted YFP under confocal microscopy. Arrows indicate reconstituted YFP on DAPI-stained DNA sites. Expression levels of protein are shown in lower panel. (B) Smad1 forms a complex with AR on ARE. LNCaP cells were cotransfected with plasmids as indicated, then treated with 10 nM DHT for 30 min, and the nuclear extracts were subjected to mobility shift analysis with 32P end-labeled ARE probe. Antibodies against AR or flag were added as indicated. (C) A schematic representation of the PSA gene enhancer/promoter showing location of AREs (modified from Kim and Coetzee, 2004). (D) Smad1 forms a complex with AR on the endogenous PSA gene promoter in response to androgen. LNCaP cells were treated with 10 nM DHT for 30 min and subjected to ChIP assays with the indicated antibodies and PCR primers. (E) MAPK inhibitors disrupt the formation of Smad1/AR complex on PSA gene promoter. Inverted gel pictures were shown (D, E).
To understand further the interaction of Smad1/AR complex with nuclear DNA, we determined whether Smad1 would be recruited to ARE in prostate cancer cells. AR and flagged Smad1 plasmids were cotransfected with caALK6 in LNCaP cells. Nuclear extracts of cells were incubated with the consensus ARE as a DNA probe in a gel-shift assay (Figure 5B). A shifted band induced by androgen was observed in the nuclear extract after 30 min of treatment with DHT (Figure 5B, lane 3). The induction by DHT of a Smad1 complex with AR on an ARE was demonstrated by supershifted bands selectively identified by antibodies against flag or AR (Figure 5B, lanes 4 and 5). This observation was further confirmed using chromatin immunoprecipitation (ChIP) assays in which lysates from LNCaP cells were treated with and without DHT and were immunoprecipitated subsequently with antibodies to AR or Smad1. The resulting precipitates were used for PCR amplification of AREs in the PSA promoter (Figure 5C) to examine formation of the AR–Smad1 complex on DNA. After 30 min exposures to androgen, endogenous AR was bound to AREII in the proximal promoter and AREIII in the distal enhancer (Figure 5D, lanes 1–2). An important observation is that Smad1 was recruited to the AREII after 30 min of DHT treatment, but not to the AREIII (Figure 5D, lanes 3 and 4), and AREII is the specific site on which AR corepressors prefer to bind in response to androgen antagonists (Shang et al, 2002).
The effect of MAPK on the formation of Smad1–AR complex on AREII was examined with MAPK inhibitors. As shown in Figure 5E, both PD98059 and SB203580 inhibited the formation of the complex on DNA. These results, together with the inhibitory effects of Smad1, demonstrate that BMP and androgen recruit Smad1 as an AR transcriptional corepressor; MAPK is required for recruitment.
Castration activates Smad1 and ERK phosphorylation and nuclear translocation
Given the essential role for MAPK in facilitating the Smad1 effect on AR, we predicted that MAPK was involved in Smad1 signaling that regulated AR activity. To test this hypothesis, we examined the activation status of Smad1 and ERK/MAPK in a xenograft model of human prostatic adenocarcinoma. This xenograft, designated CWR22, when grown in male nude mice, exhibits androgen-dependent growth and PSA secretion (Wainstein et al, 1994; Nagabhushan et al, 1996); androgen withdrawal causes these CWR22 tumors to shrink and serum PSA levels to decline dramatically (Nagabhushan et al, 1996). Importantly, the CWR22 model of prostate cancer simulates the clinical course of prostate cancer in that a significant number of CWR22 tumors relapse to an androgen-independent phenotype many months after castration (Nagabhushan et al, 1996; Myers et al, 2001) and these relapses are preceded by increasing levels of PSA (Gregory et al, 1998). A tumor derived from one such originally relapsed tumor, referred to as CWR22R, grows readily in castrated nude mice (Wainstein et al, 1994; Myers et al, 1999a).
