

Abstract
In Drosophila and mammals, insulin signalling can increase growth, progression through G1/S, cell size and tissue size. Here, we analyse the way insulin affects cell size and cell-cycle progression in two haemocyte-derived Drosophila cell lines. Surprisingly, we find that although insulin increases cell size, it slows the rate at which these cells increase in number. By using BrdU pulse-chase to label S-phase cells and follow their progression through the cell cycle, we show that insulin delays progression through G2/M, thereby slowing cell division. The ability of insulin to slow progression through G2/M is independent of its ability to stimulate progression through G1/S, so is not a consequence of feedback by the cell-cycle machinery to maintain cell-cycle length. Insulin's effects on progression through G2/M are mediated by dTOR/dRaptor signalling. Partially inhibiting dTOR/dRaptor signalling by dsRNAi or mild rapamycin treatment can increase cell number in cultured haemocytes and the Drosophila wing, respectively. Thus, insulin signalling can influence cell number depending on a balance between its ability to accelerate progression through G1/S and delay progression through G2/M.
Keywords: cell-cycle progression, cell division, G2–M transition, insulin, TOR
Introduction
Extensive work over the past 10 years has firmly established the insulin signalling pathway as a key regulator of growth (mass increase), S-phase entry, cell size and tissue size in Drosophila and mammals (Leevers and Hafen, 2004). Many of the Drosophila experiments have been carried out by genetically manipulating the activity of the pathway in developing imaginal discs, and then examining the impact on various growth parameters, either in the imaginal discs themselves or in the adult epithelial tissues that they give rise to (Leevers et al, 1996; Bohni et al, 1999; Goberdhan et al, 1999; Huang et al, 1999; Montagne et al, 1999; Verdu et al, 1999; Weinkove et al, 1999; Gao et al, 2000; Oldham et al, 2000; Zhang et al, 2000; Brogiolo et al, 2001; Ikeya et al, 2002; Radimerski et al, 2002). These experimental approaches have shown that overexpression of positive components of the insulin signalling pathway increases tissue growth and cell size, sometimes with an accompanying increase in cell number. Conversely, mutation of positive pathway components or overexpression of inhibitors of the pathway reduces tissue growth, cell size and cell number. Thus, imaginal discs have provided an excellent experimental system for establishing the in vivo function of this pathway in a developing epithelial tissue.
Although imaginal discs have been successfully used to analyse changes in cell size, cell-cycle progression and cell number, inherent biological variation makes the fine analysis of more subtle changes in these parameters difficult. Also, the overexpression of dAkt and Dp110 increases tissue and cell size, but does not increase cell number (Verdu et al, 1999; Weinkove et al, 1999) and the reason for this is not known. We have investigated the effect of insulin signalling on cell-cycle progression and cell proliferation in a cell culture system where more precise and detailed analyses are possible than in vivo. We used two haemocyte-derived Drosophila cell lines, Kc167 and S2 cells, in which bovine or human insulin can mimic the effects of Drosophila insulin-like peptides (Brogiolo et al, 2001) and increase cell size and S-phase entry (Kwon et al, 2002; Bikopoulos et al, 2004; Kim et al, 2004). We find that although insulin increases progression through the G1/S transition, it can also delay progression from S phase through G2/M, and thereby slow the rate of increase in cell number of these cultured cells. Experiments using dsRNAi and rapamycin show that dTOR/dRaptor signalling is responsible for this effect, and that under certain conditions, inhibiting insulin or dTOR signalling can increase cell number.
Results and discussion
Insulin can slow cell proliferation
We first confirmed the reported effects of insulin on Kc167 and S2 cell signalling and cell size (Kwon et al, 2002; Lizcano et al, 2003; Bikopoulos et al, 2004; Kim et al, 2004; Leevers and Hafen, 2004). In both cell types, insulin rapidly induces the phosphorylation of dAkt and dS6K on their hydrophobic motifs (S505 and T398, respectively), which is indicative of their activation. Both kinases are phosphorylated within 5 min and remain phosphorylated for at least 96 h (Supplementary Figure S1; data not shown; Lizcano et al, 2003; Miron et al, 2003). Insulin also increases cell size, assessed by forward scatter measurements (obtained by flow cytometry) and cell volume measurements (from a Coulter counter). The size of S2 cells and Kc167 cells increases within a few hours of adding insulin, is reproducibly increased by 15–20% after 48 h and can be observed at all stages of the cell cycle (Figure 1A and data not shown).
