

Abstract
HIV efficiently spreads in lymphocytes, likely through virological synapses (VSs). These cell–cell junctions share some characteristics with immunological synapses, but cellular proteins required for their constitution remain poorly characterized. We have examined here the role of ZAP-70, a key kinase regulating T-cell activation and immunological synapse formation, in HIV replication. In lymphocytes deficient for ZAP-70, or expressing a kinase-dead mutant of the protein, HIV replication was strikingly delayed. We have characterized further this replication defect. ZAP-70 was dispensable for the early steps of viral cycle, from entry to expression of viral proteins. However, in the absence of ZAP-70, intracellular Gag localization was impaired. ZAP-70 was required in infected donor cells for efficient cell-to-cell HIV transmission to recipients and for formation of VSs. These results bring novel insights into the links that exist between T-cell activation and HIV spread, and suggest that HIV usurps components of the immunological synapse machinery to ensure its own spread through cell-to-cell contacts.
Keywords: HIV, virological synapse, ZAP-70
Introduction
HIV is a fast replicating virus, infecting mostly CD4+ T cells. The state of activation of T cells governs their susceptibility to infection (Stevenson et al, 1990; Zack et al, 1990). Truly quiescent lymphocytes are refractory to the virus, whereas activation provided by TCR and IL-2 signals renders cells sensitive to productive infection (Stevenson, 2003; Chiu et al, 2005). In vivo studies of the early acute phase of AIDS demonstrated a massive infection and destruction of memory CD4+ T cells in lymphatic tissues within the intestinal tract (Haase, 2005; Li et al, 2005). These cells are not ostensibly activated but contain sufficient levels of CCR5 co-receptor, nucleotide pools and transcriptional activators to support productive infection. At later stages of the disease, a pathologic immune activation leads to an increasing proportion of activated memory CD4+ T cells, which represent the preferential targets of the virus (Haase, 2005).
A fast replication necessitates efficient means of viral propagation, which is achieved by direct transfer of the retrovirus during cell-to-cell contacts (Sato et al, 1992; Dimitrov et al, 1993; Pearce-Pratt et al, 1994; Phillips, 1994; Johnson and Huber, 2002; Igakura et al, 2003; Jolly et al, 2004; Piguet and Sattentau, 2004; Sourisseau et al, 2007). Cells naturally communicate by exchanging information through close contacts, associated with a coordinated rearrangement of receptors and other molecules at the junction region. These organized contacts, or synapses, are particularly important in immune cells, for instance promoting an adequate response of the host to pathogens (Cemerski and Shaw, 2006; Dustin et al, 2006). By analogy to the immune synapse (IS), the term virological synapse (VS) has been coined to designate a cytoskeleton- and raft-dependent adhesive junction, across which viruses are efficiently transmitted from donor to recipient cells (Igakura et al, 2003; Jolly et al, 2004; Piguet and Sattentau, 2004; Barnard et al, 2005; Jolly and Sattentau, 2005; Nejmeddine et al, 2005). Cell-to-cell HIV transfer likely occurs in secondary lymphoid organs, where 98% of lymphocytes are present and where these cells are in close contact with each other and with antigen-presenting cells (APCs).
The mechanisms of IS constitution between a lymphocyte and an APC are pretty well understood. On the lymphocyte side, numerous receptors and signal-transducing proteins are involved (Krogsgaard et al, 2003; Dustin et al, 2006). Among the latter, the role of the Syk-related tyrosine kinase ZAP-70 is of great interest. ZAP-70 is essential for lymphocyte activation through the TCR–CD3 complex. This key role has been highlighted by the identification of severe immunodeficiencies caused by mutations within the ZAP-70 gene. In humans, this disease is characterized by the absence of CD8+ T cells and the presence of CD4+ T cells unresponsive to CD2- and CD3-mediated activation (Arpaia et al, 1994; Elder et al, 1994; Meinl et al, 2000). Several models have allowed the characterization of signalling pathways controlled by ZAP-70, that is, the absence of ZAP expression in patients, the generation of ZAP-70−/− mice and the use of the P116 cell line, a Syk and ZAP-70-negative clone derived from Jurkat cells (Arpaia et al, 1994; Elder et al, 1994; Williams et al, 1998; Meinl et al, 2000). Upon TCR ligation, Lck is activated and induces ZAP-70 recruitment to the TCR zeta chain and its phosphorylation, which in turn induces ZAP-70 kinase activity, and the phosphorylation and recruitment of different substrates including SLP76, Vav and LAT (reviewed by Abraham and Weiss, 2004). This initiates intracellular calcium mobilization, cytoskeletal reorganization and cell activation. Notably, ZAP-70 signalling drives the formation of a functional IS by promoting actin cytoskeletal remodelling and microtubule-organizing center (MTOC) polarization toward the APC (Blanchard et al, 2002; Sasahara et al, 2002; Gomez et al, 2006).
The cellular proteins involved in VS formation are much less characterized. The VS forms in response to contact between infected and target cells, and contains viral antigens on one side and cellular receptors on the other, colocalized at the conjugate interface (Igakura et al, 2003; Jolly et al, 2004; Piguet and Sattentau, 2004; Barnard et al, 2005; Jolly and Sattentau, 2005; Nejmeddine et al, 2005). Adhesion molecules are found at the junction, likely stabilizing cell contacts (Hioe et al, 2001; Jolly et al, 2004; Piguet and Sattentau, 2004; Tardif and Tremblay, 2005). Whether other cellular proteins are diverted by the virus to induce the VS remains largely unknown. We hypothesized that proteins involved in the IS might also play a role during HIV replication and VS formation. We have thus examined the role of ZAP-70 in these processes. We report that HIV replication is severely impaired in ZAP-70-defective lymphoid cells. We documented this replication defect and observed that ZAP-70 is required in donor cells, and not in recipients, for efficient cell-to-cell HIV transmission and VS formation. These results bring novel insights into the links that exist between T-cell activation and HIV spread.
Results
HIV replication is impaired in ZAP-70-defective lymphocytes
We asked whether the tyrosine kinase ZAP-70, a key regulator of T-cell activation and immunological synapse formation, plays a role during HIV replication. To this aim, we compared viral growth in Jurkat lymphoid cells, and in the P116 subclone, which lacks ZAP-70 (Williams et al, 1998). To ensure that the behavior of P116 cells was due to the absence of ZAP-70, we also used P116-derived cells reconstituted with either a wild-type or a kinase-dead mutant of ZAP-70 (P116 Zwt and P116 Zdk cells, respectively) (Williams et al, 1998; Blanchard et al, 2002). Analysis of ZAP-70 levels by Western blot (Supplementary Figure 1) confirmed that P116 cells did not express ZAP-70, whereas P116 Zdk and P116 Zwt cells expressed high amounts of the protein (Blanchard et al, 2002). In these lines, HIV entry receptors CD4 and CXCR4 and adhesion molecules LFA-1, ICAM-1 and ICAM-3 were normally present at the cell surface (Supplementary Figure 1). Cell growth rates were similar with or without ZAP-70 (not shown).
Cells were infected with HIV-1 (X4 strain NL4-3) at low or high multiplicities of infection (MOI) (0.02 or 2 ng p24/106 cells, respectively; Figure 1A). At the low MOI, HIV replication occurred efficiently in parental Jurkat and in P116 Zwt cells, reaching about 1000 ng/ml of p24 in cell supernatants at day 14 post-infection (p.i.). In sharp contrast, viral growth was barely detected in P116 and P116 Zdk cells. At the higher MOI, HIV spread was much more rapid in parental Jurkat and in P116 Zwt, peaking as soon as day 7 p.i (Figure 1A). In the absence of ZAP-70, Gag p24 production became detectable in cell supernatants, but was delayed by about 7 days. In four independent experiments, in control Jurkat cells, levels of p24 in supernatants, at the day of the peak of viral production (days 6–8 p.i. for high MOI, day 14 for the low MOI), were about 15-fold higher than those measured the same days in ZAP-70-deficient cells (Figure 1B). Therefore, HIV replication is significantly impaired in the absence of ZAP-70 in Jurkat cells.