In CWR22 cells growing in intact (not castrated) nude mice, the protein expression of Smad1 was low to moderate and largely confined to the cytoplasm (Figure 6A) and the expression of C-terminal-phospholated Smad1 (P-Smad1) was almost nondetectable (Figure 6B). Castration significantly enhanced expression of Smad1 (Figure 6F) in the cytoplasm and nuclei, and the phosphorylation of Smad1 (P-Smad1) in the nuclei (Figure 6G), suggesting that androgen negatively regulates BMP signaling and castration releases this restraint. However, castration-induced Smad1 activation was not observed in the relapsed androgen-independent CWR22R tumors (Figure 6M and N), suggesting a potential role of decreased Smad1 signaling in the androgen-independent growth phase of prostate cancers. A similar observation was obtained for ERK which can directly phosphorylate Smad1 linker (Kretzschmar et al, 1997a). Specifically, CWR22 cells growing in intact male nude mice exhibited moderate expression of ERK1, ERK2, and phosphorylated ERK1/2 (P-ERK1/2); this expression was largely confined to the cytoplasm for each form of ERK. Castration enhanced expression of ERK1, ERK2, and P-ERK1/2, and with castration, the expression of P-ERK1/2 moved into the nuclei (Figure 6C–E and H–J). Importantly, nuclear accumulation of P-ERK1/2 occurred within the area associated with high levels of P-Smad1 (Figure 6K and L), indicating a simultaneous activation of Smad1 and ERK in response to castration. Like Smad1, castration-induced activation of ERK1/2 was not observed in the CWR22R tumors (Figure 6O–Q). In CWR22R cells, the expressions of Smad1, ERK2, and P-ERK1/2 actually decreased to levels similar to those seen in the CWR22 model of androgen-dependent growth although the expression was more nuclear. These results suggest that Smad1 signaling is low during the growth of prostate cancer, is activated after castration, and decreases during the growth of hormone-independent tumors; this suggests an important role for BMP/Smad1 signaling in suppression of tumor growth in response to androgen withdrawal. Importantly, the activation status of ERK/MAPK parallels Smad1 during these same states, suggesting a tight integration of ERK/MAPK signaling within the Smad1 signaling pathway in hormonally regulated control of the growth of prostate cancer.
Figure 6.
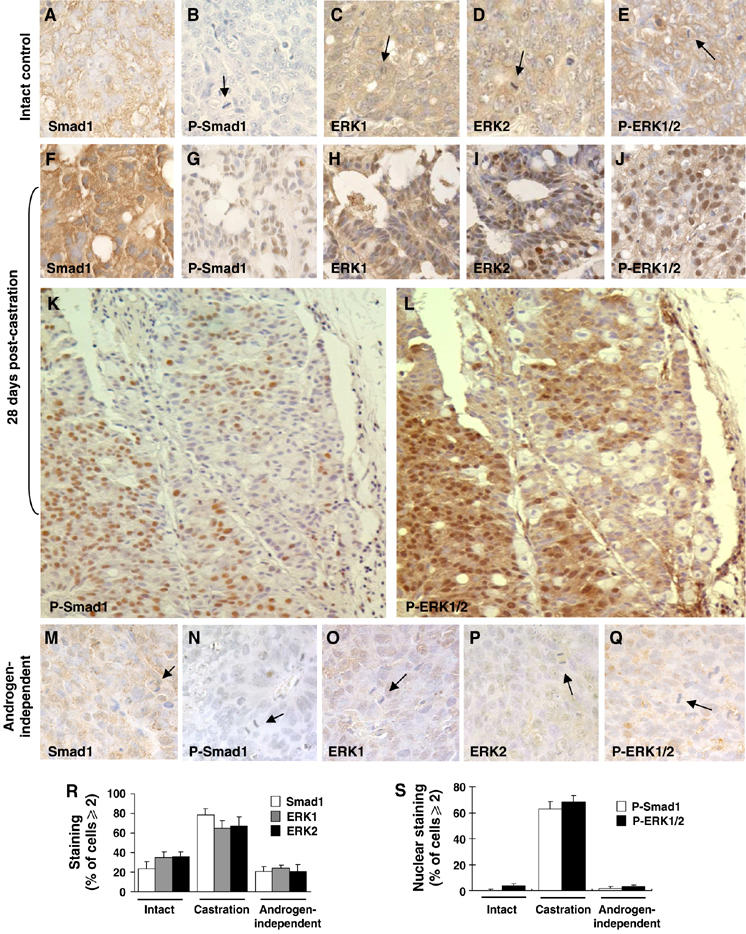
Immunohistochemical analysis of Smad1 and ERK activation statuses in CWR22 and CWR22R tumors. Smad1, P-Smad1, ERK1, ERK2, and P-ERK1/2 antigens are indicated by immunoreactivity (brown precipitate). Nuclei were lightly counterstained with hematoxylin (blue–purple areas). Arrows indicate mitotic cells. (A–E) Use of the CWR22 tumor as an intact control. (F–L) A CWR22 tumor at 28 days post-castration. (K–L) Colocalization of P-Smad1 and P-ERK1/2 in adjacent slides of a CWR22 tumor at 28 days post-castration. (M–Q) An androgen-independent CWR22R tumor. (R–S) Quantitative analysis of the expression of Smad1 and ERK, and the nuclear expression of P-Smad1 and P-ERK1/2 in CWR22 or CWR22R tumors.