Figure 1.
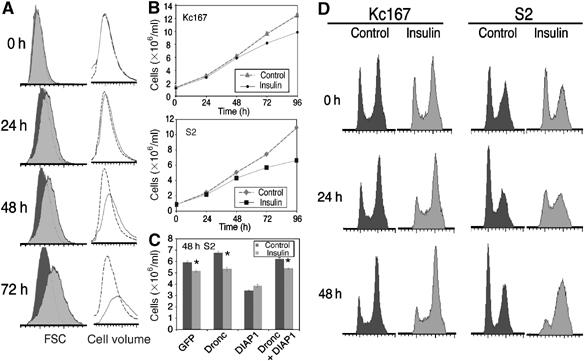
Insulin slows cell proliferation and increases the proportion of cells in S and G2 phases. (A) A 1 μM concentration of insulin increases the size of S2 cells seeded at 1 × 106 cells/ml, as detected by FSC measurements of a defined number of cells (dark grey indicates control population, light grey indicates insulin-treated population) and Coulter counter cell volume measurements of a defined volume of cells (black dotted line indicates control population, grey solid line indicates insulin-treated population). A 15–20% increase in cell volume is consistently observed by 48 h. (B) Insulin at 1 μM slows the rate of increase in cell number of Kc167 and S2 cells measured using a Coulter counter. Data represent mean±s.d. from three replicates, although the s.d.s are too small to be discernable. (C) S2 cells were treated with dsRNAi corresponding to GFP (control), Dronc or DIAP1 for 72 h, then reseeded at 1 × 106 cells/ml with fresh dsRNA and treated with 1 μM insulin for a further 48 h. dsRNAi-mediated depletion of Dronc does not affect the ability of insulin to slow the increase in S2 cell number, but prevents the reduction in cell number induced by dsRNAi-mediated depletion of DIAP1. Coulter counter cell count data represent mean±s.d. from three replicates (*P<0.01). Results shown in (C) are representative of two independent experiments. (D) Flow cytometer cell-cycle profiles of control and 1 μM insulin-treated Kc167 and S2 cells seeded at 1 × 106 cells/ml show that over 48 h, insulin reduces the proportion of cells in G1 and increases the proportion of cells in G2. Results shown in (A), (B) and (D) are from the same experiment and are representative of at least five independent experiments.
Surprisingly, we found that cultures of S2 and Kc167 cells treated with insulin increase in cell number at consistently slower rates than untreated cultures (Figure 1B). This slower increase in cell number is sometimes observed within 24 h, and a 5–15% reduction in cell number is always evident by 48 h. To investigate whether insulin slows the increase in cell number by inducing apoptosis, we examined whether the reduction in cell number could be abrogated by dsRNAi-mediated depletion of Dronc, an initiator caspase required for S2 cell apoptosis (Igaki et al, 2002; Zimmermann et al, 2002; Kiessling and Green, 2006). Depletion of Dronc by dsRNAi has no impact on the insulin-induced reduction in cell number although it completely prevents the reduction in cell number induced by dsRNAi of Drosophila inhibitor of apoptosis 1 (DIAP1; Figure 1C; Muro et al, 2002; Leulier et al, 2006). Insulin also slows the increase in cell number of S2 cells expressing the baculoviral inhibitor of apoptosis, p35, which blocks the activity of DRICE, the rate-limiting caspase for Drosophila apoptosis (data not shown; Kondo et al, 2006; Muro et al, 2006; Xu et al, 2006). Furthermore, we were unable to detect an insulin-induced increase in the sub-G1 population of S2 or Kc167 on flow cytometry profiles, which is indicative of cells undergoing apoptosis (data not shown). Together, these data demonstrate that the slowed increase in cell number of insulin-treated S2 cell cultures arises as a result of slowed cell division rather than increased apoptosis.