Figure 1.
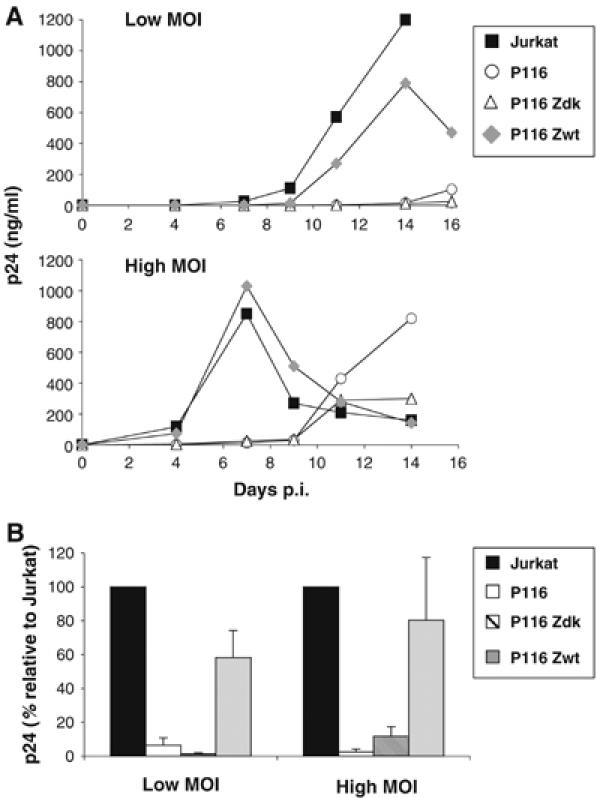
HIV replication is impaired in ZAP-70-defective Jurkat cells. (A) Kinetic analysis. Jurkat (ZAP-70+), P116 (ZAP-70−) and P116 clones reconstituted with either a wild type or a kinase-dead mutant of ZAP-70 (P116 Zwt and P116 Zdk) were exposed to the indicated HIV-1 (NL4-3 strain) inocula (0.02 and 2 ng p24/106 cells/ml). Viral replication was followed by measuring p24 release in cell supernatants, at the indicated days p.i. Data are representative of four independent experiments. (B) Viral production at the peak. A mean±s.d. of four independent experiments is depicted, with 100% corresponding to p24 values obtained in Jurkat cells at the peak (days 6–8 and 14 for the high and low MOI, respectively). With other cell clones, the % values were calculated with p24 values obtained the same day p.i. Low and high MOI corresponded to 0.02–1 and 2–10 ng p24/106 cells/ml, respectively.
Early steps of HIV replication do not require ZAP-70 activity
We sought to determine which steps of the viral cycle were affected by the absence of ZAP-70. We first assessed the early steps of viral replication. To this aim, Jurkat clones were infected with a single-cycle HIV, pseudotyped with X4 envelope glycoproteins, and expressing the luciferase reporter protein in place of Nef (HIV-Luc) (Connor et al, 1995; Nobile et al, 2005). Luciferase activity, at 48 h p.i., was similar in ZAP-70-positive (Jurkat and P116 Zwt) and -negative (P116 and P116 Zdk) cells (Figure 2A), irrespective of the MOI (not shown). We then asked whether less viruses may be produced in the absence of ZAP-70. We examined Gag expression in productively infected cells and levels of viral release upon infection with single-cycle, envelope-deleted HIV pseudotyped with VSV-G glycoproteins (HIVΔenv(VSV)). Comparable amounts of Gag-positive cells were detected by flow cytometry in P116, P116 Zdk and P116 Zwt cells (Figure 2B). Lysates from infected cells harbored similar levels of Gag p24, as measured by ELISA (not shown), and viral release in supernatants was also similar with or without ZAP-70 (Figure 2C). The efficiency of HIV DNA synthesis was then quantified by real-time PCR analysis. Jurkat derivatives were infected with replication-competent HIV (at a low MOI) and levels of pol copies were measured at various times, from 24 h to 10 days p.i (Figure 2D). At 24 h, pol products were in the range of 103 copies per 106 cells, in all Jurkat clones, with or without the kinase. However, at later time points, in cell expressing ZAP-70 (Jurkat and P116 Zwt), there was a rapid increase of pol DNA levels, reaching 1 × 106 and 5 × 108 copies per 106 cells at days 4 and 8 p.i., respectively. In contrast, viral spread was much slower in P116 and P116 Zdk cells, with a strong reduction (50- and 1000-fold decrease, at days 4 and 8 p.i., respectively) of proviral DNA. At a later time point (day 10 p.i.), these amounts reached levels detected in ZAP-70-positive cells.
Figure 2.
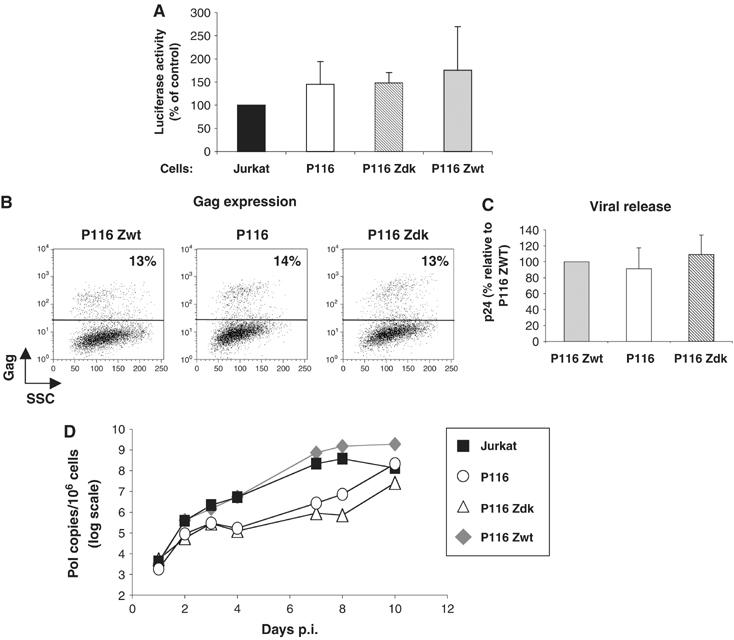
Early steps of the viral cycle and proviral DNA synthesis in ZAP-70-defective cells. (A, B) Replication of single-cycle HIV. (A) Jurkat clones were infected with single-cycle HIV, pseudotyped with X4 envelope glycoproteins, and expressing luciferase reporter protein (20 ng p24/1.5 × 106 cells). After 48 h, cell lysates were analyzed for luciferase activity (in relative light units). Data represent means±s.d. of three independent experiments, with 100% corresponding to values obtained in Jurkat cells. (B–C) HIV Gag expression and release. Jurkat clones were infected with HIV(VSV), an env-deleted HIV, pseudotyped with VSV-G glycoproteins (0.75 ng p24/1.5 × 106 cells). After 72 h, Gag expression was measured by flow cytometry (B). One out of three independent experiments is shown. No Gag signal was detected with non-infected cells (not shown). (C) Gag p24 release was measured in cell supernatants by ELISA at 72 h p.i. Data represent means±s.d. of three independent experiments, with 100% corresponding to values obtained in P116 Zwt cells. (D) HIV proviral DNA synthesis. Jurkat clones were exposed to HIV-1 NL4-3 (0.2 ng p24/ml/106 cells) for 2 h and grown at 37°C for the indicated days. Quantification of late (pol DNA) viral products was performed by real-time PCR. Data are means±s.d. of triplicates and are representative of three independent experiments.
Altogether, these results indicate that upon infection with cell-free virus, viral entry, reverse transcription, expression of viral proteins and release of Gag proteins in the supernatants do not require ZAP-70. Subsequent viral spread in the culture is much more efficient and rapid in the presence of the kinase. ZAP-70 thus likely improves other aspects of the viral cycle.