Discussion
Our results demonstrate that Smad1 modulates BMPs and MAPK signals that restrain androgen-induced mitogenic activity in the androgen-regulated control of the growth of prostate cancer. Upon the incorporation of ERK/MAPK signals at its linker region, Smad1 physically interacts with androgen-activated AR and suppresses its functions. The interaction occurs in the cytoplasmic perinuclear compartments or specific sites within the nuclear DNA. This response depends upon upstream BMPs including PDF signals, which regulate the recruitment of Smad1 to the AR/DNA complex in the nucleus as a transcriptional corepressor. The interactions in the cytoplasm also contribute to the inhibitory effect of Smad1 on AR function by occupying the active pool of AR. Therefore, we propose a simple mechanism of Smad1-mediated negative regulation of AR activity in control of prostate cell growth (Figure 7). In this working model, Smad1 is located at the center of multiple signaling pathways. BMPs restrain the mitogenic effects of androgens by inducing Smad1 translocation to the nucleus via phosphorylating its C-terminal. Linker-phosphorylated Smad1 occupies some androgen-activated AR to balance androgen-induced maintenance of prostatic cellular growth. Phosphorylation of Smad1 linker is necessary for Smad1 interaction with AR in the cytoplasm as well as the nucleus, thus MAPK signals are incorporated in the Smad1-mediated control of the growth of prostate cells.
Figure 7.
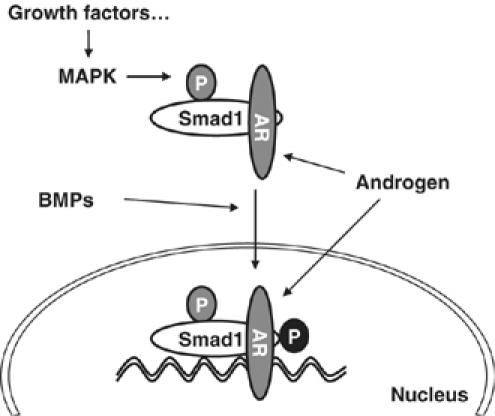
Model of the integration of BMPs and MAPK signaling pathways at the level of Smad1 in control of AR transactivation activity.
The Smad1 signaling pathway apparently exerts inhibitory effects on androgen-induced AR transactivation and inhibits the expression and secretion of PSA which is a clinical biomarker released by malignant prostate cells into interstitial spaces (Grizzle et al, 2005); thus BMP/Smad1 signaling may inhibit the androgen-dependent growth of prostate tumors. Evidence is provided by the activation of Smad1 signaling in CWR22 prostate tumors during their regression induced by the castration of the host mice. In contrast, reduction of Smad1 signaling in tumors of intact mice and in relapsed CWR22R androgen-independent tumors correlates directly with tumor growth. These in vivo data, together with in vitro studies with prostate cancer cell lines, suggest a role of BMP/Smad1 signaling in modulating the growth of prostate cells. However, the Smad pathway may not be a unique pathway by which BMPs regulate cellular growth, as other signaling pathways can either be induced by BMPs, or can modify the initial BMP-induced Smad signaling (Wan and Cao, 2005). In the progression of prostate cancer, loss of BMP receptors or Smad activities could generate a diverse outcome that is dominated mainly by signaling pathways unrelated to Smad1. The upregulation of BMPs including PDF in prostate cancer may be a cellular response due to feedback from decreased activity of BMP/Smad/signaling; this might elicit an undesired oncogenic effect (Yang et al, 2006) in contrast to the antiproliferative effect mediated by Smad1 signaling (Brubaker et al, 2004). Accordingly, high expression of TGF-β and loss of TGF-β receptor expression have been associated with a bad prognosis in prostate cancer patients (Wikstrom et al, 2001).
MAPK phosphorylation of Smad1 linker has been shown to antagonize the effects of Smad1 C-terminal phosphorylation (Kretzschmar et al, 1997a; Pera et al, 2003; Kuroda et al, 2005); however, even though ERK/MAPK is activated in the CWR22 tumors from the castrated mice, unexpectedly Smad1 is still C-terminal-phosphorylated and P-Smad1 still accumulates in nuclei (Figure 6K and L). These results support the concept that MAPK phosphorylation of Smad1 may not only serve to inhibit BMP signaling but also in some cases may be incorporated in BMP/Smad1 signaling. Mutations of the MAPK phosphorylation sites in Smad1 cause male reproductive defects in the transgenic mice (Aubin et al, 2004). Given the pivotal role of androgens in the development of the male reproductive tract and the critical role of Smad1 in coordinating the signals of AR and other growth factors that activate MAPK, the Smad1-mediated balance of androgen and MAPK signals may represent a fundamental mechanism in the development of the overall male reproductive track including the control of prostate growth.