Insulin increases the proportion of cells in S phase and G2
To investigate further this unexpected insulin-induced slowing of cell division, we used flow cytometry to examine the cell-cycle profiles of insulin-treated S2 and Kc167 cells. In Drosophila wing imaginal discs, activating the insulin signalling pathway reduces the proportion of cells in G1 and increases the proportion of cells in S and G2 phases (Weinkove et al, 1999; Gao et al, 2000). This accumulation of cells in S and G2 phases was not accompanied by a significant reduction in cell number and hence was thought to result from insulin signalling accelerating progression from G1 to S phase. We found that treating Kc167 and S2 cells with insulin also reduces the G1 cell population and increases the S- and G2-phase cell population, and that this redistribution of cells takes place over several cell divisions (Figure 1D). This effect is less pronounced in Kc167 cells, as, in the absence of insulin, a larger proportion of these cells are in G2/M at any given time (Figure 1D). Therefore, all subsequent experiments examining cell-cycle phasing were performed on S2 cells. The reduced proportion of insulin-treated cells in G1 is likely to be due in part to the previously reported accelerated progression from G1 into S phase (Kwon et al, 2002; Bikopoulos et al, 2004; Kim et al, 2004). However, our observation that insulin reduces the rate of increase in cell number raises the possibility that this change in cell-cycle distribution may also be due to slowed progression from S phase through G2 and M phases. Note that the change in cell-cycle phasing that occurs over 48 h in response to insulin treatment is not the result of cultured cells reaching confluence (Supplementary Figure S2) or a maximal culture density—both control and insulin-treated cells are still increasing in number and size after 48 h (Figure 1B).
Insulin delays progression through G2 and M phases
We sought to investigate the effect of insulin on progression from S phase through G2 and M phases in the absence of any experimental artefacts that might be introduced by cell-cycle synchronisation. A pulse of BrdU was used to specifically label the S-phase cells within an asynchronously growing population of S2 cells. These BrdU-positive cells were followed by flow cytometry as they passed from S phase into G2, through mitosis and back into G1 and then S phase (Figure 2A). Insulin was added to the medium immediately after the pulse-labelling. By 6 h, when a small number of cells have divided and reentered G1, the percentage of BrdU-positive cells in the G1 peak was smaller in insulin-treated cells compared to control cells (13% for insulin-treated cells, 23% for control cells). The ability of insulin to delay progression from S phase through G2, M and into G1 is also evident after 12 h, when 20% of insulin-treated cells are in G1 compared to 36% of control cells. Note that, although the insulin-treated cells move from S phase to G1 more slowly than the control cells, these same cells then progress rapidly through G1 and into S phase. Thus, after 15 h, the proportion of cells in S phase is higher in insulin-treated cells than in control cells (Figure 2A). Similar results were obtained with Kc167 cells (data not shown).
Figure 2.
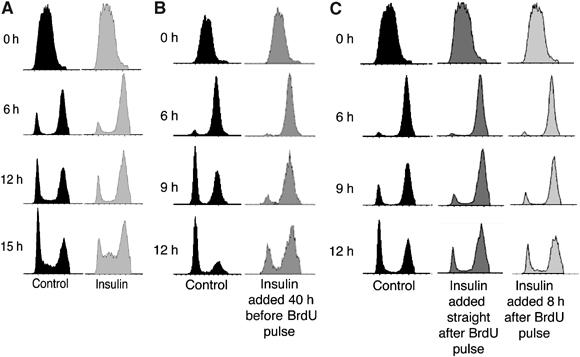
Insulin slows progression from S phase through G2/M. (A) S2 cells seeded at 1 × 106 cells/ml were pulse-labelled with BrdU for 15 min, grown in the presence or absence of 1 μM insulin and the cell-cycle profiles of BrdU-labelled cells were recorded by flow cytometry. The BrdU-labelled insulin-treated cells pass more slowly from S phase through G2 and M phases (6 and 12 h time points) and more quickly from G1 into S phase (15 h time point). Results shown are representative of four independent experiments. (B) S2 cells seeded at 1 × 106 cells/ml were grown in 1 μM insulin for 40 h before labelling with a pulse of BrdU for 15 min. Passage of the BrdU-labelled cells was followed for a further 12 h. The ability of insulin to delay progression through G2/M was apparent after 6, 9 and 12 h. (C) S2 cells seeded at 1 × 106 cells/ml were labelled with a pulse of BrdU for 15 min and then 1 μM insulin was added immediately or after 8 h. Passage of the BrdU-labelled cells was followed for 12 h. Even 9 h after BrdU labelling, treatment with insulin for 1 h delayed progression of cells from S phase through G2/M. Results in (B) and (C) are representative of two independent experiments.