Impact of ZAP-70 on HIV infectivity and cell-to-cell transmission
We then examined how late steps of viral replication occurred in the presence or absence of ZAP-70. We measured the infectivity of virions released from the various Jurkat clones and also asked whether the kinase may affect direct cell-to-cell transmission of the infection. Western blot analysis of virions issued from ZAP-70-positive and -negative Jurkat cells did not reveal any obvious difference in Gag and Env content and processing (not shown). Infectivity of viral particles was then measured by using a single-cycle assay. Supernatants from Jurkat cells were normalized for Gag p24 content and tested on P4 indicator cells, an HeLa-CD4 derivative carrying an HIV-LTR Lac-Z cassette activated by Tat upon HIV-1 infection. There was no significant difference when comparing virions released by parental Jurkat and P116 cells (Figure 3A), indicating that ZAP-70 does not significantly impact infectivity of cell-free viral particles. Results were more variable when analyzing infectivity of virions produced in P116 Zdk and P116 Zwt cells. There was a decrease of infectivity with the kinase-dead mutant and an increase with the wild-type kinase. Of note, in these two cell lines, the two proteins are overexpressed when compared to endogenous levels found in parental cells (Supplementary Figure 1), and this overexpression may explain these slight variations in viral infectivity. Overall, we conclude that endogenous ZAP-70 does not significantly influence the infectivity of viral particles released from Jurkat cells.
Figure 3.
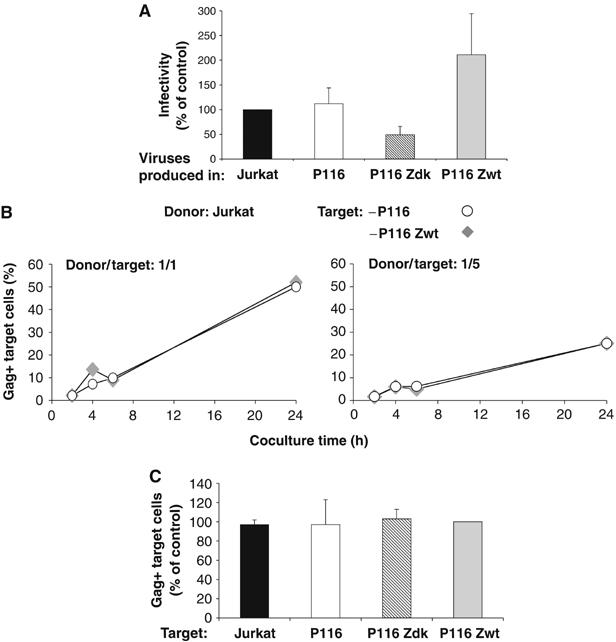
Impact of ZAP-70 on infectivity of cell-free virions and on cell-to-cell HIV transmission. (A) Infectivity of virions produced in ZAP-70-defective cells. Viruses produced in the indicated Jurkat clones was assayed in an infectivity assay. Supernatants were harvested at the pre-peak and peak of viral production (60–500 ng p24/ml). P4 reporter cells were exposed to HIV (NL4-3, 5 ng p24) and infection was assessed 24 h later, by measuring β-galactosidase activity in cell extracts. Data are means±s.d. of five independent experiments, with 100% corresponding to values obtained in Jurkat cells. (B, C) HIV cell-to-cell transfer does not require ZAP-70 in target cells. Productively HIV-1-infected Jurkat cells (20% Gag+ cells at the beginning of the assay) were co-cultivated with CFSE+ P116 or P116 Zwt target cells, at a 1/1 or a 1/5 donor/target ratio. The % of Gag+ cells among targets (CFSE+) is shown at the indicated times of coculture. (B) A representative experiment is shown. (C) A mean±s.d. of five independent experiments (24 h time point) is depicted, with 100% corresponding to values obtained in P116 Zwt cells.
Cell-to-cell viral transmission is a rapid and potent phenomenon (Sato et al, 1992; Kok et al, 1993; Phillips, 1994; Davis et al, 1997). We recently reported that HIV propagation in cultures involves mostly cell-associated virus, whereas free virions play a marginal role in this process (Sourisseau et al, 2007). We have developed a quantitative flow-cytometry-based assay that selectively monitors cell-to-cell viral transfer (Sourisseau et al, 2007). Briefly, acutely infected donor cells are mixed with CFSE-labelled target lymphocytes and viral production is followed by measuring Gag levels in targets at different times. We therefore examined the role of ZAP-70 during cell-to-cell viral transmission. We used different combinations of infected donors and recipients, expressing or not an active kinase.
We first assessed the importance of ZAP-70 in recipient cells. Parental Jurkat cells were first productively infected with HIV. After 2 days, about 20% of the cells were Gag+. These cells were used as donors and co-cultivated with CFSE-labelled P116 Zwt or P116 cells (Figure 3B). With P116 Zwt recipients (at a 1/1 donor/target ratio), productive viral transfer was efficacious, with about 10% of the targets expressing Gag at 4–6 h post coculture. This fraction increased rapidly reaching 50% of Gag+ cells at 24 h. Similar kinetics were observed with P116 targets (Figure 3B). With a 1/5 donor/target ratio, the efficacy of transfer was reduced, and again no difference was detected between P116 Zwt and P116 cells (Figure 3B). Similar results were obtained with Jurkat and P116 Zdk cells (Figure 3C), confirming that ZAP-70 in targets does not influence cell-to-cell HIV transfer.
We then compared the behavior of Jurkat and P116 as donors in the viral transmission assay. These cells were first productively infected with HIV. A higher viral inoculum was used with P116, as they are partly restrictive to HIV (see Figure 1). With these different inocula, about 25% of Jurkat and P116 cells were Gag+, at day 3 p.i (Figure 4C). These donors were then cocultured with CFSE+ Jurkat as recipients. As expected, parental Jurkat efficiently transmitted HIV to these targets with about 55% of Gag+ cells at 24 h post coculture (donor/target ratio 1/1) (Figure 4A). In contrast, P116 donors less potently transmitted the infection, with only 4 and 20% of targets being Gag+ at 6 and 24 h, respectively (Figure 4A). A similar loss of viral transfer occurred at a lower donor/target ratio (1/6) (Figure 4A). ZAP-70 itself facilitates cell-to-cell transfer as P116 Zwt behaved like parental Jurkat, whereas P116 Zdk were poor transmitters (Figure 4B). In six independent experiments, there was a 2–2.5-fold decrease in the efficiency of viral transfer from ZAP-70-defective cells (Figure 4B).
Figure 4.
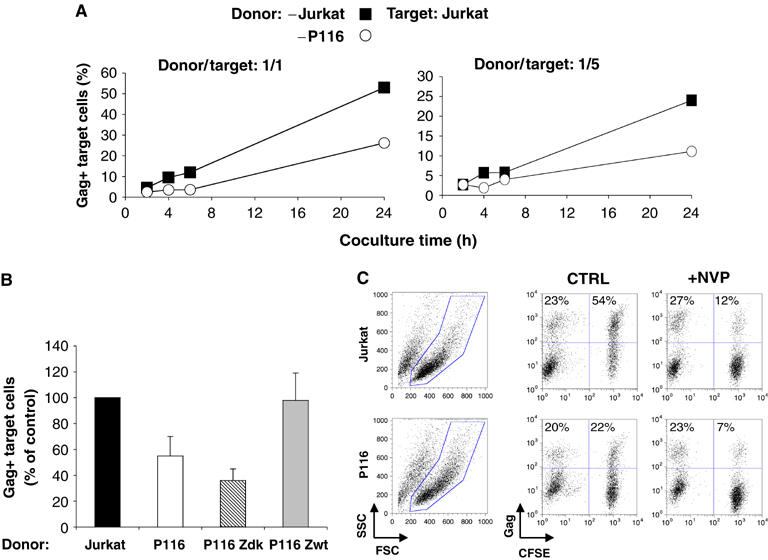
ZAP-70 facilitates cell-to-cell HIV transfer to recipient cells. (A) Productively HIV-infected Jurkat and P116 cells (about 25% Gag+) were cocultivated with target CFSE+ Jurkat cells, at a 1/1 or a 1/5 ratio. The % of Gag+ cells among targets (CFSE+) is shown at the indicated times of coculture. (B) A mean±s.d. of six independent experiments (24 h time point) is depicted, with 100% corresponding to values obtained in Jurkat cells. (C) HIV productive transfer is sensitive to NVP. HIV-infected Jurkat and P116 cells (25% of Gag+ cells at the beginning of the assay) were cocultivated with target CFSE+ Jurkat cells, with or without NVP. The % of Gag+ cells among donors and targets, at 24 h post coculture, is indicated. Data are representative of three independent experiments.