Our results indicate that the cytoplasmic Smad1/AR interaction induced by DHT occur in specific perinuclear compartments (Figure 5A). Given that RAS/MAPK signaling is activated at intracellular compartments to which MAPK substrates are recruited including the Golgi apparatus as well as the ER complex and the endosomes (Chiu et al, 2002; Mor and Philips, 2006; Quatela and Philips, 2006), these organelles may act as a scaffold to which MAPK recruits the AR–Smad1 complex. This cytoplasmic interaction not only may account for the inhibitory effect of Smad1 to AR transactivation but also this pattern suggests an antagonistic action of androgen to the BMP/Smad1 signaling due to androgen-induced interaction occupying a large proportion of the cytoplasmic pool of Smad1. Specifically, we observed that androgen strongly inhibited nuclear translocation of Smad1 and Smad1-mediated transcriptional activation (data not shown). Consistently, DHT has been shown to inhibit TGFβ/Smad3 signaling in prostate cancer cells (Chipuk et al, 2002).
Given that a BMP-induced Smad1–AR complex inhibits the function of AR in the nucleus, androgens may reduce this inhibition by blocking BMP/Smad1 signaling and causing an accumulation of Smad1 in the cytoplasm. If this antagonism occurs and if it is mediated by MAPK, then MAPK should be present in the nucleus when BMP/Smad1 signaling is activated and in the cytoplasm when androgen is suppressing BMP/Smad1 signaling. As we observed in vivo in the CWR22 xenograft model, Smad1 was distributed in the cytoplasm of androgen-dependent CWR22 cells (Figure 6A) accompanying the inactivation of BMP/Smad1 signaling (Figure 6B); correspondingly, ERK was moderately active in the cytoplasm (Figure 6C–E), supporting that ERK/MAPK mediates Smad1/AR interaction in the cytoplasm to suppress BMP/Smad1 signaling and facilitating androgen-modulated growth. These observations in the cytoplasm correlate with the opposing activities of BMP and Ras/ERK/MAPK at the level of Smad1 phosphorylation (Kretzschmar et al, 1997a). In contrast, when BMP/Smad1 signaling was strongly activated by castration, both P-Smad1 and P-ERK1/2 were expressed in the nuclei (Figure 6K and L). These observations support our proposed mechanism (Figure 7) that ERK/MAPK signals modulate Smad1 signaling to regulate AR function in the nucleus. Also, it has been reviewed that transcriptional factors are important ERK/MAPK targets in the nucleus (Chang and Karin, 2001). Although the observations from tissues do not necessarily mean that the Smad1 linker is phosphorylated, they provide the evidence that ERK/MAPK expression parallels with Smad1 signaling in the nucleus to repress tumor growth following androgen withdrawal.
Our results indicate that castration activates ERK and Smad1 in the CWR22 human prostate cancer xenograft; however, when a CWR22 tumor relapses after castration and its growth becomes androgen-independent; P-Smad1 and P-ERK decreases to levels similar to those observed in androgen-dependent tumors before castration. These results suggest a potential role of ERK/MAPK and Smad1 in the development of androgen independence of prostate cancers. Androgen appears to be required in the control of the coordination among BMPs, androgen, and MAPK signals. Lowering the androgen level by castration breaks the balance and results in the simultaneous elevation of ERK and Smad1 signaling (Figure 6K and L). Reconstitution of the balance may diminish the requirement for androgen to support the growth of prostate cancer cells. Constitutive activation of MAPK signaling may play a causal role in moving prostate cancer cells towards androgen-independent growth (Gioeli et al, 2001; Weber and Gioeli, 2004). Once the tumor begins to grow in a low-androgen environment, Smad1 loses its function as an AR corepressor and the need for incorporation of MAPK signals is reduced. Subsequently, the hormone-independent growth of the tumor is characterized by reactivation of AR (Chen et al, 2004) and increasing levels of PSA (Gregory et al, 1998). Thus, increasing the recruitment of Smad1 to AR in hormone-independent tumors might become a therapeutic and/or diagnostic approach for hormonally independent prostate cancer.
