

Abstract
Ubiquitination is known to regulate early stages of intracellular vesicular transport, without proteasomal involvement. We now show that, in yeast, ubiquitination regulates a late-stage, membrane fusion, with proteasomal involvement. A known proteasome mutant had a vacuolar fragmentation phenotype in vivo often associated with vacuolar membrane fusion defects, suggesting a proteasomal role in fusion. Inhibiting vacuolar proteasomes interfered with membrane fusion in vitro, showing that fusion cannot occur without proteasomal degradation. If so, one would expect to find ubiquitinated proteins on vacuolar membranes. We found a small number of these, identified the most prevalent one as Ypt7 and mapped its two major ubiquitination sites. Ubiquitinated Ypt7 was linked to the degradation event that is necessary for fusion: vacuolar Ypt7 and vacuolar proteasomes were interdependent, ubiquitinated Ypt7 became a proteasomal substrate during fusion, and proteasome inhibitors reduced fusion to greater degree when we decreased Ypt7 ubiquitination. The strongest model holds that fusion cannot proceed without proteasomal degradation of ubiquitinated Ypt7. As Ypt7 is one of many Rab GTPases, ubiquitin–proteasome regulation may be involved in membrane fusion elsewhere.
Keywords: fusion, membrane, proteasome, ubiquitination, Ypt7
Introduction
The proteasome consists of a cylindrical core particle (CP)—a multi-protein complex that contains proteolytic active sites—and two regulatory particles (RP), one at each end of the cylinder. Its most important function is to recognize and degrade proteins that have been tagged with a multiubiquitin chain (Glickman and Ciechanover, 2002). Ubiquitination is a multi-stage process in which a succession of enzymes activate a single ubiquitin unit and then attach it either directly to a protein, or to an ubiquitin unit or chain that has already been attached to a protein (Glickman and Ciechanover, 2002). Thus, the ubiquitination process can result in a protein tagged with a single ubiquitin or, alternatively, a multiubiquitin chain. Proteins tagged with a single ubiquitin are not degraded by the proteasome and have important functions in endocytosis control and in the sorting of membrane proteins within late endosomes (also known as multivesicular bodies) (Hicke and Dunn, 2003; Dupre et al, 2004; Bowers and Stevens, 2005).
Membrane fusion and vesicular transport are crucial for maintaining a cell's vesicular compartmentalization, which is a defining feature of eukaryotes. It is already known that ubiquitination regulates the initiation of vesicular transport: monoubiquitination leads to the collection of cargo proteins into a vesicle and then to its subsequent release. The vesicle next travels through the cell to its target acceptor membrane, with which it fuses. We present here data which indicate that, in yeast, ubiquitination also regulates the termination of transport—membrane fusion—and does so together with the proteasome, which was not previously known to have a role in fusion. We discovered that fusion of vacuoles cannot proceed unless proteasomal degradation occurs, and that ubiquitinated Ypt7 is a substrate during fusion. Our strongest model suggests that fusion cannot be completed without degradation, by the proteasome, of ubiquitinated Ypt7.
To see if the ubiquitin–proteasome system has a role in membrane fusion, we used an in vitro assay for homotypic vacuolar (that is, vacuole-to-vacuole) fusion (Wickner and Haas, 2000; Mayer, 2002; Wickner, 2002). (Lysosomes of Saccharomyces cerevisiae are called ‘vacuoles'). In this model system, AAA-ATPase Sec18/NSF initiates membrane fusion as follows. Sec18 breaks the bonds between the proteins that constitute cis-SNARE complexes, which are located in a membrane, to form free SNARE proteins that are no longer bonded to each other but are still in the membrane. Simultaneously, Sec18/NSF activates Ypt7, a Rab-like GTPase (Haas et al, 1995), via the HOPS chaperone complex; Ypt7 then activates two other GTPases, Rho1 and Cdc42 (Eitzen et al, 2000, 2001; Muller et al, 2001). Docking, an intermediate step in fusion, now occurs: free SNARE proteins on one membrane bond with free SNARE proteins on an opposing membrane, thereby forming trans-SNARE complexes that link opposing membranes together. After vacuoles have docked, Ca2+ release from the vacuolar lumen induces fusion. Many mechanistic details of fusion are still unclear, but it is thought to occur at the vertex ring domain at which the two tightly bound vacuoles meet. Other processes such as vacuolar acidification are also involved in the fusion process, as well as other proteins and lipids such as actin, the Vtc protein complex, vacuolar H+-ATPase, phosphatidylinositol 4,5-biphosphate and ergosterol.
Results
Indications that the proteasome has a role in vacuolar membrane fusion
A multi-copy suppressor screen performed with a proteasomal mutation suggested the hypothesis that the proteasome might be involved in membrane fusion (data not shown). To start evaluating this hypothesis, we studied the in vivo vacuolar morphology of different proteasome mutants to look for evidence of fusion impairment. Using the vital stain FM4-64, we found that one mutant, rpt1-K256S, has an in vivo vacuolar fragmentation phenotype (Figure 1A). Rpt1 is one of six ATPases present in the RP. A point mutation in Rpt1's ATP-binding motif (K256S) causes a slow-growth phenotype with a G1 cell cycle delay (Rubin et al, 1998). Fragmented vacuolar morphology is often observed where membrane fusion is impaired. Thus, the fact that there is a proteasomal mutant with fragmented vacuoles—and so, presumably, impaired fusion—suggests that the proteasome is involved in membrane fusion in vivo.
Figure 1.
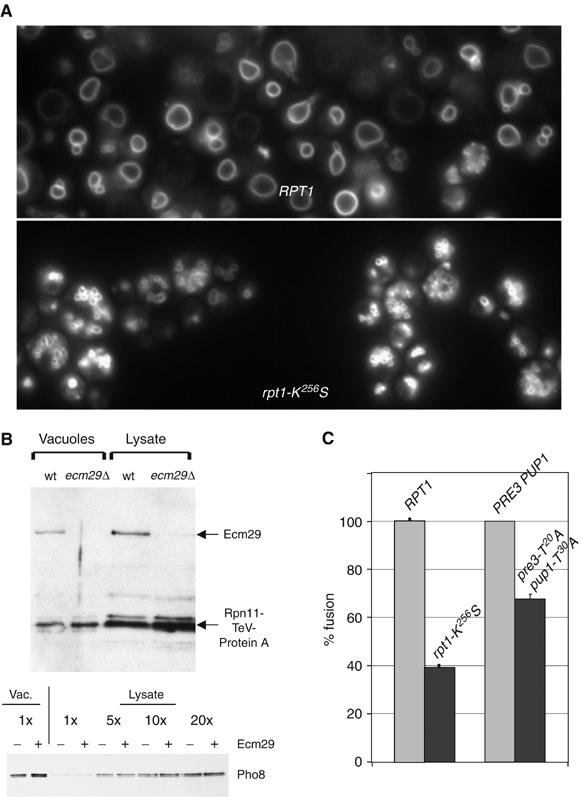
Indications that the proteasome has a role in vacuolar membrane fusion. (A) RPT1 or mutant rpt1-K256S yeast cells were incubated for 1 h with FM4-64 (10 μM), chased for 1.5 h in YPD, and visualized by immunofluorescence microscopy (DY85, DY106). (B) Equal protein amounts of vacuoles and total cell lysate (from wild-type or ecm29Δ cells) were probed with antibodies to visualize the proteasomal Rpn11 and Ecm29 subunits. Vacuoles were washed twice before gel analysis (sMK-172, sMK-173). The bottom panel shows enrichment for the Pho8 marker in both wild-type and ecm29Δ vacuolar preparations (sMK-186, sMK-187). (C) pho8Δ and pep4Δ prb1Δ vacuoles purified from either wild-type or proteasome mutant strains were tested for in vitro fusion activity. We used rpt1-K256S (sMK-191, sMK-193, sMK-220, sMK-230) and pre3-T20A pup1-T30A (sMK-245, sMK-247, sMK-248, sMK-251). Data represent percentage Pho8 activity relative to that from fusion of wild-type vacuoles. Absolute fusion values of RPT1 and PRE3 PUP1 reactions: 0.68 U, 1.67 U.