After 24 h of coculture, viability was similar for Jurkat and P116 cells (Figure 4C and not shown). When cells were incubated with the reverse transcriptase inhibitor nevirapine (NVP), the fraction of Gag+ cells among recipients was strongly decreased in both cell types (Figure 4C), indicating that the Gag signal mostly corresponded to de novo synthesis. Furthermore, only a few syncytia were formed with this experimental setting, without a significant difference between P116 and Jurkat cells (not shown; Sourisseau et al, 2007).
Altogether, these results indicate that ZAP-70 facilitates, in productively infected cells, viral transmission to targets, through direct cell-to-cell contacts.
ZAP-70 facilitates formation of the VS
We hypothesized that ZAP-70 may regulate HIV cell-to-cell transfer by facilitating VS formation (Jolly et al, 2004). We first examined the localization of Env and Gag proteins in infected cells, by confocal microscopy (Figure 5A). In the absence or presence of ZAP-70, Env staining gave a typical pattern, with a signal enriched in an intracellular compartment, likely corresponding to newly synthesized proteins in the Golgi or accumulating in an endocytic compartment (Miranda et al, 2002; Blot et al, 2003; Pelchen-Matthews et al, 2003). The Gag signal did not significantly overlap with Env and was heterogeneous. In parental Jurkat cells, Gag proteins were present in the cytoplasm, with bright spots often observed at the cell cortex (Figure 5A). These spots probably represent Gag proteins accumulating in multivesicular body (MVB)-like compartments or present at discrete regions of the plasma membrane (Grigorov et al, 2006; Nydegger et al, 2006; Perlman and Resh, 2006). In P116 cells, the Gag pattern was often more diffuse, with less intense signal at the plasma membrane (Figure 5A). In ZAP-70+ cells (Jurkat or P116 Zwt), about 60% of the cells harbored this discrete Gag signal near or at the plasma membrane (Figure 5B). This punctuate pattern was more rarely observed in ZAP-70-deficient cells (about 20% of P116 and P116 Zdk cells).
Figure 5.
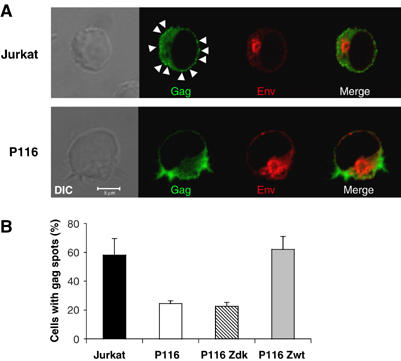
Localization of HIV Gag and Env proteins in ZAP-70-defective cells. (A) Jurkat and P116-infected cells (about 50% Gag+) were fixed and stained with anti-Gag or anti-Env mAbs. Scale bar, 5 μm. A representative single medial optical section is shown in the fluorescence picture. Gag and Env stainings were negative in non-infected cells (not shown and Figure 7). Arrowheads point to discrete Gag spots. (B) Quantification of cells with discrete Gag spots. The indicated cells were infected with HIV and stained with anti-Gag antibodies. The % of infected cells with discrete bright Gag+ spots was measured by examining the cells with a fluorescence microscope. About 600 cells were analyzed for each condition, in four independent experiments. A mean±s.d. of four independent experiments is shown.
We then examined how infected cells formed VS. We measured by immunofluorescence the recruitment of Gag proteins at the contact zone between donors and recipients, a hallmark of VS formation (Jolly et al, 2004). Parental Jurkat were used as recipients and were stained with CFSE, before being incubated for 1 h with HIV-1-infected ZAP-70-positive and -negative cells. The percentage of Gag+ cells forming conjugates with CFSE+ cells was similar with the various Jurkat derivatives (about 20%) (Figure 6B). With parental Jurkat, as well as P116 Zwt cells, a polarization of Gag at the interface zone with recipient cells was detected (Figure 6A). About 30% of conjugates displayed a polarization of Gag at the junction (Figure 6C). The situation was different with P116 and P116 Zdk cells, in which the diffuse Gag staining was generally less clustered at the cell interface. Less than 15% of conjugates harbored polarized Gag patches (Figure 6C). Altogether, these data show that the ability of infected lymphocytes to conjugate with targets does not require ZAP-70. However, in the absence of an active kinase, Gag proteins do not correctly localize within infected cells. This results in an impaired constitution of VS, when ZAP-70-deficient infected cells encounter uninfected targets.
Figure 6.
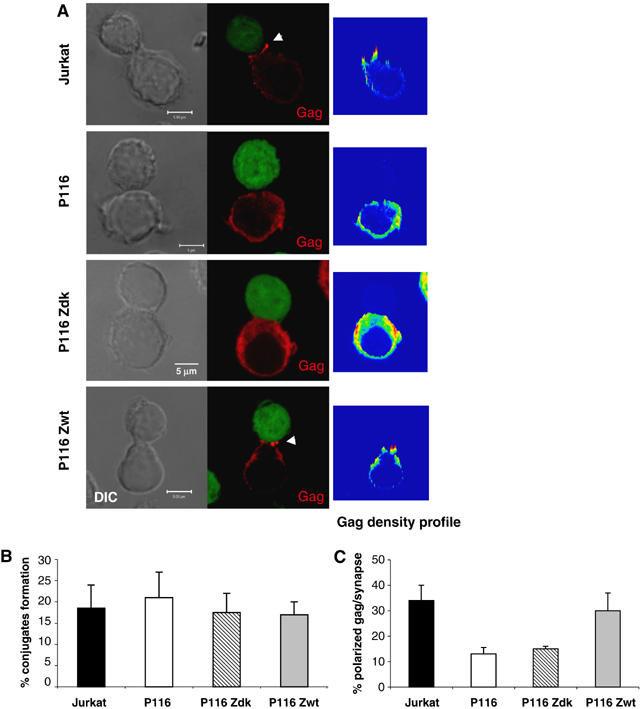
ZAP-70 facilitates formation of the VS. (A) Subcellular localization of Gag displayed by T cell conjugates. The indicated HIV-infected Jurkat derivatives (with about 50–60% of Gag+ cells) were incubated for 1 h with CFSE+ Jurkat cells, fixed and stained with anti-Gag mAbs. Scale bar, 5 μm. A representative single medial optical section is shown in the fluorescence picture. Arrowheads point to polarized Gag staining at the cell–cell junction. Right panels represent the density profiles of Gag fluorescence obtained from a XY projection of three medial optical sections. Color scale goes from blue (zero) to yellow (intermediate) to red (maximal). (B) Quantitative analysis of conjugates formation among Gag+ cells. Cells were treated as in (A) and the % of infected (Gag+) cells forming conjugates with target CFSE+ cells was quantified. A total number of about 600 cells were analyzed for each cell type condition, in four independent experiments. A mean±s.d. of four independent experiments is shown. (C) Quantitative analysis of Gag polarization at the cell–cell junction. Cells were treated as in (A) and the % of Gag+/CFSE+ cell conjugates harboring a polarized Gag staining at the cell interface was measured. A total number of about 200 conjugates were analyzed for each clone. A mean±s.d. of four independent experiments is shown.