Materials and methods
Plasmids and antibodies
The plasmids, Flag-Smad1/pRK5, Flag-Smad2/pRK5 (Zhang et al, 1996), Flag-Smad6/pcDNA3, Flag-Smad7/pcDNA3 (Bai et al, 2000), YPF1-zipper/pcDNA3.1, zipper-YFP2/pcDNA3.1 (Remy et al, 2004), AR/pCMV (Lubahn et al, 1988), HA-caALK6/pcDNA3 (Wieser et al, 1995), and luciferase reporters PSA6.1 (Cleutjens et al, 1997) have been described previously. GFP, Flag-Smad1, and AR HA-caALK6 were introduced into pMSCVneo retroviral expression vectors (BD Biosciences). The mutants Flag-LM-Smad1 and Flag-CM-Smad1 were generated by primer-mediated PCR mutagenesis and verified via DNA sequencing. YFP1-CM-Smad1, YFP1-LM-Smad1, YFP1-WT-Smad1, and AR-YFP2 were generated by replacing the zippers in YFP1-zipper or zipper-YFP2. Protein expression was examined using antibodies specific to Flag (Sigma), HA (Covance), Smad1, AR, PSA, α-Tubulin (Santa Cruz), and P-Smad1 (Ser463/465) (Cell Signaling).
Cells, transfection, and reagents
The human prostate cancer cell lines, LNCaP, PC3, and RWPE-1, were obtained from American Type Culture Collection (ATCC) and maintained in RPM1 1640 supplemented with 10% FBS (Invitrogen). Human embryonic kidney HEK293 cells, also obtained from ATCC, were maintained in DMEM with 10% FBS. PC3-AR and LNCaP-Smad1 cells were generated by stable expression of AR and Smad1 with retrovirus transduction. The transfection of DNA plasmids were performed with Lipofectamine reagent (Invitrogen) using the conditions recommended by the manufacturer. For DHT (Sigma) stimulation, transfected cells were grown in phenol red-free medium with 10% charcoal-dextran-stripped FBS (Germini). The cells were treated with 10 nM DHT for 1 h before harvest. Ethanol was used as the control for DHT. Treatment of 200 ng/ml Noggin (Sigma) was performed 2 h before the addition of DHT.
Cell growth, viability, and FACS analysis
To establish the cell-growth curves, LNCaP or PC3 cells were plated on day 1 in six-well plates (5 × 104 cells/well) with 10% FBS RPMI 1640. After 24 h, the cells were infected with the retroviruses overnight between days 2 and 3, and then allowed to grow in fresh medium on day 3. Up to 95% efficiency of infection was observed in the GFP control on day 5. Beginning on day 3, cell numbers were counted daily for 4 days using a hemocytometer. For the cell viability assay, LNCaP or PC3-AR cells were plated in 96-well plates (3 × 103 cells/well) and the viability of cells was then determined on day 5 with the cell proliferation XTT II kit according to the manufacturer's instructions (Roche). To reduce the endogenous AR in LNCaP cells, AR-siRNA and control-siRNA (Santa Cruz) were transfected on day 2 before the infection of retroviruses using the Lipofectamine siRNA transfection protocol (Invitrogen). For FACS analysis, cells collected on day 5 were stained with propidium iodide (BD Biosciences) and cell cycle distribution was then determined using a FACSCalibur instrument (BD Biosciences, San Jose, CA).
Immunohistochemistry
Immunohistochemical analyses of sections from paraffin-embedded tissue from CWR22 xenografts from intact or castrated nude mice, and the CWR22R (relapsed tumor) were performed as described in previous work (Myers et al, 1999a, 1999b, 2001). Three 4 μm adjacent sections from each of four different paraffin blocks for each tumor were stained with Smad1 (Upstate, rabbit 1:20), ERK1 (Santa Cruz, Rabbit 1:20), ERK2 (Santa Cruz, mouse 1:100), P-Smad1 (Cell Signaling, rabbit 1:50), or P-ERK1/2 (Santa Cruz, mouse 1:30) as described previously. Immunostaining was evaluated by determining the percentage of cells in three random × 400 fields that demonstrated strong staining or nuclear staining (⩾2 in intensity) on a scale of 0–4 (Grizzle et al, 1998a, 1998b).
Immunoprecipitation, immunoblotting analysis, luciferase reporter assay, and ELISA
Immunoprecipitation and immunoblotting analyses of cell lysates were performed as described previously (Wu et al, 2003). Immunoblots were characterized by using the SuperSignal West Femto system (Pierce). Luciferase activities were assayed with the Dual-Luciferase assay kit (Promega) by following the manufacturer's instructions. PSA expression and secretion were analyzed with the human PSA ELISA kit (Anogen). The values for the luciferase assays and ELISA shown in the figures are representative of transfection experiments performed in triplicate in at least two independent experiments.