To test if proteasomes are present on vacuoles, we purified vacuoles by equilibrium flotation. (We first verified that equilibrium flotation, our standard procedure for making vacuolar preparations, generates preparations that are highly enriched for vacuoles. We can infer that the procedure does generate such preparations from the behavior of vacuolar protein marker Pho8 (Figure 1B, bottom panel) (Haas, 1995). After washing vacuolar membranes twice, we found them to be rich in proteasomes and in the proteasome-associated Ecm29 protein (Leggett et al, 2002) (Figure 1B; data not shown). The mass of proteasome per unit protein in vacuolar preparations was roughly equivalent to that in total lysates, even though proteasomes are highly abundant in cytosol and nuclear compartments. We incubated vacuoles with the proteasome-specific fluorogenic substrate LLVY-AMC and observed cleavage activity (data not shown), which we would expect to observe if proteasomes were present. As proteasome presence in these preparations depended on the presence of a key vacuolar marker protein (Figure 5), we inferred that proteasomes in the vacuolar preparations are associated with vacuoles, not with nonvacuolar membranes that are contaminants. As it has been proposed that mammalian Ecm29 links proteasomes to membranes (Gorbea et al, 2004), we tested if Ecm29 is a link, in yeast, between proteasomes and vacuoles. We found that it is not: proteasomes were present in equal quantities on wild-type and ecm29Δ vacuoles (Figure 1B). The presence of Ecm29 did not affect vacuolar enrichment (bottom panel, Figure 1B).
Figure 5.
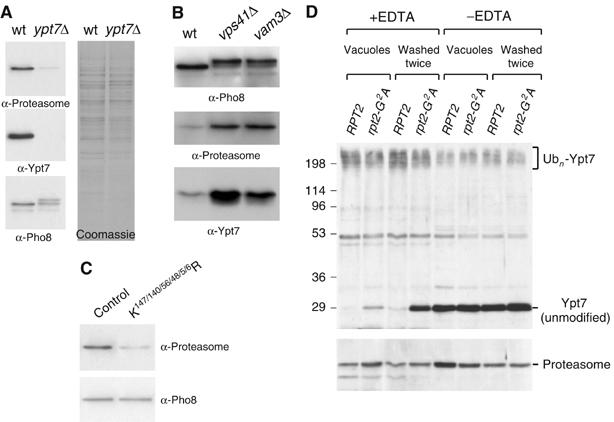
Relationships between Ypt7 species and proteasomes in the context of a vacuolar membrane. (A) Equal protein amounts of purified vacuoles from wild-type and ypt7Δ strains (RG-wt, RG-ypt7Δ) were probed for proteasomes, Ypt7 and Pho8; they were also Coomassie-stained. (B) Vacuoles from wild-type, vps41Δ and vam3Δ strains (RG-wt, RG-vps41Δ, RG-vam3Δ) were probed for Pho8, proteasomes and Ypt7, as in (A). (C) Vacuoles from 3HA-ypt7 tagged strains—wild-type control and K147/140/56/48/5/6R (sMK-413, sMK-421)—were probed for proteasome and Pho8 levels, as in (A). (D) Vacuoles were purified from a wild-type or a proteasomal rpt2-G2A myristoylation site mutant strain. The vacuoles, generated either in the presence or absence of EDTA, were pelleted and either harvested directly, or resuspended and pelleted twice more at 4°C before harvesting. The membranes were analyzed for the presence of both Ypt7 and the proteasome. Indicated are unmodified Ypt7 and high molecular weight Ypt7 ubiquitin conjugates (sMK-309, -310).
We next tested if the rpt1-K256S proteasome mutation, which causes fragmented vacuoles in vivo, interfered with membrane fusion in vitro. We used an assay to compare the degree of fusion that occurs between wild-type vacuoles to that which occurs between mutant vacuoles (Haas, 1995; Wickner and Haas, 2000; Mayer, 2002; Wickner, 2002). This assay causes pep4Δ prb1Δ vacuoles to fuse with pho8Δ vacuoles. Pho8 is initially an inactive pro-enzyme, but it becomes an active alkaline phosphatase when its carboxy-terminus is clipped off by vacuolar proteinases A (Pep4) or B (Prb1). The more fusion activity occurs, the more alkaline phosphatase activity results. The degree of phosphatase activity can then be measured to quantify the degree of membrane fusion (Haas, 1995). This assay showed that there was less fusion activity between the mutant vacuoles (61% less than wild-type vacuoles; Figure 1C), suggesting that the mutant's defect, a defect in the proteasome which leads to fragmented vacuoles in vivo, was physically associated with purified vacuoles in vitro.
Although the data suggested a role for the proteasome in fusion, we did not yet know which of its functions is involved. To test if degradation—the proteasome's main function—is involved in fusion, we tested vacuoles purified from a mutant proteasome strain pre3-T20A pup1-T30A (Arendt and Hochstrasser, 1999). This mutant lacks two of the three proteolytic active sites: its trypsin-like and caspase-like sites. Cells containing proteasomes with this mutation have only slight growth impairment, as the proteasomes' major chymotrypsin site is still intact (Arendt and Hochstrasser, 1999). Such cells, with their mild phenotype, did not show in vivo vacuolar fragmentation (data not shown). However, vacuoles purified from these cells exhibited 35% less in vitro vacuolar fusion activity than wild-type vacuoles (Figure 1C). Because less fusion activity occurs when proteasomes are less able to degrade proteins, proteasomal degradation is likely to have a role in fusion.
Inhibition of proteasomal degradation interferes with fusion
Because the proteasome is essential to life, genetic approaches are limited to studying mutations with partial loss-of-function mutations such as pre3-T20A pup1-T30A. In order to test whether the proteasome's role in fusion includes degradation, it was necessary to interfere more drastically with proteasomes' degradation function, so we switched from genetic to biochemical techniques. The in vitro homotypic vacuolar membrane fusion assay, mentioned earlier, was used to study the effect of proteasome inhibitors on fusion. A typical fusion assay is shown in Figure 2A. Pho8Δ vacuoles were mixed with pep4Δ prb1Δ vacuoles at a fusion-promoting temperature (27°C). This assay measures the level of fusion activity as the difference between the levels of alkaline phosphatase activity that result when pho8Δ and pep4Δ prb1Δ vacuoles are mixed at 27°C and at 4°C (Haas, 1995). The assay worked properly: alkaline phosphatase activity correlated with the amount of clipped mature Pho8 present (Figure 2A), and such activity occurred only when vacuoles of both types were mixed together. Some background alkaline phosphatase activity was observed when pep4Δ prb1Δ vacuoles were incubated alone at 27°C, due to cleavage of pro-enzyme Pho8 from other vacuolar proteases. This background alkaline phosphatase activity did not prevent the assay from measuring fusion because the level of activity was low, correlated with fusion levels and was also sensitive to fusion inhibitors (see below).
Figure 2.
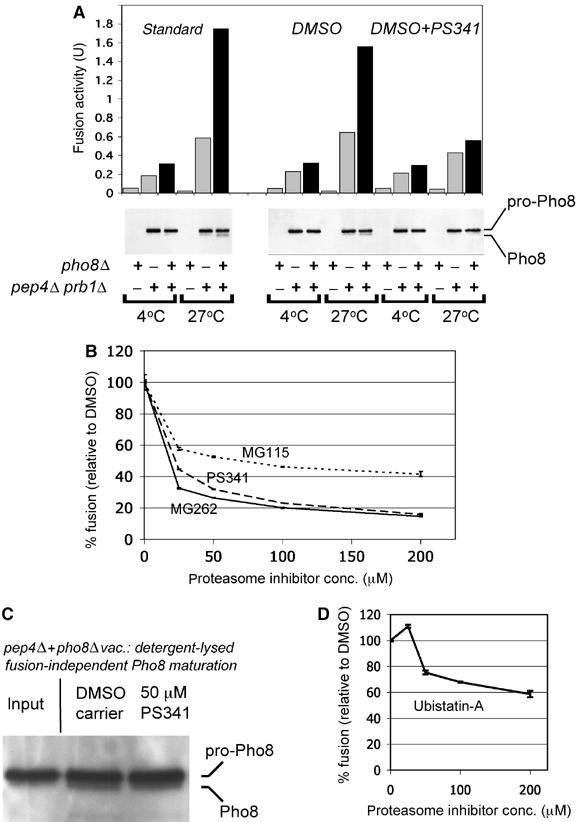
Inhibition of proteasomal degradation interferes with homotypic vacuolar membrane fusion. (A) Vacuoles from a pho8Δ and a pep4Δ prb1Δ strain (sMK-172, sMK-215) were added to a fusion reaction either alone, or mixed (lanes with black bar). Pho8 activity is depicted in fusion activity units (U). Vacuoles (20 μg) were added to each fusion reaction: either 20 μg of one type of vacuole, or 10 μg each of two types of vacuole. Parallel samples were analyzed by SDS–PAGE for Pho8 maturation. The right part of the panel shows the effects of adding 100 μM proteasome inhibitor PS341, versus carrier DMSO. (B) Three proteasome inhibitors, PS341, MG262 and MG115, were each added to the fusion reaction and compared to the addition of DMSO. In each case, the mixed vacuolar sample's fusion activity is the difference between Pho8 level at 4 and 27°C (sMK-172, sMK-215). Absolute fusion values of reactions without inhibitors: 1.83 U (PS341), 1.66 U (MG262), 1.49 U (MG115). (C) pep4Δ and pho8Δ vacuoles were mixed, lysed in detergent buffer in the presence of either DMSO or 50 μM PS341, incubated for 10 min at 27°C and probed for Pho8 (sMK-395, sMK-404). (D) The proteasome inhibitor Ubistatin-A, which targets multiubiquitin chains on substrates rather than proteasomal active sites, was added to the fusion reaction. Fusion was measured against the DMSO control (sMK-172, sMK-215). Absolute fusion value of reaction without UbistatinA: 1.64 U.