ZAP-70 promotes polarization of the MTOC at the immunological synapse (Blanchard et al, 2002). On the other hand, during HTLV-I propagation, the MTOC is reorientated at the junction zone between infected and naïve lymphocytes (Igakura et al, 2003; Barnard et al, 2005; Nejmeddine et al, 2005). We thus examined the localization of the MTOC in Jurkat and P116 cells, upon contact with Jurkat cells. With uninfected donor cells, irrespective of the presence or absence of the kinase, there was a random localization of the MTOC (visualized with an anti-centrin mAb), with about 35% of cells displaying staining at the contact zone (Figure 7). Upon HIV infection of ZAP-70-positive donors (Jurkat and P116 Zwt cells), we observed an increased reorientation of the MTOC at the VS (in about 55% of cell conjugates; Figure 7). Interestingly, this was not the case with HIV-infected P116 and P116 Zdk donor cells (Figure 7). Therefore, the MTOC tends to be reorientated at the VS during HIV cell-to-cell spread, and this phenomenon is improved by the presence of ZAP-70 in donor cells.
Figure 7.
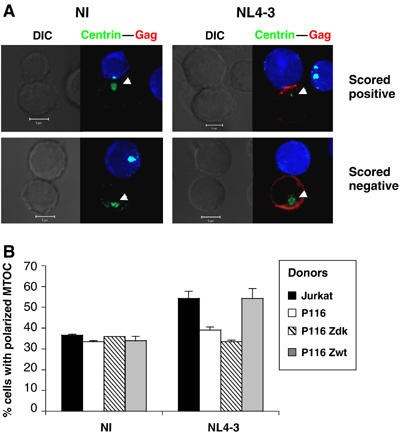
MTOC localization in VSs. (A) Non-infected (NI) or HIV-infected Jurkat cells were incubated with Cell Trace-labelled target Jurkat cells (blue staining) for 1 h. Cells were then fixed and labelled with anti-Gag (red) and anti-centrin3 antibodies (green). Localization of MTOC in donor cells within conjugates was evaluated. Cells were scored positive for MTOC localization if the centrin labelling (pointed by a white arrowhead) was located in proximity to the cell–cell junction zone. (B) A mean±s.d. of three independent experiments is shown with the indicated cell clones. At least 400 conjugates were blindly scored for each cell type.
ZAP-70 is required for efficient HIV replication in primary lymphocytes
We then examined the role of ZAP-70 in primary cells. We used CD4+ lymphocytes from three ZAP-70-deficient infants, suffering from a severe combined immunodeficiency syndrome (Meinl et al, 2000; Noraz et al, 2000). The absence of ZAP-70 was verified by Western blot (Supplementary Figure 1). Surface levels of HIV receptors and a panel of adhesion molecules were normal in ZAP-70-deficient cells, when compared to cells from control donors (Supplementary Figure 1). Cells were infected with the X4 isolate NL4-3 and the R5 strain NLAD8, at two MOIs (2 and 20 ng p24/106 cells, respectively). One representative experiment is depicted in Figure 8A. Both viruses efficiently replicated in control cells, but not in ZAP-70-defective cells. In four independent experiments with cells from three ZAP-70-deficient donors, there was a 5–10-fold decrease in viral production, when compared to control cells from three donors (Figure 8B). We further documented this defect. Infectivity of NL4-3 and NLAD8 virions produced from deficient cells was normal (Figure 8C). We then used single-cycle HIVΔenv(VSV) viruses. Once productively infected, ZAP-deficient cells expressed and released levels of Gag proteins equivalent to control lymphocytes (Figure 8D). Altogether, these experiments demonstrate that the crucial role of ZAP-70 in HIV replication is a general phenomenon, not restricted to transformed Jurkat cells.
Figure 8.
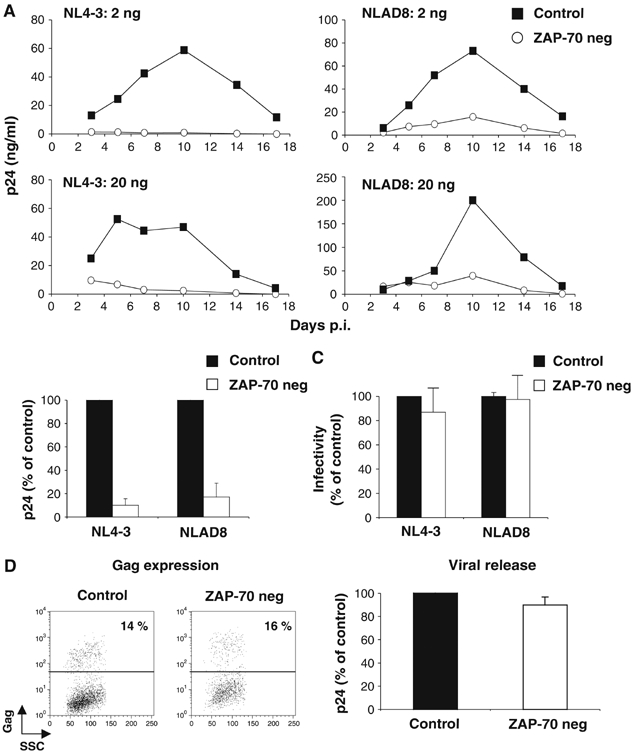
HIV replication in primary ZAP-70-deficient CD4+ T cells. (A) Kinetic analysis. HIV replication in primary CD4+ T cells from one control and one ZAP-70-negative donor. The X4 NL4-3 and R5 NLAD8 strains (2 and 20 ng p24/ml/106 cells) were used. Data are representative of four independent experiments. (B) Viral production at the peak. A mean±s.d. of four experiments, with cells from three controls and three ZAP-70-negative individuals, is depicted, with 100% corresponding to p24 values obtained in control cells at the peak. (C) Infectivity of virions. Viruses produced in the indicated primary cells were assayed in a P4 infectivity assay, as described in Figure 3. Data are means±s.d. of three independent experiments, with 100% corresponding to values obtained with ZAP-70+ cells. To produce high amounts of HIV in primary cells, a high MOI was used (100 ng p24/106 cells). (D–E) HIV Gag expression and release. Primary cells were infected with HIV(VSV), an env-deleted HIV, pseudotyped with VSV-G glycoproteins (100 ng p24/1.5 × 106 cells). After 72 h, Gag expression was measured by flow cytometry (D). One out of three independent experiments is shown. No Gag signal was detected with non-infected cells (not shown). (E) Gag p24 release was measured in cell supernatants by ELISA at 72 h p.i. Data represent means±s.d. of three independent experiments, with 100% corresponding to values obtained in P116 Zdk cells.