Gel-shift and ChIP assays
See Supplementary data for detailed description for gel-shift and ChIP assays.
YFP PCA analysis and imaging
The YFP PCA analysis was performed as described previously (Remy et al, 2004). See Supplementary data for detailed description.
Statistical analysis
Statistical differences between two groups of data were analyzed using the Student's t-test. The data are presented as mean±s.d. (standard deviation).
Supplementary Material
Supplementary data
Acknowledgments
We thank Dr SW Michnick and Odyssey Thera Inc. for the generous gift of YFP-based PCA constructs; Dr J Trapman for PSA6.1 luciferase reporter; Mr M Chamberlain for polishing the manuscript. This work was supported by the National Institutes of Health (NIH) Grant DK60913 (to X Cao) and by the Early Detection Research Network (EDRN) Reference Laboratory CA086359-07 (to WE Grizzle).
References
- Adachi-Yamada T, Nakamura M, Irie K, Tomoyasu Y, Sano Y, Mori E, Goto S, Ueno N, Nishida Y, Matsumoto K (1999) p38 mitogen-activated protein kinase can be involved in transforming growth factor beta superfamily signal transduction in Drosophila wing morphogenesis. Mol Cell Biol 19: 2322–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin J, Davy A, Soriano P (2004) In vivo convergence of BMP and MAPK signaling pathways: impact of differential Smad1 phosphorylation on development and homeostasis. Genes Dev 18: 1482–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Shi X, Yang X, Cao X (2000) Smad6 as a transcriptional corepressor. J Biol Chem 275: 8267–8270 [DOI] [PubMed] [Google Scholar]
- Barnes J, Anthony CT, Wall N, Steiner MS (1995) Bone morphogenetic protein-6 expression in normal and malignant prostate. World J Urol 13: 337–343 [DOI] [PubMed] [Google Scholar]
- Bentley H, Hamdy FC, Hart KA, Seid JM, Williams JL, Johnstone D, Russell RG (1992) Expression of bone morphogenetic proteins in human prostatic adenocarcinoma and benign prostatic hyperplasia. Br J Cancer 66: 1159–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottner M, Suter-Crazzolara C, Schober A, Unsicker K (1999) Expression of a novel member of the TGF-beta superfamily, growth/differentiation factor-15/macrophage-inhibiting cytokine-1 (GDF-15/MIC-1) in adult rat tissues. Cell Tissue Res 297: 103–110 [DOI] [PubMed] [Google Scholar]
- Brubaker KD, Corey E, Brown LG, Vessella RL (2004) Bone morphogenetic protein signaling in prostate cancer cell lines. J Cell Biochem 91: 151–160 [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40 [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL (2004) Molecular determinants of resistance to antiandrogen therapy. Nat Med 10: 33–39 [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Cornelius SC, Pultz NJ, Jorgensen JS, Bonham MJ, Kim SJ, Danielpour D (2002) The androgen receptor represses transforming growth factor-beta signaling through interaction with Smad3. J Biol Chem 277: 1240–1248 [DOI] [PubMed] [Google Scholar]
- Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, Cox AD, Philips MR (2002) Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 4: 343–350 [DOI] [PubMed] [Google Scholar]
- Cleutjens KB, van der Korput HA, Ehren-van Eekelen CC, Sikes RA, Fasciana C, Chung LW, Trapman J (1997) A 6-kb promoter fragment mimics in transgenic mice the prostate-specific and androgen-regulated expression of the endogenous prostate-specific antigen gene in humans. Mol Endocrinol 11: 1256–1265 [DOI] [PubMed] [Google Scholar]
- Danielpour D (2005) Functions and regulation of transforming growth factor-beta (TGF-beta) in the prostate. Eur J Cancer 41: 846–857 [DOI] [PubMed] [Google Scholar]
- Engel ME, McDonnell MA, Law BK, Moses HL (1999) Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem 274: 37413–37420 [DOI] [PubMed] [Google Scholar]
- Gioeli D, Zecevic M, Weber MJ (2001) Immunostaining for activated extracellular signal-regulated kinases in cells and tissues. Methods Enzymol 332: 343–353 [DOI] [PubMed] [Google Scholar]
- Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, French FS (1998) Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res 58: 5718–5724 [PubMed] [Google Scholar]