To test whether proteasome-mediated degradation is involved in vacuolar fusion, we used this assay to measure the effect of proteasome inhibitors on fusion. First, we employed PS341, a selective and potent proteasome inhibitor that belongs to the boronate family and is approved for clinical use. Levels of alkaline phosphatase activity and of mature Pho8 protein were lower when the reaction mixture also contained PS341 (Figure 2A), suggesting that PS341 inhibits vacuolar membrane fusion. Next, we used three distinct lines of experimentation (1–3 below) to confirm that the inhibitor inhibits fusion by targeting the proteasome.
(1) To verify that PS341 does not decrease the read-out by targeting the Pep4 and Prb1 proteases, we repeated the experiment described in the previous paragraph with five proteasome inhibitors from a total of three different classes: two boronate inhibitors (PS341, MG262), two aldehyde inhibitors (MG115, MG132) and clasto-lactacystin β-lactone (Figure 2B; data not shown). Inhibitors of different classes all target the proteasome's active sites, which are responsible for degradation, but they use different chemistry to do so. All five were found to interfere with fusion (Figure 2B; data not shown).
We used inhibitor concentrations such as 25 or 50 μM that are within the normal range of concentrations used when studying protein substrates. Admittedly, the literature suggests a different range of concentrations, e.g. a range of nM Ki values for boronate inhibitors. However, the ranges suggested in the literature were obtained by the use of peptide substrates: substrates that can be cleaved despite the fact they are not ubiquitinated. In contrast, we worked with protein substrates, which can be degraded only when ubiquitinated. Another difference between such substrates is that it is easier to inhibit degradation of peptides than of proteins, because peptides are usually cleaved by a particular active site of the proteasome, whereas proteins can be cleaved by any of the three (Kisselev et al, 2006).
A second and third experiment also suggested that PS341 did not decrease the read-out by targeting the Pep4 and Prb1 proteases. In the second experiment, pep4Δ prb1Δ vacuoles were mixed with pho8Δvacuoles, in the presence of a proteasome inhibitor, by detergent lysis rather than by membrane fusion. In this case, the inhibitor did not prevent Pho8 from maturing (Figure 2C), indicating that the inhibitor did not knockout Pep4 and Prb1. In the third, we pretreated the pep4Δ prb1Δ vacuoles, but not the pho8Δ vacuoles, with the irreversible inhibitor clasto-lactacystin β-lactone. When we mixed these vacuoles in the fusion assay, a low read-out resulted, suggesting that fusion was inhibited (data not shown). Fusion inhibition cannot have been caused by the knockout of the Pep4 and Prb1 proteases, at least in this case, as the pho8Δ vacuoles were not pretreated.
(2) If proteasome inhibitors interfere with fusion by inhibiting the proteasome, one would predict that genetic mutations that make the proteasome more susceptible to inhibitors also make membrane fusion more susceptible to inhibitors. To test this prediction, we examined vacuoles from the proteasome mutant pre3-T20A pup1-T30A, which lacks two of its three active sites, for fusion sensitivity to proteasome inhibitors. As predicted, a larger percentage drop in fusion activity was observed when proteasome inhibitors were added to mutant vacuoles than to wild-type vacuoles. For instance, when 25 μM MG132 inhibitor was added to wild-type vacuoles, this reduced fusion activity to 85% of its preinhibited value. By contrast, when the same inhibitor of the same concentration was added to the mutant vacuoles, this reduced fusion activity to 77% of its preinhibited value. Thus, there was an 8% difference between the percentage drops of fusion activity in the wild-type and mutant vacuoles. This 8% difference was observed when the procedure was performed with each of the aldehyde inhibitors MG132 and MG115, using concentrations between 25 and 200 μM. The addition of the boronate inhibitor PS341 resulted in a 5% difference. Similarly, treatment with the irreversible proteasome inhibitor clasto-lactacystin β-lactone resulted in a 17% difference.
(3) Experiment (1) used a variety of proteasome inhibitors, each of which works by inhibiting the proteasome's active sites. Experiment (3) instead used a proteasome inhibitor (Ubistatin-A) that works by targeting multiubiquitin chains on substrates, thereby preventing the proteasome from recognizing and degrading them (Verma et al, 2004). We added Ubistatin-A to the in vitro homotypic vacuolar membrane fusion assay. Like the inhibitors used in experiment (1), Ubistatin-A interfered with in vitro vacuolar membrane fusion (Figure 2D). Because the mechanism by which it inhibits is fundamentally different from that of a classic proteasome inhibitor, this is further evidence that the proteasome's function in fusion includes degradation.
Discovery and identification of ubiquitin-modified proteins on vacuolar membranes
If proteasomal degradation is required for vacuolar fusion, we would expect to find ubiquitin-modified proteins on vacuolar membranes, because virtually all proteasome substrates are ubiquitinated. Using mass spectrometry, we identified such proteins on vacuolar membranes derived from a strain that carries tagged ubiquitin.
In more detail, we used a yeast strain in which all four endogenous ubiquitin genes had been replaced with an extrachromosomal plasmid that expresses tagged His6-cMyc-ubiquitin at physiological levels (Finley et al, 1994). This tag allowed us to extract ubiquitinated proteins from purified vacuoles. We analyzed these proteins by mass spectrometry and identified two GTPases, Ypt7 and Rho1, known to have a role in homotypic vacuole fusion (Haas et al, 1995; Eitzen et al, 2000, 2001; Muller et al, 2001).
To confirm that Ypt7 and Rho1 exist on vacuoles as multiubiquitinated conjugates, each protein was 3HA-tagged at its amino-terminus and expressed from a GAL promoter in the His6-cMyc-ubiquitin strain. Purified vacuoles from the double-tagged Ypt7 or Rho1 strains were solubilized in detergent and used to perform an α-HA immunoprecipitation (Figure 3A). A substantial portion of the Ypt7 and Rho1 migrated as high molecular weight species, which also tested positive for the cMyc-ubiquitin tag.
Figure 3.