Discussion
We report here that ZAP-70 positively regulates HIV replication in lymphocyte cultures. In ZAP-70-negative P116 cells, as well as in primary CD4+ lymphocytes from ZAP-deficient patients, viral growth was significantly impaired. In P116 cells, re-introduction of wild-type ZAP-70, but not of a catalytically inactive mutant, restored normal viral replication. We determined which steps of the viral cycle were affected in the absence of ZAP-70. Early steps, from entry of cell-free virus to LTR-mediated HIV transcription, were similar in ZAP-70-negative and -positive Jurkat cells. Quantification of HIV DNA levels over time indicated that in ZAP-70-defective cells, the first infection cycle with free virions occurred normally, whereas subsequent viral propagation was strongly reduced. The infectivity of viral particles released from Jurkat and P116 cells was similar, opening the possibility that cell-to-cell viral transfer may be altered in the absence of ZAP-70. We indeed demonstrate that the presence of ZAP-70 in productively infected donor cells facilitates viral transfer to targets. Without the kinase, this inefficient viral transfer was associated with an abnormal localization of HIV Gag proteins in infected cells and with an impaired formation of VS between donor and recipient cells, as assessed by an altered polarization of Gag at the cell–cell junction. The key role of ZAP-70 in the signalling cascade that accompanies TCR-mediated lymphocyte activation is well documented (Hivroz and Fischer, 1994; Abraham and Weiss, 2004). Recognition by a T cell of its specific ligand at the surface of an APC provokes dramatic cytoskeletal changes and MTOC polarization toward the APC (Roumier et al, 2001; Barda-Saad et al, 2005; Cemerski and Shaw, 2006). This phenomenon allows the relocalization of the T-cell's secretory and recycling machineries and a polarized delivery of lymphokines and proteins at the IS (Das et al, 2004; Dustin et al, 2006). These events rely on ZAP-70, which controls T-cell polarization and the supply of the synapse with signalling molecules like PKC-theta, LAT and WASP (Blanchard et al, 2002; Sasahara et al, 2002). ZAP-70 exerts additional effects in lymphocytes, controlling the reactivity of the cells to other stimuli, such as chemoattraction to CXCL12 or RANTES, LFA-1-dependent lymphocyte attachment and migration, or response to oxidative stress (Ticchioni et al, 2002; Bacon et al, 1996; Griffith et al, 1998; Soede et al, 1999; Goda et al, 2004).
How do these effects of ZAP-70 on lymphocyte biology relate to the positive role of the kinase in HIV cell-to-cell transfer? Viruses hijack cellular machineries of activation, internalization and exocytosis to replicate. The early steps of the viral cycle are generally regulated by virus-induced signalling and endocytic events, as well as by cytoskeleton reorganization (Pelkmans, 2005; Marsh and Helenius, 2006). Viral release often requires the endosomal compartment, as well as the actin or microtubule cytoskeleton, for the transport of viral components to assembly and budding sites. Manipulation of these cellular machineries by the virus allows for a polarized egress and efficient cell-to-cell spread of numerous viral species, such as herpes and vaccinia viruses, HIV and HTLV (Johnson and Huber, 2002). For instance, an HTLV-1-infected cell polarizes its MTOC toward the cell–cell junction, where Gag proteins accumulate before transfer (Barnard et al, 2005; Nejmeddine et al, 2005). Cytoskeletal reorganization is also required for HIV transfer through the VS (Pearce-Pratt et al, 1994; Jolly et al, 2004). One attractive hypothesis is that the ability of ZAP-70 to control lymphocyte polarization is exploited by HIV to ensure its own spread, by facilitating accumulation of Gag proteins at the right place. This hypothesis is supported by our observation that MTOC polarization, as well as Gag localization, is different with or without ZAP-70. In the absence of the kinase, Gag staining was more diffuse and less punctuated, and this abnormal pattern was associated with an impaired polarization, when naïve recipients were added to the cultures. The trafficking of Gag precursors is well characterized (Morita and Sundquist, 2004; Demirov and Freed, 2004). Gag proteins usurp the cellular machinery that normally creates MVBs (Demirov and Freed, 2004; Morita and Sundquist, 2004; Grigorov et al, 2006; Perlman and Resh, 2006). Moreover, Gag proteins are enriched in subcellular microdomains, where lipid rafts and specific proteins like tetraspanins also concentrate (Chazal and Gerlier, 2003; Garcia et al, 2005; Nydegger et al, 2006). It will be worth dissecting further the impact of ZAP-70 on the spatial and temporal arrangements of cellular or viral proteins and lipids, leading to efficient HIV cell-to-cell transfer.
Similarities between VS and IS include high concentration of adhesion molecules and talin, which links the adhesion rings to the actin cytoskeleton, and polarization of the MTOC toward the synapse (Morita and Sundquist, 2004; Piguet and Sattentau, 2004). In the IS, these events require ZAP-70. Our work demonstrates that HIV diverts components of the IS to facilitate its spread through cell–cell contacts.
Env localization looked normal in ZAP-70-defective lymphocytes. Formation of cell–cell conjugates as well as surface levels of various adhesion molecules that are central to cell-to-cell viral transfer and VS formation (Hioe et al, 2001; Piguet and Sattentau, 2004; Tardif and Tremblay, 2005), was not obviously affected by the kinase. Therefore, our results suggest that ZAP-70 does not regulate Env binding to its receptors, nor interactions between adhesion molecules during viral transfer.
It will be now worth determining whether viral proteins known to influence T-cell activation and IS formation, such as Nef (Simmons et al, 2001; Thoulouze et al, 2006), are also involved in viral transfer through VS. Of note, replication of Nef-deleted HIV was more affected than that of wild-type virus in P116 cells (not shown). Moreover, HIV infection did not compensate the defects in IS formation and T-cell activation associated with the absence of ZAP-70 (not shown). Experiments are currently performed in our laboratory to decipher the role of Nef, in the absence or presence of the kinase.
Our findings have implications concerning HIV-1 pathogenesis. We have identified here a novel limiting step in infected, quiescent lymphocytes. These cells probably inefficiently transmit viral infection to bystander cells, owing to the lack of active ZAP-70. This kinase is activated by TCR triggering, but also by stimulation through CD2, chemokine receptors or LFA-1, or during an oxidative stress (Griffith et al, 1998; Soede et al, 1999; Meinl et al, 2000). Our results suggest that signalling through these pathways will promote cell-to-cell viral spread. During the acute phase of HIV and SIV infection, viral replication occurs mostly in phenotypically resting lymphocytes (Haase, 2005; Li et al, 2005). It is tempting to speculate that in these cells, ZAP-70 or related proteins are sufficiently activated to support cell-to-cell viral transfer.
Materials and methods
Cells, viruses and infections
Jurkat (E6.1 clone), P4 (HeLa-CD4+ HIV LTR-LacZ), and P116 derivatives were grown as described (Williams et al, 1998; Blanchard et al, 2002; Nobile et al, 2005; Thoulouze et al, 2006). Human primary CD4+ T lymphocytes were isolated from three healthy donors and three ZAP-70-deficient infants, as described previously (Meinl et al, 2000; Noraz et al, 2000). For activation, two protocols were used, with similar results. In the first protocol, primary T cells were treated with phorbol myristate acetate (20 ng/ml) and ionomycin (5 × 10−7 M) for 48 h and cultured in the presence of IL-2 (50 IU/ml; Chiron). In the second protocol, cells were stimulated with PHA and irradiated feeder cells as described (Noraz et al, 2000) and cultivated with IL-2. After stimulation, cells were generally expended for 10 days before infection.
Production of HIV (NL4-3 or NLAD8 strains) and HIVΔenv(VSV) has been described (Maréchal et al, 1998; Petit et al, 2001). For HIV replication, cells were exposed to the indicated virus for 2 h 30 min, washed and cultured in 96-well plates (2 × 105 cells/well, in triplicate). Viral input varied from 0.02 to 20 ng p24/ml/106 cells, leading to 1–20% of Gag+ Jurkat cells after 48 h of infection (in single-cycle titration experiments in P4 cells, 1 ng p24 corresponded to 100–1000 PFU; Schwartz et al, 1995). Viral release was monitored by measuring Gag p24 production in the supernatants by ELISA (Perkin-Elmer Life Science). For cell-to-cell HIV transfer or immunofluorescence studies, Jurkat derivatives were infected with NL4-3 (2–50 ng p24/ml/106 cells) and used a few days later, when about 20–60% of the cells were Gag+. Single-cycle infections of P4 cells were performed as described (Maréchal et al, 1998). Infectivity was measured 36 h after viral exposure. For experiments with single-cycle viruses carrying the luciferase gene (HIV-Luc, 20 ng p24/ml/1.5.106 cells) or with HIVΔenv(VSV) (0.5–5 and 100 ng p24/ml/1.5.106 cells for Jurkat derivatives or primary cells, respectively), cells were harvested 48 h p.i. Luciferase activity in cell lysates was measured using a kit (Promega) and a luminometer (Perkin-Elmer).