- Grizzle WE, Myers RB, Manne U, Srivastava S (1998a) Immunohistochemical evaluation of biomarkers in prostatic and colorectal Neoplasia. In John Walker's Methods in Molecular Medicine—Tumor Marker Protocols, Margaret H, Zbigniew W (ed) pp 143–160. Totowa, NJ: Humana Press Inc [Google Scholar]
- Grizzle WE, Myers RB, Manne U, Stockard CR, Harkins LE, Srivastava S (1998b) Factors affecting immunohistochemical evaluation of biomarker expression in Neoplasia. In John Walker's Methods in Molecular Medicine—Tumor Marker Protocols, Margaret H, Zbigniew W (ed) pp 161–179. Totowa, NJ: Humana Press Inc [Google Scholar]
- Grizzle WE, Semmes OJ, Bigbee W, Zhu L, Malik G, Oelschlager DK, Manne B, Manne U (2005) The need for review and understanding of SELD/MALDI mass spectroscopy data prior to analysis. Cancer Informatics 1: 86–97 [PMC free article] [PubMed] [Google Scholar]
- Hamdy FC, Autzen P, Robinson MC, Horne CH, Neal DE, Robson CN (1997) Immunolocalization and messenger RNA expression of bone morphogenetic protein-6 in human benign and malignant prostatic tissue. Cancer Res 57: 4427–4431 [PubMed] [Google Scholar]
- Hata A, Lagna G, Massague J, Hemmati-Brivanlou A (1998) Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev 12: 186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath LG, Henshall SM, Kench JG, Turner JJ, Golovsky D, Brenner PC, O'Neill GF, Kooner R, Stricker PD, Grygiel JJ, Sutherland RL (2004) Loss of BMP2, Smad8, and Smad4 expression in prostate cancer progression. Prostate 59: 234–242 [DOI] [PubMed] [Google Scholar]
- Hromas R, Hufford M, Sutton J, Xu D, Li Y, Lu L (1997) PLAB, a novel placental bone morphogenetic protein. Biochim Biophys Acta 1354: 40–44 [DOI] [PubMed] [Google Scholar]
- Ide H, Yoshida T, Matsumoto N, Aoki K, Osada Y, Sugimura T, Terada M (1997) Growth regulation of human prostate cancer cells by bone morphogenetic protein-2. Cancer Res 57: 5022–5027 [PubMed] [Google Scholar]
- Kim IY, Lee DH, Ahn HJ, Tokunaga H, Song W, Devereaux LM, Jin D, Sampath TK, Morton RA (2000) Expression of bone morphogenetic protein receptors type-IA, -IB and -II correlates with tumor grade in human prostate cancer tissues. Cancer Res 60: 2840–2844 [PubMed] [Google Scholar]
- Kim IY, Lee DH, Lee DK, Ahn HJ, Kim MM, Kim SJ, Morton RA (2004) Loss of expression of bone morphogenetic protein receptor type II in human prostate cancer cells. Oncogene 23: 7651–7659 [DOI] [PubMed] [Google Scholar]
- Kim J, Coetzee GA (2004) Prostate specific antigen gene regulation by androgen receptor. J Cell Biochem 93: 233–241 [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massague J (1997a) Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 389: 618–622 [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Liu F, Hata A, Doody J, Massague J (1997b) The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev 11: 984–995 [DOI] [PubMed] [Google Scholar]
- Kuroda H, Fuentealba L, Ikeda A, Reversade B, De Robertis EM (2005) Default neural induction: neuralization of dissociated Xenopus cells is mediated by Ras/MAPK activation. Genes Dev 19: 1022–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamm ML, Podlasek CA, Barnett DH, Lee J, Clemens JQ, Hebner CM, Bushman W (2001) Mesenchymal factor bone morphogenetic protein 4 restricts ductal budding and branching morphogenesis in the developing prostate. Dev Biol 232: 301–314 [DOI] [PubMed] [Google Scholar]
- Lubahn DB, Joseph DR, Sar M, Tan J, Higgs HN, Larson RE, French FS, Wilson EM (1988) The human androgen receptor: complementary deoxyribonucleic acid cloning, sequence analysis and gene expression in prostate. Mol Endocrinol 2: 1265–1275 [DOI] [PubMed] [Google Scholar]
- Mor A, Philips MR (2006) Compartmentalized ras/mapk signaling. Annu Rev Immunol 24: 771–800 [DOI] [PubMed] [Google Scholar]
- Myers RB, Oelschlager D, Manne U, Coan PN, Weiss H, Grizzle WE (1999a) Androgenic regulation of growth factor and growth factor receptor expression in the CWR22 model of prostatic adenocarcinoma. Int J Cancer 82: 424–429 [DOI] [PubMed] [Google Scholar]