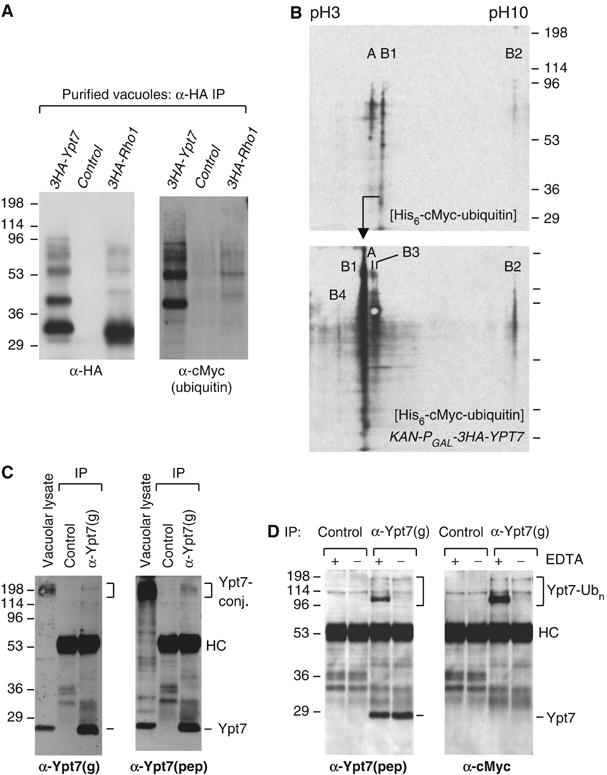
Ubiquitinated species of Ypt7 and Rho1 are present in vacuolar preparations. (A) Vacuoles from untagged or 3HA-tagged Ypt7 and Rho1 strains in a His6-cMyc-ubiquitin background (SUB592, sMK-303, sMK-307) were purified, subjected to an α-HA immunoprecipitation and probed for both HA and cMyc. (B) His-tagged ubiquitin material from vacuoles (untagged or 3HA-tagged Ypt7 strain: SUB592, sMK-303) was subjected to 2D IEF-SDS–PAGE, and probed for ubiquitin conjugates using an α-cMyc antibody. The top panel (untagged Ypt7) shows three ubiquitin-positive products, labeled A, B1 and B2. The bottom panel uses the 3HA-tagged Ypt7 strain. The products labeled B in the bottom panel are HA-reactive, whereas A is not (data not shown). (C) Vacuoles were purified from a wild-type strain that has unmodified endogenous YPT7- and ubiquitin genes. A fraction of such vacuoles was lysed directly in SDS sample buffer for analysis by SDS–PAGE. This sample lane is labeled ‘vacuolar lysate'. The remaining vacuoles were lysed in NP-40 detergent buffer, and the lysate was used to immunoprecipitate endogenous Ypt7 using the α-Ypt7(g) antibody. Ypt7 was detected by probing with the same α-Ypt7(g) antibody used for immunoprecipitation, as well as with a second, unrelated Ypt7 antibody, α-Ypt7(pep) (sMK-310). (D) Two vacuolar preparations were made from a strain with an unmodified YPT7 gene in a His6-cMyc-ubiquitin background (SUB592). EDTA was either present or absent during the purification from spheroplasting onward. These vacuoles were lysed and subjected to an α-Ypt7(g) immunoprecipitation. These were probed with the α-Ypt7(pep) serum and the cMyc antibody detecting ubiquitin.
To determine the number of different ubiquitin-conjugated protein species associated with vacuoles, we analyzed a vacuolar preparation of the His6-cMyc-ubiquitin strain. Ubiquitinated proteins were first eluted from the Ni2+-NTA beads and then separated on a two-dimensional (2D) isoelectric focusing (IEF) (pH 3–10)-SDS–PAGE system. Next, we visualized them by the use of a cMyc-specific antibody. Only a small number of ubiquitin-conjugated species were detected (top panel Figure 3B). However, other ubiquitinated species may exist undetected if they do not focus in the pH 3–10 range or if our protocol did not capture them efficiently. As no proteasome inhibitors were used during vacuole purification, these data suggest that a small number of protein species that are stably multiubiquitinated are associated with vacuolar membranes.
To verify that Ypt7 was one of these few ubiquitinated species, we analyzed vacuoles from the 3HA-tagged Ypt7 strain (Figure 3B, bottom panel). A probe of the membrane using an α-HA antibody revealed several ubiquitinated Ypt7 species (products labeled B in Figure 3B, bottom panel) and one species unrelated to Ypt7 (product labeled A in Figure 3B, bottom panel) (data not shown). Product B1 was more abundant in the bottom panel than in the top, consistent with GAL promoter overexpression, and its IEF point was shifted as a result of the fact that B1 became tagged. Product B2 was also more abundant in the bottom than the top panels. Two new species were present in the gel represented by the bottom panel, but not in the gel represented by the top one. Similar results were obtained by the use of an α-ubiquitin antibody and several HA-tagged Ypt7 and Rho1 strains that each expressed wild-type ubiquitin (data not shown). In sum, the 2D analysis showed that Ypt7 is the most prevalent ubiquitinated protein that is present on purified vacuoles.
We also identified ubiquitinated Ypt7 species in vacuolar preparations that, like the preparation shown in the top panel of Figure 3B, did not overexpress Ypt7 (Figure 3C). For instance, when we used a preparation from a strain with wild-type ubiquitin and an unmodified YPT7 gene, two α-Ypt7 antibodies independently detected high molecular weight Ypt7 conjugates in vacuolar lysates (Figure 3C). To verify that the conjugates identified by the two different antibodies were both Ypt7, we immunoprecipitated Ypt7 with one antibody and probed the membrane with both. We observed that both antibodies recognized identical 200 kDa Ypt7-positive material, despite the fact that these large conjugates immunoprecipitate inefficiently.
The fragility of some ubiquitinated Ypt7 species in some contexts may help to explain why their existence and their presence on membranes have not hitherto been noted. Even though ubiquitinated Ypt7 is stable in the absence of proteasome inhibitors, it can lose its multiubiquitin chain, or split up proteolytically, during the vacuolar purification procedure or subsequent handling. When we performed an experiment using a cMyc-tagged ubiquitin strain that has an unmodified YPT7 gene, we observed that, when EDTA was not present from the spheroplasting stage onwards, some Ypt7 ubiquitinated species were not retained (Figure 3D), although they were retained when EDTA, which has a stabilizing effect, was present. The fact that some, but not all, species of ubiquitinated Ypt7 were EDTA-dependent suggests that they are heterogeneous, probably because their multiubiquitin chains are attached to different sites (see Figure 4). Note that the fraction of Ypt7 that is ubiquitinated was even larger than Figure 3D suggests, because large conjugates precipitate less efficiently than unmodified Ypt7 (Figure 3C).
Figure 4.
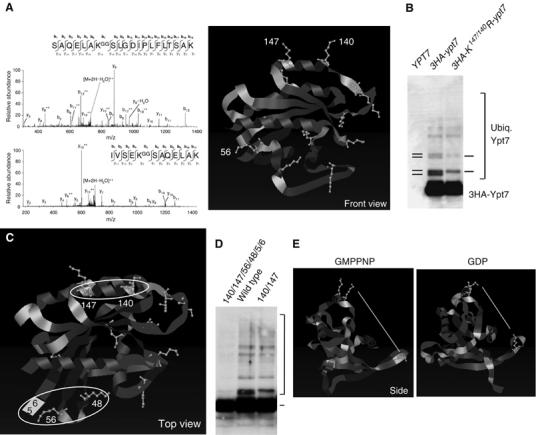
Identification and description of Two Ypt7 ubiquitination sites. (A) Vacuoles were purified from a 3HA-ypt7 strain (sMK-303). Spectra of two tryptic peptides carrying the -GG modification on lysines K147 and K140 were identified. Lysines K147 and K140 are highlighted in the structure of Ypt7 complexed with GMPP(N)P (Constantinescu et al, 2002). (B) Vacuoles from an untagged (sMK-186), a 3HA-ypt7 tagged (sMK-325) and a 3HA-K147/140R-ypt7 strain (sMK-329) were purified, subjected to an α-HA immunoprecipitation, and probed with an α-Ypt7 antibody. (C) A top view of the Ypt7 structure shown in (A). White circles indicate the two ubiquitination loci on Ypt7. (D) Vacuoles from 3HA-ypt7-tagged strains (K147/140/56/48/5/6R, wild type, K147/140R) were purified, subjected to an α-HA immunoprecipitation and probed with an α-Ypt7 antibody (sMK-395, -325, -329). The bracket indicates ubiquitinated Ypt7 species. (E) This is a side view of the Ypt7 structure presented in (A). The left panel shows Ypt7 complexed with the GTP-analog GMPPNP, the right shows it complexed with GDP. The lysine residues 147 and 140 are highlighted in yellow at the top and 56 near the bottom. The white line shows the distance between the primary amines of lysines 147 and 56 (top panel 35.40 Å, bottom panel 25.46 Å).
In sum, we found a small number of ubiquitin-modified proteins on vacuolar membranes. We identified these as the GTPases Ypt7 and Rho1, which are stably multiubiquitinated in most but not all contexts. We decided to focus, in subsequent experiments, on the role of Ypt7 ubiquitination in vacuolar membrane fusion, for several reasons: Ypt7 is necessary for fusion and is the Rab GTPase active in homotypic vacuolar membrane fusion (Haas et al, 1995). Furthermore, unlike Rho1, Ypt7 is a member of a large family of GTPases (Rab GTPases) that regulate membrane fusion events elsewhere in the cell.