Flow cytometry, confocal microscopy and Western blot analysis
Cell surface stainings were performed at 4°C for 30 min using mAbs directed against the following molecules: CD4 (13B8.2-APC. Beckman Coulter), CXCR4 and CCR5 (12G5 and 2D7, NIH AIDS research and reference reagent program), CD11a (TS1/22, ATCC), CD18 (TS1/18, ATCC), ICAM-1 and ICAM-3 (F10.2 and CBRR-IC3//1). Gag p24 expression was measured on permeabilized cells with anti-Gagp24 (FITC- or RD1-coupled) mAb (KC57, Coulter). Isotype-matched mAbs were used as negative controls. Samples were analyzed by flow cytometry using a FacsCalibur (Becton Dickinson), with CellQuest software. For immunofluorescence, cells were fixed, permeabilized and stained with anti-Gag mAbs (Hybridolabs, Pasteur). Double Gag/Env stainings were performed with rabbit anti-p24 from NIH AIDS reagents program (reference 384) and anti-Env mAb (110.H, Hybridolabs, Pasteur). Rabbit anti-centrin3 Ab has been described (Blanchard et al, 2002). Confocal microscopy analysis was carried out on a Zeiss LSM510 using a × 63 objective as described (Thoulouze et al, 2006). Z-series of optical sections were performed at 0.2–0.5 μm increments for qualitative analysis. Green and red fluorescence were acquired sequentially. Anti ZAP-70 (clone 29, Pharmingen) and anti-Vav (clone 30, a kind gift from J Griffin, Boston) mAbs were used in the Western blot analysis.
Analysis of HIV proviral DNA levels by quantitative PCR
Jurkat derivatives were exposed to HIV NL4-3 (0.2 ng p24/ml/106 cells) for 2 h 30 min and cultured for the indicated periods of time. DNA was prepared from infected cells and levels of pol DNA were measured with a Light Cycler as described (Nobile et al, 2005).
Analysis of cell-to-cell HIV transfer by flow cytometry
Donor cells were infected with HIV NL4-3 and used a few days later, when 20–40% of the cells were Gag+. Cell-to-cell HIV transfer was performed as described (Sourisseau et al, 2007). Target cells were labelled with CFSE (2.5 μM; Molecular Probes) for 10 min at 37°C. Donor and target cells were then mixed at the indicated ratio, in 96-well plates, at a final concentration of 1 × 106/ml, in a final volume of 200 μl. At the indicated time points, cells were stained for intracellular Gag and analyzed by flow cytometry. When stated, NVP (12.5 nM) was added 0.5 h before coculture and maintained during the assay.
VS formation
Donor cells were infected with HIV NL4-3 and used a few days later, when about 50% of the cells were Gag+. Donors and CFSE-labelled target cells were then mixed at a 1/1 ratio, in 1.5 ml vials, at a final concentration of 1 × 106/ml, in 200 μl. After 1 h at 37°C, cells were gently harvested and loaded on a polylysine-coated cover slip. Cells were then fixed and stained. The % of infected cells carrying Gag bright spots and the % of infected (Gag+) cells forming conjugates with target CFSE+ cells were determined by visual examination of cells present in representative fields. About 600 cells were analyzed for each experimental condition, in four independent experiments. For quantitative analysis of Gag polarization, the % of Gag+/CFSE+ cell conjugates harboring a polarized Gag staining at the cell–cell interface was measured on numerous representative fields. About 200 conjugates were analyzed for each point, in four independent experiments. When stated, targets were labelled with Cell Trace Far Red DDAO-SE (Molecular Probes).
Supplementary Material
Supplementary Material
Acknowledgments
We thank N Casartelli for critical reading of the manuscript, M Bornens, J Griffin and the NIH AIDS research and reference reagent program for the kind gift of reagents and Pascal Roux and Emmanuelle Perret (Plateforme d'Imagerie Dynamique Institut Pasteur) for expert assistance with microscopy imaging. We thank the patients for the gift of cell samples. This work was supported by grants from ANRS, SIDACTION, Fondation de France, CNRS, the European Community and Institut Pasteur.
References
- Abraham RT, Weiss A (2004) Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol 4: 301–308 [DOI] [PubMed] [Google Scholar]
- Arpaia E, Shahar M, Dadi H, Cohen A, Roifman CM (1994) Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell 76: 947–958 [DOI] [PubMed] [Google Scholar]
- Bacon KB, Szabo MC, Yssel H, Bolen JB, Schall TJ (1996) RANTES induces tyrosine kinase activity of stably complexed p125FAK and ZAP-70 in human T cells. J Exp Med 184: 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE (2005) Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol 6: 80–89 [DOI] [PubMed] [Google Scholar]
- Barnard AL, Igakura T, Tanaka Y, Taylor GP, Bangham CR (2005) Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood 106: 988–995 [DOI] [PubMed] [Google Scholar]
- Blanchard N, Di Bartolo V, Hivroz C (2002) In the immune synapse, ZAP-70 controls T cell polarization and recruitment of signaling proteins but not formation of the synaptic pattern. Immunity 17: 389–399 [DOI] [PubMed] [Google Scholar]
- Blot G, Janvier K, Le Panse S, Benarous R, Berlioz-Torrent C (2003) Targeting of the human immunodeficiency virus type 1 envelope to the trans-Golgi network through binding to TIP47 is required for env incorporation into virions and infectivity. J Virol 77: 6931–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cemerski S, Shaw A (2006) Immune synapses in T-cell activation. Curr Opin Immunol 18: 298–304 [DOI] [PubMed] [Google Scholar]
- Chazal N, Gerlier D (2003) Virus entry, assembly, budding, and membrane rafts. Microbiol Mol Biol Rev 67: 226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC (2005) Cellular APOBEC3G restricts HIV-1 infection in resting CD4(+) T cells. Nature 435: 108–114 [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR (1995) Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206: 935–944 [DOI] [PubMed] [Google Scholar]
- Das V, Nal B, Dujeancourt A, Thoulouze MI, Galli T, Roux P, Dautry-Varsat A, Alcover A (2004) Activation-induced polarized recycling targets T cell antigen receptors to the immunological synapse; involvement of SNARE complexes. Immunity 20: 577–588 [DOI] [PubMed] [Google Scholar]
- Davis AJ, Li P, Burrell CJ (1997) Kinetics of viral RNA synthesis following cell-to-cell transmission of human immunodeficiency virus type 1. J Gen Virol 78 (Part 8): 1897–1906 [DOI] [PubMed] [Google Scholar]
- Demirov DG, Freed EO (2004) Retrovirus budding. Virus Res 106: 87–102 [DOI] [PubMed] [Google Scholar]
- Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA (1993) Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol 67: 2182–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Tseng SY, Varma R, Campi G (2006) T cell-dendritic cell immunological synapses. Curr Opin Immunol 18: 512–516 [DOI] [PubMed] [Google Scholar]
- Elder ME, Lin D, Clever J, Chan AC, Hope TJ, Weiss A, Parslow TG (1994) Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science 264: 1596–1599 [DOI] [PubMed] [Google Scholar]
- Garcia E, Pion M, Pelchen-Matthews A, Collinson L, Arrighi JF, Blot G, Leuba F, Escola JM, Demaurex N, Marsh M, Piguet V (2005) HIV-1 trafficking to the dendritic cell-T-cell infectious synapse uses a pathway of tetraspanin sorting to the immunological synapse. Traffic 6: 488–501 [DOI] [PubMed] [Google Scholar]
- Goda S, Quale AC, Woods ML, Felthauser A, Shimizu Y (2004) Control of TCR-mediated activation of beta 1 integrins by the ZAP-70 tyrosine kinase interdomain B region and the linker for activation of T cells adapter protein. J Immunol 172: 5379–5387 [DOI] [PubMed] [Google Scholar]
- Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, Zhu P, Freedman BD, Clark MR, Rawlings DJ, Billadeau DD, Burkhardt JK (2006) HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity 24: 741–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith CE, Zhang W, Wange RL (1998) ZAP-70-dependent and -independent activation of Erk in Jurkat T cells. Differences in signaling induced by H2o2 and Cd3 cross-linking. J Biol Chem 273: 10771–10776 [DOI] [PubMed] [Google Scholar]