A change in the length of one cell-cycle phase is often accompanied by a compensatory adjustment in another phase of the cycle. For example, overexpression of the Drosophila Cdk1 inhibitory kinase, dWee1, in the wing imaginal disc lengthens G2. These cells then compensate to maintain the same cell division time by shortening G1 (Reis and Edgar, 2004). Thus, the elongation of G2/M observed in insulin-treated S2 cells might conceivably result from a compensation for the insulin-induced shortening of G1. Importantly, this is not the case; insulin was added after the S-phase cells were pulse-labelled with BrdU and so the BrdU-positive cells that were followed through G2 and M phases had gone through a normal G1.
To rule out the possibility that ‘insulin shock' is responsible for the effect on progression from G2 through M and into G1, similar experiments were carried out in cells that were grown in insulin for 40 h before labelling with a pulse of BrdU. Under these conditions, insulin treatment results in the same delay in progression from S phase through G2/M (Figure 2B). We also examined how quickly insulin is able to delay progression through G2/M, by treating cells with insulin 8 h after the BrdU pulse and analysing their cell-cycle profile 1 h later. Even 1 h of insulin treatment is able to produce a detectable delay in progression from S phase through G2/M (Figure 2C, 9 h timepoint). Thus, insulin is able to slow the progression of S2 cells from S phase through G2/M within 1 h of its addition and after the cultures have been incubated with insulin for 40 h. Taken together, the data in Figures 1 and 2 lead to the surprising and previously unreported conclusion that insulin can reduce the ability of S2 cells to progress from S phase through G2 and M phases.
Reducing dTOR signalling promotes progression through G2 and M phases
Next, we investigated which molecules on the insulin signalling pathway might mediate the ability of insulin to delay progression from S phase through G2 and M phases. Various pathway components were partially depleted using dsRNAi, and a ‘mini-screen' was carried out for dsRNAs that can prevent the insulin-induced accumulation of cells in G2 (data not shown). dTOR dsRNAi was most effective and within 48 h significantly reduced the proportion of both control and insulin-treated cells in G2 (Figure 3B). Depleting dTOR might reduce the proportion of cells in G2 both by delaying progression through G1/S and by accelerating progression through G2/M. Importantly, and consistent with the latter, over a 48-h period, the partial depletion of dTOR also abrogates the insulin-induced reduction in cell number (Figure 3A).
Figure 3.
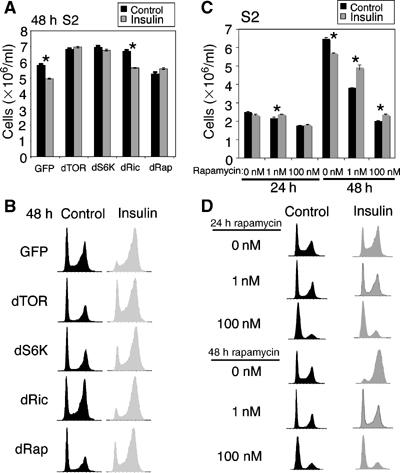
Reducing dTOR signalling abrogates the ability of insulin to reduce cell number and increase the proportion of cells in G2. (A, B) S2 cells were treated with dsRNA corresponding to GFP (control), dTOR, dS6K, dRictor or dRaptor for 72 h and then the cells were reseeded at 1 × 106 cells/ml with fresh dsRNA. The cells were grown in the presence or absence of 1 μM insulin for a further 48 h, and then Coulter counter cell count (A) and flow cytometer cell-cycle distribution (B) data were recorded. Depletion of dTOR, dRaptor or dS6K inhibits the insulin-induced reduction in cell number and increase in the proportion of cells in G2. Data shown in (A) and (B) are representative of five independent experiments. (C, D) S2 cells seeded at 1 × 106 cells/ml were treated with DMSO (0 nM), 1 nM rapanycin or 100 nM rapamycin for 1 h, then grown with or without 1 μM insulin for 24 or 48 h and Coulter counter cell count (C) and flow cytometer cell-cycle distribution (D) data were recorded. Coulter counter cell count data shown in (A) and (C) represent mean±s.d. from three replicates (*P<0.01). Data shown in (C) and (D) are representative of three independent experiments.