Identification and description of two Ypt7 ubiquitination sites
Although Ypt7 has a known role in vacuolar fusion and we discovered that vacuolar Ypt7 is ubiquitinated (Figure 3), we still did not know if Ypt7's function in fusion depends on whether or not, and the extent to which, Ypt7 is ubiquitinated. The effect of Ypt7 ubiquitination on fusion cannot be investigated without further knowledge of Ypt7's ubiquitination sites. We next identified and described these sites, and located them on the Ypt7 backbone using a combination of mass spectrometry and information on Ypt7's structure.
To obtain material to analyze by mass spectrometry, vacuoles were isolated from the HA-Ypt7 strain and solubilized in detergent. Ypt7 was immunoprecipitated with α-HA antibody and subjected to SDS–PAGE. The gel region predicted to contain HA-Ypt7-Ubn (n=1–7) was excised. Mass spectrometry then identified two Ypt7 lysines modified with ubiquitin, K147 and K140, as well as many tryptic peptides from Ypt7 (nine peptides) and ubiquitin (six peptides). In more detail, MS/MS spectra matching the sequences SAQELAKGGSLGDIPLFLTSAK (K147) and IVSEKGGSAQELAK (K140) were detected, where KGG represents GG-modified lysine (+114.0429 amu) (Figure 4A). This GG-modified lysine is produced when trypsin cleaves a conjugated molecule of ubiquitin between R74 and G75. GG-modified ubiquitin peptides, which were derived from ubiquitin lysines 48 and 63, were also identified (data not shown), indicating that Ypt7 multiubiquitin chains were made up of both K48- and K63-ubiquitin linkages. Together, these mass spectrometry results confirmed independently the presence of stable, ubiquitinated Ypt7 species in vacuolar preparations.
Our analysis also suggested that these two lysines, K147 and K140, make up a single ubiquitination site. On the Ypt7 structure (Constantinescu et al, 2002), the two lysines are close together in helix α4 and opposite Ypt7's GTP-binding site (Figure 4A). Their side chains point into space in the same direction, because the two lysines are separated by two whole turns of the helix. To test if lysines K147 and K140 comprise a single ubiquitination site, we generated a K147/140R ypt7 double mutant and compared it to wild type. The double mutation did not affect the overall levels of Ypt7 protein, but did change the ubiquitination pattern (Figure 4B). Certain mono- and di-ubiquitinated species that have a slightly faster migration pattern on SDS–PAGE were absent from the K147/140R mutant (Figure 4B). In contrast, when we examined a single Ypt7 K147R mutation, no differences were seen in the Ypt7 ubiquitination pattern (data not shown), reflecting, perhaps, a redundancy between lysines K147 and K140. As Ypt7 remained ubiquitinated even when it lost certain types of ubiquitinated species, these experiments also suggested that at least two sites on the Ypt7 protein surface are ubiquitinated. An experiment with a triple mutation confirmed this. The triple mutant lacked not only the lysine pair missing on the double mutant but also the only other lysine close to the pair (K101), which is present in an adjacent α-helix. However, ubiquitination levels were the same for the double and triple mutants (data not shown).
We next mapped the second Ypt7 ubiquitination site. Mass spectrometry was used to analyze mutant vacuolar Ypt7 protein purified from the K147/140/101R strain. We identified an additional GG-modified lysine peptide, evidence of ubiquitination on K56 (data not shown). K56 and K147/140 are located on different parts of Ypt7 (Figure 4A, C and E). Three other lysine residues (K48, K5 and K6) are located close to K56. K48 is in the same loop as K56, whereas K5 and K6 are outside of the loop but align with K56 as in a β-sheet (Figure 4C). Lysines 5 and 6 have been drawn into the structure shown in Figure 4C to show these lysines' expected proximity to K56. (The structure is shown from residue 7 onwards.)
To discover which of the four lysines constitute the second site, we mutated them into arginine in three different combinations: K56R, K56/48R and K56/5/6R (data not shown). We found that ubiquitination did not decrease significantly with these combinations. However, it did when all four lysines were knocked out (Figure 4D), suggesting that the second site consists of all four. Figure 4D also shows that these are the two major sites and that most but not all ubiquitination occurs on these: a small amount of ubiquitination on the sextuplet mutant was detected on longer immunoblot exposure, but much less than what occurs on the two major sites (Figure 4D).
Our discovery of the second site's location is interesting, because it is in a part of Ypt7's backbone that, according to Constantinescu et al (2002), undergoes a minor conformational change during the GTP/GDP cycle. We discovered that this change makes a large difference to the second ubiquitination site's orientation and location: the K56 side chain is rotated upwards and inwards in the GDP-bound structure (Figure 4A, C and E); consequently, the conformational change reduces the distance between the primary amines of K56 and K147 by 10 Å (from 35.40 to 25.46 Å) (Figure 4E). This conformational change, which repositions Ypt7's ubiquitination site, may underlie the function of ubiquitinated Ypt7 during fusion.
Relationships between Ypt7 Species and proteasomes in the context of a vacuolar membrane
We discovered that Ypt7 is not only ubiquitinated but also a major ubiquitinated species on vacuolar membranes (Figure 3). However, we had as yet no data, which suggested that the proteasome can recognize ubiquitinated Ypt7 in the context of a vacuolar membrane, which it would need to be able to do if ubiquitinated Ypt7 is a substrate of the proteasome during fusion. We next investigated if proteasomes and ubiquitinated Ypt7 influence each other when both are in a vacuolar membrane micro-environment. We found that the level of proteasomes on vacuoles varies according to whether or not Ypt7 is present, and that a particular proteasome mutation destabilizes the ubiquitination of Ypt7.
Vacuoles purified from a strain that lacks the vacuolar Rab GTPase Ypt7 had a substantially lower level of proteasomes than wild-type vacuoles (Figure 5A). The same reduction was observed in vacuoles purified from a strain in which Ypt7 expression had been downregulated by the GAL promoter (data not shown). Vacuoles purified from a ypt7Δ strain were fusion-impaired (Haas et al, 1995) and consequently contained immature vacuolar alkaline phosphatase Pho8 of higher molecular weight. The mature form was also present (see Figure 5A) (Wichmann et al, 1992).
Vacuoles can fuse only when Ypt7 is present. To eliminate the possibility that proteasome levels depend on capacity to fuse, rather than on the presence of Ypt7, we tested vacuoles from two unrelated vacuolar protein marker strains that, like the ypt7Δ strain just mentioned, are both fusion-impaired. One strain lacked Vps41, a HOPS chaperone complex component; the other lacked the SNARE protein Vam3. We observed immature Pho8 in these vacuoles but not in wild type (Figure 5B, top panel), which confirmed that the vacuoles are fusion-impaired. Proteasomes were observed (Figure 5B, middle panel) despite the fact that these vacuoles could not fuse. Their presence eliminates the possibility that proteasome levels depend on vacuolar capacity to fuse. The fact that these vacuoles have higher levels of both proteasomes and Ypt7 protein than wild-type vacuoles (Figure 5B, bottom panel) strengthens Figure 5A's implication that proteasome levels on vacuoles correlate with presence of Ypt7.
To investigate if the ubiquitin that is attached to Ypt7 contributes to proteasome-membrane linkage, control vacuoles were compared with vacuoles from the ubiquitination-impaired ypt7 mutant (Figure 4D: K140/147/56/48/5/6R ypt7). Mutant ypt7 vacuoles were observed to carry a lower level of proteasomes (Figure 5C), suggesting that Ypt7 ubiquitination at least contributes to proteasome-membrane linkage.
Having seen that Ypt7 presence affects proteasome levels, we next investigated if the proteasome affects Ypt7. We observed that, when the proteasome was mutated, ubiquitination of Ypt7 became unstable. In more detail, we generated a new point mutation, rpt2-G2A, which targets a known myristoylation site in the proteasomal subunit Rpt2 (Kimura et al, 2003). Wild-type and rpt2-G2A vacuoles were purified in the presence of EDTA, to best preserve Ypt7 ubiquitination. After purification, most of the Ypt7 on wild-type vacuoles was present as high molecular weight ubiquitinated Ypt7, which is labeled ‘Ubn-Ypt7' (first lane, Figure 5D). In contrast, some of the Ypt7 on the mutant vacuoles lacked ubiquitination (second lane). We then washed each batch of vacuoles. Washing did not change the ubiquitination status of Ypt7 on wild-type vacuoles (third lane). In contrast, a high proportion of remaining ubiquitinated Ypt7 on mutant vacuoles lost its ubiquitination, resulting in the appearance of a strong unmodified Ypt7 band (fourth lane). (The appearance of unmodified Ypt7, which is visible in the bottom band as one moves from lanes 3 and 4, is more clearly observed than the disappearance of Ubn-Ypt7, which is visible in the top smear as one moves from lanes 3 and 4. This is because a difference in the amount of protein present in two lanes gives rise to a smaller difference in signal intensities in a smear than in a band.) In the absence of EDTA, premature loss of Ypt7 ubiquitination during purification masked the rpt2-G2A phenotype. The rpt2-G2A mutant did not have a fragmented vacuolar phenotype (data not shown).