- Myers RB, Oelschlager DK, Coan PN, Frost AR, Weiss HL, Manne U, Pretlow TG, Grizzle WE (1999b) Changes in cyclin dependent kinase inhibitors p21 and p27 during the castration induced regression of the CWR22 model of prostatic adenocarcinoma. J Urol 161: 945–949 [PubMed] [Google Scholar]
- Myers RB, Oelschlager DK, Weiss HL, Frost AR, Grizzle WE (2001) Fatty acid synthase: an early molecular marker of progression of prostatic adenocarcinoma to androgen independence. J Urol 165: 1027–1032 [PubMed] [Google Scholar]
- Nagabhushan M, Miller CM, Pretlow TP, Giaconia JM, Edgehouse NL, Schwartz S, Kung HJ, Vere White RW, Gumerlock PH, Resnick MI, Amini SB, Pretlow TG (1996) CWR22: the first human prostate cancer xenograft with strongly androgen-dependent and relapsed strains both in vivo and in soft agar. Cancer Res 56: 3042–3046 [PubMed] [Google Scholar]
- Nakamura T, Scorilas A, Stephan C, Yousef GM, Kristiansen G, Jung K, Diamandis EP (2003) Quantitative analysis of macrophage inhibitory cytokine-1 (MIC-1) gene expression in human prostatic tissues. Br J Cancer 88: 1101–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paralkar VM, Vail AL, Grasser WA, Brown TA, Xu H, Vukicevic S, Ke HZ, Qi H, Owen TA, Thompson DD (1998) Cloning and characterization of a novel member of the transforming growth factor-beta/bone morphogenetic protein family. J Biol Chem 273: 13760–13767 [DOI] [PubMed] [Google Scholar]
- Pera EM, Ikeda A, Eivers E, De Robertis EM (2003) Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev 17: 3023–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quatela SE, Philips MR (2006) Ras signaling on the Golgi. Curr Opin Cell Biol 18: 162–167 [DOI] [PubMed] [Google Scholar]
- Remy I, Montmarquette A, Michnick SW (2004) PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol 6: 358–365 [DOI] [PubMed] [Google Scholar]
- Settle S, Marker P, Gurley K, Sinha A, Thacker A, Wang Y, Higgins K, Cunha G, Kingsley DM (2001) The BMP family member Gdf7 is required for seminal vesicle growth, branching morphogenesis, and cytodifferentiation. Dev Biol 234: 138–150 [DOI] [PubMed] [Google Scholar]
- Shang Y, Myers M, Brown M (2002) Formation of the androgen receptor transcription complex. Mol Cell 9: 601–610 [DOI] [PubMed] [Google Scholar]
- Thomas R, Anderson WA, Raman V, Reddi AH (1998) Androgen-dependent gene expression of bone morphogenetic protein 7 in mouse prostate. Prostate 37: 236–245 [DOI] [PubMed] [Google Scholar]
- Wainstein MA, He F, Robinson D, Kung HJ, Schwartz S, Giaconia JM, Edgehouse NL, Pretlow TP, Bodner DR, Kursh ED (1994) CWR22: androgen-dependent xenograft model derived from a primary human prostatic carcinoma. Cancer Res 54: 6049–6052 [PubMed] [Google Scholar]
- Wan M, Cao X (2005) BMP signaling in skeletal development. Biochem Biophys Res Commun 328: 651–657 [DOI] [PubMed] [Google Scholar]
- Weber MJ, Gioeli D (2004) Ras signaling in prostate cancer progression. J Cell Biochem 91: 13–25 [DOI] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massague J (1995) GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J 14: 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom P, Damber J, Bergh A (2001) Role of transforming growth factor-beta1 in prostate cancer. Microsc Res Tech 52: 411–419 [DOI] [PubMed] [Google Scholar]
- Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, Liu Z, Lu C, Mao Z, Cao X (2003) Smad4 as a transcription corepressor for estrogen receptor alpha. J Biol Chem 278: 15192–15200 [DOI] [PubMed] [Google Scholar]
- Yang S, Lim M, Pham LK, Kendall SE, Reddi AH, Altieri DC, Roy-Burman P (2006) Bone morphogenetic protein 7 protects prostate cancer cells from stress-induced apoptosis via both Smad and c-Jun NH2-terminal kinase pathways. Cancer Res 66: 4285–4290 [DOI] [PubMed] [Google Scholar]
- Yang S, Zhong C, Frenkel B, Reddi AH, Roy-Burman P (2005) Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res 65: 5769–5777 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng X, We R, Derynck R (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 383: 168–172 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data