Recent work has shown that TOR functions in two distinct complexes, TORC1 and TORC2, each of which is likely to have several targets (Loewith et al, 2002; Dann and Thomas, 2006). TORC1contains Raptor and mediates the phosphorylation and activation of S6K (Hara et al, 2002; Kim et al, 2002), TORC2 contains Rictor and mediates the phosphorylation and activation of Akt (Sarbassov et al, 2005). Thus, we also examined the effect of dsRNAi depletion of dRaptor, dS6K and dRictor on insulin-stimulated changes in S2 cell-cycle profiles and cell number. To ensure that the predicted proteins were depleted by the dsRNA treatments, cell lysates were analysed by immunoblotting with antibodies against the phosphorylated hydrophobic motifs of dAkt (Ser505) and dS6K (Thr398; Supplementary Figure S3). Consistent with the ability of TORC1 to activate dS6K, depletion of dTOR and dRaptor reduces phospho-dS6K levels (Lizcano et al, 2003; Leevers and Hafen, 2004; Sarbassov et al, 2005). Consistent with the ability of TORC2 to phosphorylate dAkt on S505, depletion of dTOR and dRictor reduces phospho-dAkt levels (Supplementary Figure S3; Sarbassov et al, 2005). Finally, consistent with the negative feedback of S6K on molecules higher up the insulin signalling pathway, depletion of dS6K slightly increased dAkt phosphorylation (Harrington et al, 2005). Depletion of dRaptor and, to a lesser extent, dS6K mimics the effect of dTOR depletion and reduces the insulin-stimulated accumulation of cells in G2 and reduction in cell number. In contrast, depletion of dRictor has little effect (Figure 3A and B). Together, these data imply that dTORC1, possibly via its activation of dS6K, can delay progression from S phase through G2/M, whereas dTORC2 cannot.
To investigate further the influence of dTOR on progression through G2/M, we treated cells with rapamycin, which inhibits the activity of TORC1 (Dann and Thomas, 2006) and thus abolishes dS6K phosphorylation in S2 cells (data not shown; Lizcano et al, 2003). Cells were treated with rapamycin for 1 h, then insulin was added for 48 h and cell-cycle profiles and cell number were assessed. In contrast to what happens when dTOR signalling is partially inhibited by dTOR dsRNAi, cells treated with 100 nM rapamycin go through an initial round of cell division, nearly doubling their cell number at 24 h (Figure 3C), and then by 48 h most of these cells accumulate in G1 and cell number does not increase further (Figure 3C and D). Non-rapamycin-treated cells, with and without insulin, continue to increase in cell number and do not accumulate in G1 (Figure 3C and D).
We examined whether lower doses of rapamycin (1 nM) would inhibit dTOR more weakly and increase cell number at 48 h, thereby mimicking the effect of dTOR dsRNAi. Whereas the ability of 1 nM rapamycin to arrest cells in G1 and inhibit proliferation was reduced at both 24 and 48 h compared to 100 nM rapamycin, 1 nM rapamycin did not increase cell number relative to the control as was observed with dTOR dsRNAi (Figure 3C and D). Treatment with even lower doses of rapamycin was ineffective and had no effect on signalling or proliferation (data not shown). Our inability to observe the same changes in cell number and cell-cycle distribution in cells treated with dTOR dsRNA and rapamycin may be due to the molecular, temporal and kinetic complexities of the TORC1 and TORC2 signalling complexes and their sensitivity to rapamycin (for example, see Harrington et al, 2005; Sarbassov et al, 2006).
Nevertheless, we sought to examine whether rapamycin-mediated inhibition of dTOR impairs the ability of insulin to delay progression from S phase through G2 and M phases during the initial cell cycle that occurs immediately before cells treated with 100 nM rapamycin accumulate in G1. Changes in the cell-cycle profiles of BrdU pulse-labelled cells treated with or without 100 nM rapamycin and insulin were followed over 15 h. Importantly, we found that the BrdU-labelled cells pass more quickly from S phase through G2/M when treated with rapamycin, in the presence or absence of insulin (Figure 4A, compare G1 population in DMSO-and rapamycin-treated cells at 9 and 15 h).