Ubiquitinated Ypt7 is a proteasomal substrate important for fusion
Our data suggested that Ypt7 may be a proteasome substrate during fusion: proteasomal degradation is required for fusion (Figure 2), Ypt7 is a major ubiquitinated species on vacuolar membranes (Figure3), and the proteasome and ubiquitinated Ypt7 influence each other (Figure 5). To test if it is a proteasome substrate during fusion, we tracked the fate of ubiquitinated Ypt7, which is normally stable during vacuolar purification, during membrane fusion.
Multiubiquitinated Ypt7 species were observed to disappear under conditions in which fusion proceeds (Figure 6A). Because a proteasome inhibitor prevented their disappearance (Figure 6A), it is likely that proteasomes degraded the ubiquitinated Ypt7 conjugates. We eliminated the possibility that other proteases degraded the conjugates instead of the proteasome by adding a general protease inhibitor cocktail to all samples. Because the same conditions that enable fusion also lead to proteasomal degradation of ubiquitinated Ypt7 species, we hypothesized that ubiquitinated Ypt7 species were degraded as part of the fusion process. This hypothesis was substantiated by the experiments reported below.
Figure 6.
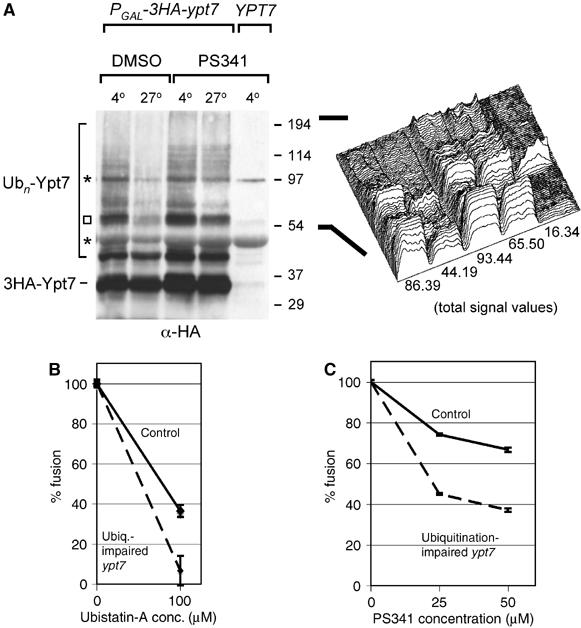
Ubiquitinated Ypt7 is a proteasomal substrate important for fusion. (A) Vacuoles from an HA-tagged or untagged Ypt7 strain (sMK-303, SUB592) were purified and allowed to fuse at 27 or 4°C with DMSO or 200 μM PS341 proteasome inhibitor, and probed for the HA-tag. A protease inhibitor cocktail was present in all reactions. Asterisks indicate background bands also present in the untagged Ypt7 lane. To better judge the fate of the many ubiquitinated Ypt7 species, which are heterogeneous with regard to size, the lanes from the square upwards to the top of the gel were quantified using Scion Image (Scion Corporation). In the right panel, this quantification is graphically displayed in a surface plot. The number under each lane is the value of the total signal present in that lane. (B) Control vacuoles and vacuoles from the ubiquitination-impaired ypt7 mutant (Figure 4D: K140/147/56/48/5/6R ypt7) were compared for fusion sensitivity to the proteasome inhibitor Ubistatin-A. The percentage fusion remaining when adding 100 μM Ubistatin-A over DMSO is depicted (sMK-319, -403, -413, -421). Absolute fusion values of reactions without inhibitor: 0.25U (control), 0.18U (mutant). (C) Comparison of the fusion sensitivity of control and ubiquitination-impaired ypt7 mutant vacuoles (Figure 4D: K140/147/56/48/5/6R ypt7) to the PS341 proteasome inhibitor (sMK-319, -403, -413, -421). Absolute fusion values of reactions without inhibitor: 0.21 U (control), 0.29 U (mutant).
Having discovered that proteasomal degradation is necessary for fusion to occur (Figure 2), the question arose: is proteasomal degradation of ubiquitinated Ypt7 necessary for fusion to occur? To answer it, we analyzed vacuoles purified from the ubiquitination-impaired ypt7 mutant strain (Figure 4D: K140/147/56/48/5/6R ypt7). This mutant is a functional enzyme: vacuoles that carried mutant Ypt7 were not fusion-impaired and this mutation of Ypt7 did not result in a fragmented vacuolar phenotype (data not shown). We then conducted an experiment with two different classes of proteasome inhibitor: Ubistatin-A, which targets ubiquitination, and PS341, which targets the proteasome's active sites. Whichever inhibitor was used, when it was added to the fusion reaction, the mutant vacuoles' fusion activity decreased more than that of the wild-type vacuoles. When we added 100 μM Ubistatin-A, fusion activity of the mutant vacuoles dropped to 6% of total capacity versus 36% with wild type (Figure 6B). A similar difference between the reductions in fusion activity of wild-type and mutant vacuoles was observed when we added PS341 (Figure 6C). This experiment showed that reducing the degree of Ypt7's ubiquitination makes fusion more sensitive to proteasome inhibitors. Therefore, Ypt7 is linked to the proteasomal degradation event that is required for fusion. In the following discussion, we argue that the most plausible explanation of this observation, in the context of our entire data set, is that fusion cannot go to completion without proteasomal degradation of ubiquitinated Ypt7.
Discussion
Structure of our argument
Our data suggest a previously unknown role for the ubiquitin–proteasome system: it regulates the fusion of yeast's vacuolar membranes. First, several observations show that proteasomes play a role in vacuolar fusion. Living cells that carry a known proteasome mutation were discovered to have an in vivo phenotype of fragmented vacuoles. Proteasomes were found on purified vacuoles. Proteasome mutations, as well as proteasome inhibitors, were observed to interfere with vacuolar membrane fusion in vitro. Second, we identified putative proteasomal substrates, one of which is ubiquitinated Ypt7. We observed a small number of stably ubiquitinated species in vacuolar preparations and identified the predominant ones as Ypt7 and Rho1, proteins known to have roles in vacuolar fusion. We exhaustively mapped and analyzed Ypt7's ubiquitination sites and multiubiquitin chains. Third, we presented data that link proteasomal degradation during vacuolar fusion to ubiquitinated Ypt7. Having identified several putative substrates, we identified interdependencies between one, ubiquitinated Ypt7, and membrane-bound vacuolar proteasomes. We showed that the proteasome degrades multiubiquitinated Ypt7 species during fusion. Importantly, reduction of Ypt7 ubiquitination increased fusion sensitivity to the proteasome inhibitors PS341 and Ubistatin-A, linking the proteasomal degradation event required for membrane fusion to Ypt7 ubiquitination.
Models
The simplest and, to our minds, the most plausible model is that membrane fusion cannot go to completion unless proteasomes degrade ubiquitinated Ypt7. This model is supported by our data: fusion sensitivity to Ubistatin-A showed that the degradation that is necessary for fusion involves an ubiquitinated substrate (Figure 2D). We found that Ypt7 is one of the few ubiquitinated proteins on vacuoles, and that it is degraded by the proteasome during fusion. Finally, we discovered that the proteasomal degradation event necessary for fusion is linked to Ypt7: reduction of Ypt7 ubiquitination made fusion more sensitive to proteasome inhibitors. An alternative, second model is that ubiquitination-impaired Ypt7 reduces proteasome levels on vacuoles (Figure 5C), which then inhibits the degradation of an unrelated substrate X; and it is degradation of this substrate (X), rather than of ubiquitinated Ypt7, that is necessary for fusion to occur. Although we cannot exclude this model, it is less plausible than the first because it does not explain why proteasome inhibitors have a greater effect on the fusion of mutant than wild-type vacuoles. By contrast, the first model does, as decreases in Ypt7's ubiquitination status change Ypt7 into a worse proteasome substrate. Assuming that fusion activity correlates with degree of proteasomal degradation, enzyme kinetics can describe mathematically the observed inhibition of fusion by a proteasome inhibitor. According to enzyme kinetics, a competitive inhibitor (I) competes with a substrate (S) for being processed by enzyme (E); furthermore, when fusion becomes more sensitive to the same concentration of I, S must have become changed either in concentration or in KM/KS, if E remains unmodified. (Note that E's status as modified or unmodified does not depend on E's concentration.) As we know that the KM/KS of Ypt7 changed, it is likely that Ypt7 is the main substrate S. A third model is that multiubiquitin chains themselves inhibit fusion and must be removed for fusion to occur. However, we would then expect that reducing Ypt7 ubiquitination status would reduce, not enhance, the sensitivity of fusion to proteasome inhibitors. In sum: our data set suggest that proteasomal degradation of ubiquitinated Ypt7 species is necessary for the fusion process to go to completion.