- Grigorov B, Arcanger F, Roingeard P, Darlix JL, Muriaux D (2006) Assembly of infectious HIV-1 in human epithelial and T-lymphoblastic cell lines. J Mol Biol 359: 848–862 [DOI] [PubMed] [Google Scholar]
- Haase AT (2005) Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol 5: 783–792 [DOI] [PubMed] [Google Scholar]
- Hioe CE, Chien PC Jr, Lu C, Springer TA, Wang XH, Bandres J, Tuen M (2001) LFA-1 expression on target cells promotes human immunodeficiency virus type 1 infection and transmission. J Virol 75: 1077–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hivroz C, Fischer A (1994) Immunodeficiency diseases. Multiple roles for ZAP-70. Curr Biol 4: 731–733 [DOI] [PubMed] [Google Scholar]
- Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR (2003) Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299: 1713–1716 [DOI] [PubMed] [Google Scholar]
- Johnson DC, Huber MT (2002) Directed egress of animal viruses promotes cell-to-cell spread. J Virol 76: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Kashefi K, Hollinshead M, Sattentau QJ (2004) HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med 199: 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Sattentau QJ (2005) Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J Virol 79: 12088–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok T, Li P, Burrell C (1993) Cell-to-cell transmission of human immunodeficiency virus infection induces two distinct phases of viral RNA expression under separate regulatory control. J Gen Virol 74 (Part 1): 33–38 [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Huppa JB, Purbhoo MA, Davis MM (2003) Linking molecular and cellular events in T-cell activation and synapse formation. Semin Immunol 15: 307–315 [DOI] [PubMed] [Google Scholar]
- Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT (2005) Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434: 1148–1152 [DOI] [PubMed] [Google Scholar]
- Maréchal V, Clavel F, Heard JM, Schwartz O (1998) Cytosolic Gag p24 as an index of productive entry of human immunodeficiency virus type 1. J Virol 72: 2208–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M, Helenius A (2006) Virus entry: open sesame. Cell 124: 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinl E, Lengenfelder D, Blank N, Pirzer R, Barata L, Hivroz C (2000) Differential requirement of ZAP-70 for CD2-mediated activation pathways of mature human T cells. J Immunol 165: 3578–3583 [DOI] [PubMed] [Google Scholar]
- Miranda LR, Schaefer BC, Kupfer A, Hu Z, Franzusoff A (2002) Cell surface expression of the HIV-1 envelope glycoproteins is directed from intracellular CTLA-4-containing regulated secretory granules. Proc Natl Acad Sci USA 99: 8031–8036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI (2004) Retrovirus budding. Annu Rev Cell Dev Biol 20: 395–425 [DOI] [PubMed] [Google Scholar]
- Nejmeddine M, Barnard AL, Tanaka Y, Taylor GP, Bangham CR (2005) Human T-lymphotropic virus, type 1, tax protein triggers microtubule reorientation in the virological synapse. J Biol Chem 280: 29653–29660 [DOI] [PubMed] [Google Scholar]
- Nobile C, Petit C, Moris A, Skrabal K, Abastado JP, Mammano F, Schwartz O (2005) Covert human immunodeficiency virus replication in dendritic cells and in DC-SIGN-expressing cells promotes long-term transmission to lymphocytes. J Virol 79: 5386–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noraz N, Schwarz K, Steinberg M, Dardalhon V, Rebouissou C, Hipskind R, Friedrich W, Yssel H, Bacon K, Taylor N (2000) Alternative antigen receptor (TCR) signaling in T cells derived from ZAP-70-deficient patients expressing high levels of Syk. J Biol Chem 275: 15832–15838 [DOI] [PubMed] [Google Scholar]
- Nydegger S, Khurana S, Krementsov DN, Foti M, Thali M (2006) Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J Cell Biol 173: 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce-Pratt R, Malamud D, Phillips DM (1994) Role of the cytoskeleton in cell-to-cell transmission of human immunodeficiency virus. J Virol 68: 2898–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelchen-Matthews A, Kramer B, Marsh M (2003) Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol 162: 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans L (2005) Viruses as probes for systems analysis of cellular signalling, cytoskeleton reorganization and endocytosis. Curr Opin Microbiol 8: 331–337 [DOI] [PubMed] [Google Scholar]
- Perlman M, Resh MD (2006) Identification of an intracellular trafficking and assembly pathway for HIV-1 gag. Traffic 7: 731–745 [DOI] [PubMed] [Google Scholar]
- Petit C, Buseyne F, Boccaccio C, Abastado JP, Heard JM, Schwartz O (2001) Nef is required for efficient HIV-1 replication in cocultures of dendritic cells and lymphocytes. Virology 286: 225–236 [DOI] [PubMed] [Google Scholar]
- Phillips DM (1994) The role of cell-to-cell transmission in HIV infection. Aids 8: 719–731 [DOI] [PubMed] [Google Scholar]
- Piguet V, Sattentau Q (2004) Dangerous liaisons at the virological synapse. J Clin Invest 114: 605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier A, Olivo-Marin JC, Arpin M, Michel F, Martin M, Mangeat P, Acuto O, Dautry-Varsat A, Alcover A (2001) The membrane-microfilament linker ezrin is involved in the formation of the immunological synapse and in T cell activation. Immunity 15: 715–728 [DOI] [PubMed] [Google Scholar]
- Sasahara Y, Rachid R, Byrne MJ, de la Fuente MA, Abraham RT, Ramesh N, Geha RS (2002) Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol Cell 10: 1269–1281 [DOI] [PubMed] [Google Scholar]
- Sato H, Orenstein J, Dimitrov D, Martin M (1992) Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 186: 712–724 [DOI] [PubMed] [Google Scholar]
- Schwartz O, Maréchal V, Danos O, Heard JM (1995) Human immunodeficiency virus type 1 Nef increases the efficiency of reverse transcription in the infected cell. J Virol 69: 4053–4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons A, Aluvihare V, McMichael A (2001) Nef triggers a transcriptional program in T cells imitating single-signal T cell activation and inducing HIV virulence mediators. Immunity 14: 763–777 [DOI] [PubMed] [Google Scholar]
- Soede RD, Driessens MH, Ruuls-Van Stalle L, Van Hulten PE, Brink A, Roos E (1999) LFA-1 to LFA-1 signals involve zeta-associated protein-70 (ZAP-70) tyrosine kinase: relevance for invasion and migration of a T cell hybridoma. J Immunol 163: 4253–4261 [PubMed] [Google Scholar]
- Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O (2007) Inefficient HIV replication in mobile lymphocytes. J Virol (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson M (2003) HIV-1 pathogenesis. Nat Med 9: 853–860 [DOI] [PubMed] [Google Scholar]
- Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA (1990) HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9: 1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif MR, Tremblay MJ (2005) LFA-1 is a key determinant for preferential infection of memory CD4+ T cells by human immunodeficiency virus type 1. J Virol 79: 13714–13724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoulouze MI, Sol-Foulon N, Blanchet F, Dautry-Varsat A, Schwartz O, Alcover A (2006) Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 24: 547–561 [DOI] [PubMed] [Google Scholar]
- Ticchioni M, Charvet C, Noraz N, Lamy L, Steinberg M, Bernard A, Deckert M (2002) Signaling through ZAP-70 is required for CXCL12-mediated T-cell transendothelial migration. Blood 99: 3111–3118 [DOI] [PubMed] [Google Scholar]
- Williams BL, Schreiber KL, Zhang W, Wange RL, Samelson LE, Leibson PJ, Abraham RT (1998) Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol Cell Biol 18: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen ISY (1990) HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61: 213–222 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material