Figure 4.
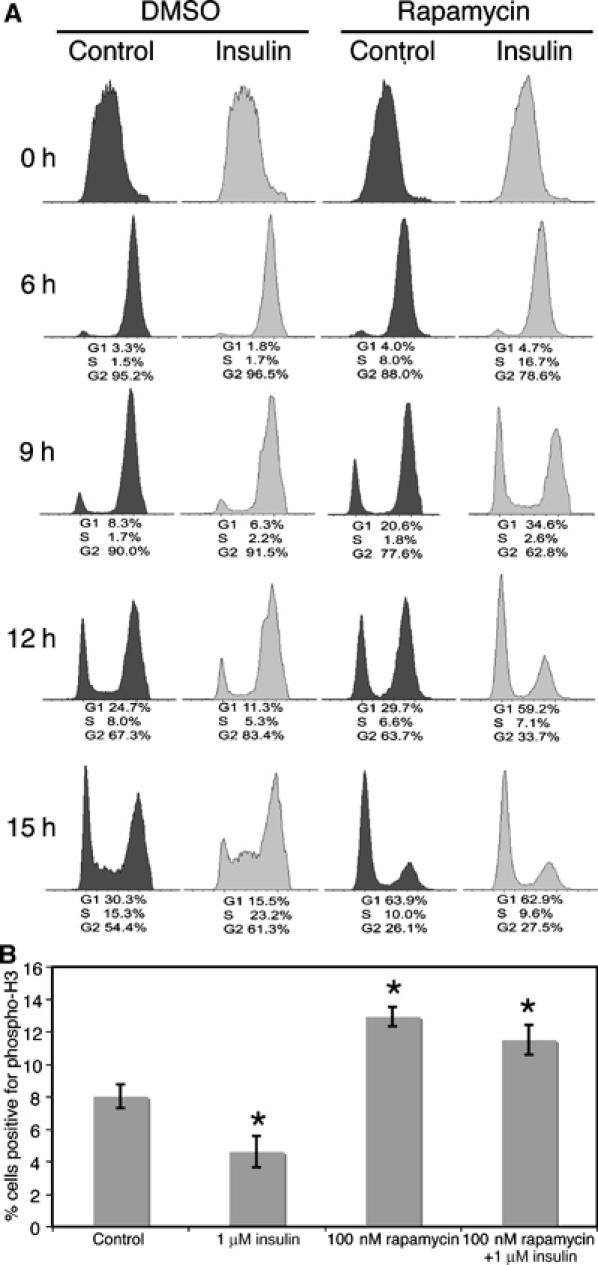
Inhibiting the dTOR/dRaptor complex with rapamycin promotes progression from S phase through G2/M phase. (A) S2 cells seeded at 1 × 106 cells/ml were pulse-labelled with BrdU for 15 min and then grown in the presence or absence of 100 nM rapamycin and 1 μM insulin. The cell-cycle profiles of the BrdU-labelled cells fixed at the time points shown were recorded by flow cytometry. The ability of rapamycin to accelerate progression from S phase through G2 and M phases and into G1 is evident after 9 h and more marked by 12 h. Data are indicative of three independent experiments. (B) Cells were seeded at 1 × 106 cells/ml, then grown in the presence or absence of 100 nM rapamycin and 1 μM insulin for 9 h and immunostained with anti-phospho-H3. The percentage of cells in mitosis for each condition was determined by counting the number of phospho-H3 positive cells and total number of cells in six fields of view (n=at least 250). Insulin treatment reduces and rapamycin treatment increases the percentage of cells in M phase. Data shown represent mean data±s.d. from three replicates (*P<0.01) and are representative of two independent experiments.
Histone H3 is phosphorylated at its amino terminus late in G2 and is required for chromosome condensation during mitosis (Hendzel et al, 1997). Because histone H3 becomes completely dephosphorylated by telophase, its phosphorylation state is a strong and precise marker for mitosis and can be used to determine a mitotic index (the percentage of cells in a population in mitosis at any one time; Tapia et al, 2006). To confirm that insulin treatment inhibits and rapamycin treatment accelerates the progression of cells from S phase into G2/M, S2 cells were treated as described above, then fixed and stained with anti-phosphohistone H3 after 9 h (when the changes in cell-cycle progression first become obvious in the BrdU pulse-chase experiment). After 9 h, the percentage of S2 cells in mitosis decreases from approximately 8.0% in control cells to approximately 4.6% in insulin-treated cells. Furthermore, rapamycin increased the percentage of cells in mitosis to approximately 12.9 and 11.5% in control and insulin-treated cells, respectively (Figures 4B). Together, our data demonstrate that insulin and TOR signalling have two distinct effects on cell-cycle progression, and independently accelerate progression from G1 to S phase and slow progression from S phase through G2 and M phases.