Explanation of the surprising stability of ubiquitinated Ypt7 and Rho1
Degradation substrates are usually hard to detect. Our data showed that ubiquitinated-Ypt7 and -Rho1 are stable before fusion begins. What is the explanation of the unexpected stability? One possibility is that their multiubiquitin chains contain K63 linkages, which are thought not to be signals for proteasomal degradation. However, we observed that Ypt7 multiubiquitin chains missing this linkage are also stable (data not shown). A second possibility is that ubiquitinated Ypt7 and -Rho1 are stable before fusion starts but unstable during fusion, because they exist in a stable conformation before fusion but change to an unstable conformation when fusion starts. We know that Ypt7 has two conformations and that at least the second ubiquitin site shifts when Ypt7 undergoes a conformational change (Figure 4E). This change may enable Ypt7 thereby to become recognizable, and thus degradable, by the proteasome.
Generalization
Our results are about fusion between yeast vacuoles. Are they also true of other fusion events in yeast and/or other organisms? If Rab GTPases other than Ypt7 can also become ubiquitinated, this would suggest that our results generalize. Rab GTPases control fusion in multiple locations in the cells of many organisms. For instance, yeast has 11 Rab GTPases, each of which controls a particular membrane fusion event. It should be investigated whether ubiquitination occurs on other Rab GTPases, and whether this regulates Rab protein function during membrane fusion or other events elsewhere in the cell. Ubiquitinated lysines on Ypt7 may not align with those on other Rab GTPases: we found that ubiquitin chains on Ypt7 did not always attach to the same lysine at a particular site.
Further evidence of an ubiquitin/proteasome role in membrane fusion
VPS11 and VPS18 are HOPS complex components, without which vacuolar fusion cannot occur. They contain RING domains (SGD database), which are structural core domains for ubiquitin ligases (Glickman and Ciechanover, 2002), and could therefore be the ubiquitin ligases that ubiquitinate Ypt7 and Rho1. Dong et al (2004) reported an interaction between the proteasomal α-subunit XAPC7 and the mammalian Ypt7 counterpart, Rab7. Similarly, we observed pre-fusion relationships between the proteasome and ubiquitinated Ypt7 (Figure 5). Finally, a recent article on a mammalian model also suggests that ubiquitin has a role in membrane fusion (Wang et al, 2004). The authors report that the activity of a deubiquitinating enzyme, VCIP135, is necessary for mitotic Golgi fragments to reassemble. In further research, we will seek to identify other proteasomal substrates and other roles for the proteasome-ubiquitin system in fusion, and to explain the physical processes that underlie these roles.
Materials and methods
Strains and antibodies
See Table I for yeast strains, and Supplementary data for strain construction details. Polyclonal antibodies were generated against Ecm29 and the proteasome (α-Rpn12; α-Rpn8). The Pho8 monoclonal antibody was purchased from Molecular Probes (A-6458), and Rpt1, Rpt6 and the ubiquitin protein conjugate (UG9510) polyclonal antibodies came from Affiniti. The monoclonal α-HA antibody (F7) and an α-cMyc rabbit polyclonal were produced by Santa Cruz Biotechnology (CA). α-Ypt7 (g), raised against an Ypt7-GST fusion protein, and α-Ypt7 (pep), raised against an Ypt7 peptide, were kindly provided by Dr W Wickner.
Table 1.
Yeast strains
| SUB62 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 |
| RG-wt | MATa his3D1 leu2D0 met15D0 ura3D0 (Research Genetic collection) |
| RG-ypt7Δ | MATa his3D1 leu2D0 met15D0 ura3D0 ypt7Δ∷KAN (Research Genetic collection) |
| RG-vps41Δ | MATa his3D1 leu2D0 met15D0 ura3D0 vps41Δ∷KAN (Research Genetic collection) |
| RG-vam3Δ | MATa his3D1 leu2D0 met15D0 ura3D0 vam3Δ∷KAN (Research Genetic collection) |
| sMK-50 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) |
| sMK-60 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) ecm29Δ∷TRP1 |
| sMK-168 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 pho8Δ∷CLONNAT |
| sMK-170 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 pho8Δ∷CLONNAT ecm29Δ∷TRP1 |
| sMK-172 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) pho8Δ∷CLONNAT |
| sMK-173 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) pho8Δ∷CLONNAT ecm29Δ∷TRP1 |
| sMK-186 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) pep4Δ∷CLONNAT |
| sMK-187 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) pep4Δ∷CLONNAT ecm29Δ∷TRP1 |
| sMK-215 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3)pep4Δ∷CLONNAT prb1Δ∷KAN |
| sMK-227 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpn11∷RPN11-TEVProA (HIS3) pep4Δ∷CLONNAT ecm29Δ∷TRP1 prb1Δ∷KAN |
| sMK-235 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 pep4Δ∷CLONNAT ecm29Δ∷TRP1 prb1Δ∷KAN |
| sMK-237 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 pep4Δ∷CLONNAT prb1Δ∷KAN |
| DY85 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpt1Δ∷HIS3 [Ycplac111-RPT1] |
| DY106 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpt1Δ∷HIS3 [Ycplac111-rpt1K256S] |
| sMK-191, -193 | DY85 pho8Δ∷CLONNAT, DY106 pho8Δ∷CLONNAT |
| sMK-220, -230 | DY85 pep4Δ∷CLONNAT prb1Δ∷KAN, DY106 pep4Δ∷CLONNAT prb1Δ∷KAN |
| MHY-1177 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 pre3-Δ2∷HIS3 pup1Δ∷leu2∷HIS3 [pRS317-PUP1] [Ycplac22-PRE3] |
| MHY-1178 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 pre3-Δ2∷HIS3 pup1Δ∷leu2∷HIS3 [pRS317-pup1-T30A] [Ycplac22-pre3-T20A] |
| sMK-245, -247 | MHY-1177 pho8Δ∷CLONNAT, MHY-1178 pho8Δ∷CLONNAT |
| sMK-248, -251 | MHY-1177 pep4Δ∷CLONNAT prb1Δ∷KAN, MHY-1178 pep4Δ∷CLONNAT prb1Δ∷KAN |
| SUB280 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 ubi1∷TRP1 ubi2-Δ2∷ura3 ubi3-Δub2 ubi4-Δ2∷LEU2 [pUB39= wild-type ubiquitin] [pUB100] |
| SUB413 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 ubi1∷TRP1 ubi2-Δ2∷ura3 ubi3-Δub2 ubi4-Δ2∷LEU2 [pUB176=K63R ubiquitin] [pUB100] |
| sMK-260, -263 | SUB280 pho8Δ∷CLONNAT, SUB413 Δpho8∷CLONNAT |
| sMK-270, -273 | SUB280 pep4Δ∷CLONNAT prb1Δ∷KAN, SUB413 pep4Δ∷CLONNAT prb1Δ∷KAN |
| sMK-291 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 ypt7Δ∷PGAL-3HA-YPT7 (TRP1) |
| SUB592 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 ubi1∷TRP1 ubi2-Δ2∷ura3 ubi3-Δub2 ubi4-Δ2∷LEU2 [pUB221: His6-cMyc-Ubiquitin] [pUB100] |
| sMK-303, -307 | SUB592 ypt7∷PGAL-3HA-YPT7 (KAN), SUB592 rho1∷PGAL-3HA-RHO1 (KAN) |
| sMK-309, -310 | MATa lys2-801 leu2-3, 2-112 ura3-52 his3-Δ200 trp1-1 rpt2Δ∷HIS3 [RPT2∷LEU/CEN] [rpt2G2A∷LEU/CEN] |
| sMK-319, -325 | sMK-172 ypt7∷PGAL-3HA-YPT7 (KAN), sMK-186 ypt7∷PGAL-3HA-YPT7 (KAN) |
| sMK-323, -329 | sMK-172 ypt7∷PGAL-3HA-K147/140R-ypt7 (KAN) sMK-186 ypt7∷PGAL-3HA-K147/140R-ypt7 (KAN) |
| sMK-403, -395 | sMK-172 ypt7∷PGAL-3HA-K147/140/56/5/6/48R-ypt7(KAN) sMK-186 ypt7∷PGAL-3HA-K147/140/56/5/6/48R-ypt7 (KAN) |
| sMK-413, -421 | sMK-325 prb1Δ∷TRP1 sMK-395 prb1Δ∷TRP1 |
Vacuole purification and homotypic vacuole fusion
We followed established purification procedures (Haas, 1995) (W Wickner, personal communication). See Supplementary data for a detailed description. Freshly purified vacuoles were used in all experiments, obviating the need to add cytosol to fusion reactions.
Biochemical reagents and immunoprecipitations
PS341 was kindly provided by Millennium Pharmaceuticals. MG262 was purchased from Calbiochem; MG115 and MG132 from Peptide Institute. Ubistatin-A (Verma et al, 2004) was kindly provided by Dr R King. Ubiquitin aldehyde and β-lactone were obtained from Boston Biochem. Epoxomicin was incapable of inhibiting proteasomes in lipid-rich vacuolar preparations, whereas β-lactone had to be used at 500 μM. This is probably due to the hydrophobic properties of both these reagents. FM4-64 was obtained from Molecular Probes. See Supplementary data for the immunoprecipitation protocol.
Ubiquitin conjugate isolation from purified vacuoles
SUB592 (Finley et al, 1994) vacuoles were dissolved in a denaturing buffer B (8 M urea, 0.1 M NaH2PO4, 0.01 M Tris (pH 8)), containing 5 mM imidazole. Ni2+NTA resin (Qiagen) was added to capture tagged ubiquitin, washed in buffer C (pH 6.3), and eluted in IEF sample buffer (8 M urea, 2% CHAPS, 0.5% (v/v) ampholytes (pH 3–10), 0.002% Bromophenol Blue) containing 200 mM imidazole. The eluate was subjected to 2D IEF/SDS–PAGE (Invitrogen ZOOM IPG Runner), transferred to PVDF membrane, and probed for cMyc to detect tagged ubiquitin (Figure 3B). SUB592 was also used to N-terminally tag Ypt7 and Rho1 with a 3HA tag under the control of the GAL promoter (sMK-303, sMK-307). Here, yeast was grown in YEP medium supplemented with 2% raffinose/2% galactose instead of 2% dextrose.
Mass spectrometry analysis
To identify vacuolar ubiquitin conjugates from Ni2+-purified samples, protein was eluted from Ni2+NTA resin in IEF sample buffer. Samples were prepared from SDS–PAGE gels by in-gel digestion as previously described (Peng and Gygi, 2001), and analyzed by an LTQ-FT hybrid linear ion trap mass spectrometer (ThermoElectron). Data were searched using the SEQUEST algorithm against the yeast protein database, and ubiquitination sites identified as previously described (Peng et al, 2003). All matching spectra were validated manually.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Daniel Finley for the freedom he gave MFK to pursue this project, and his guidance and support, while MFK was a postdoctoral fellow in his laboratory at Harvard Medical School. We extend our warm thanks to William Wickner for invaluable discussions, reagents and technical assistance. We thank Randall King, Hidde Ploegh, Alexei Kisselev and Mark Hochstrasser for advice on proteasome inhibition, and for providing the core mutant (MH). We are grateful to lab colleagues and Neema Sofaer for their insightful comments. This work was supported by a fellowship from the Charles A King Trust, Fleet National Bank, a Bank of America Company, Co-Trustee (Boston, MA) to MFK and by three NIH grants: GM65592 to Daniel Finley and HG3456 and GM67945 to SPG.
References
- Arendt C, Hochstrasser M (1999) Eukaryotic 20S proteasome catalytic subunit propeptides prevent active site inactivation by N-terminal acetylation and promote particle assembly. EMBO J 18: 3575–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K, Stevens TH (2005) Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1744: 438–454 [DOI] [PubMed] [Google Scholar]
- Constantinescu AT, Rak A, Alexandrov K, Esters H, Goody RS, Scheidig AJ (2002) Rab-subfamily-specific regions of Ypt7p are structurally different from other RabGTPases. Structure 10: 569–579 [DOI] [PubMed] [Google Scholar]
- Dong J, Chen W, Welford A, Wandinger-Ness A (2004) The proteasome α-subunit XAPC7 interacts specifically with Rab7 and late endosomes. J Biol Chem 279: 21334–21342 [DOI] [PubMed] [Google Scholar]
- Dupre S, Urban-Grimal D, Haguenauer-Tsapis R (2004) Ubiquitin and endocytic internalization in yeast and animal cells. Biochim Biophys Acta 1695: 89–111 [DOI] [PubMed] [Google Scholar]
- Eitzen G, Thorngren N, Wickner W (2001) Rho1p and Cdc42p act after Ypt7p to regulate vacuole docking and fusion. EMBO J 20: 5650–5656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitzen G, Will E, Gallwitz D, Haas A, Wickner W (2000) Sequential action of two GTPases to promote vacuole docking and fusion. EMBO J 19: 6713–6720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Sadis S, Monia BP, Boucher P, Ecker DJ, Crooke ST, Chau V (1994) Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol 14: 5501–5509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Gorbea C, Goellner G, Teter K, Holmes R, Rechsteiner M (2004) Characterization of mammalian Ecm29, a 26S proteasome-associated protein that localizes to the nucleus and membrane vesicles. J Biol Chem 279: 54849–54861 [DOI] [PubMed] [Google Scholar]
- Haas A (1995) A quantitative assay to measure homotypic vacuole fusion in vitro. Methods Cell Sci 17: 283–294 [Google Scholar]
- Haas A, Schleglmann D, Lazar T, Gallwitz D, Wickner W (1995) The GTPase Ypt7 of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J 14: 5258–5270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Dunn R (2003) Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol 19: 141–172 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Saeki Y, Yokosawa H, Polevoda B, Sherman F, Hirano H (2003) N-terminal modifications of the 19S regulatory particle subunits of the yeast proteasome. Arch Biochem Biophys 409: 341–348 [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Callard A, Goldberg AL (2006) Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem 281: 8582–8590 [DOI] [PubMed] [Google Scholar]
- Leggett DS, Hanna J, Borodovsky A, Crossas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D (2002) Multiple associated proteins regulate proteasome structure and function. Mol Cell 10: 495–507 [DOI] [PubMed] [Google Scholar]
- Mayer A (2002) Membrane fusion in eukaryotic cells. Annu Rev Cell Dev Biol 18: 289–314 [DOI] [PubMed] [Google Scholar]
- Muller O, Johnson D, Mayer A (2001) Cdc42p functions at the docking stage of intracellular membrane fusion. EMBO J 20: 5657–5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Gygi SP (2001) Proteomics: the move to mixtures. J Mass Spectrom 36: 1083–1091 [DOI] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias J, Thoreen C, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi S (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotech 8: 921–926 [DOI] [PubMed] [Google Scholar]
- Rubin D, Glickman M, Larsen C, Dhruvakumar S, Finley D (1998) Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J 17: 4909–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW (2004) Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 306: 117–120 [DOI] [PubMed] [Google Scholar]
- Wang Y, Satoh A, Warren G, Meyer HH (2004) VCIP135 acts as a deubiquitinating enzyme during p97-p47-mediated reassembly of mitotic Golgi fragments. J Cell Biol 164: 973–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann H, Hengst L, Gallwitz D (1992) Endocytosis in yeast: evidence for the involvement of a small GTP-binding protein (Ypt7p). Cell 71: 1131–1142 [DOI] [PubMed] [Google Scholar]
- Wickner W (2002) Yeast vacuoles and membrane fusion pathways. EMBO J 21: 1241–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W, Haas A (2000) Yeast homotypic vacuole fusion: a window on organelle trafficking mechanisms. Annu Rev Biochem 69: 247–275 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information