Insulin is less able to delay progression through G2 and M phases when nutrients are low
The molecular connections on the insulin and TOR signalling pathways are complex, involving positive and negative feedback loops, and the activity of TOR is influenced by both insulin and amino-acid levels (Leevers and Hafen, 2004; Dann and Thomas, 2006). Thus, we hypothesise that the TOR-mediated delay in progression from S phase through G2 and M phases may only have a detectable impact on cell number under certain experimental conditions. For example, the partial depletion of dTOR, and presumably dTORC1 and dTORC2, by dsRNAi for 48 h increases cell number presumably because it accelerates progression through G2/M more than it delays progression through G1/S (Figure 3A and B). In contrast, more complete inhibition of dTORC1 signalling with rapamycin reduces cell number, presumably because although progression through G2/M is accelerated, the delayed progression through G1/S is more pronounced (Figure 3C and D).
To investigate further whether different experimental manipulations of dTOR signalling can have different outcomes for cell number, we examined whether the ability of insulin to slow the increase in S2 cell number can be altered by different nutrient conditions. The S2 cell culture medium was serially diluted with PBS to deplete it of amino acids and other nutrients, thereby reducing dTOR signalling. The cells were allowed to adapt to the new medium for 24 h, then insulin was added for a further 48 h. As expected, diluting the growth medium reduces basal and insulin-stimulated dS6K phosphorylation (Figure 5A; Radimerski et al, 2002). Importantly, under these conditions of reduced nutrients and dTOR signalling, the ability of insulin to slow the increase in cell number of S2 cell cultures was reduced (Figure 5B, compare control versus insulin cell number in 25 or 100% Schneider's medium). Thus, the ability of dTOR to delay progression through G2/M and thereby reduce cell number in S2 cells is detectable only when high nutrient levels synergise with insulin to maximally activate dTOR signalling.
Figure 5.
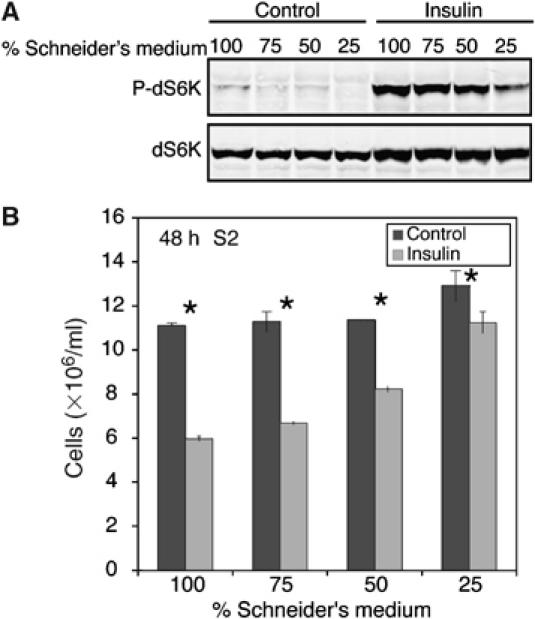
Insulin is less able to delay progression through G2 and M phases when nutrients are low. S2 cells seeded at 1 × 106 cells/ml were grown in Schneider's medium that was serially diluted with PBS (before adding 10% FBS) for 24 h. (A) Insulin was added for 15 min and then cell lysates were prepared and analysed by immunoblotting to demonstrate that nutrient depletion reduces basal and insulin-stimulated dS6K phosphorylation. (B) The cells were grown for a further 48 h in 1 μM insulin and then Coulter counter cell count data were recorded. Data shown represent mean data±s.d. from three replicates (*P<0.01) and are representative of three independent experiments.
Inhibiting dTOR signalling can increase cell number in the Drosophila wing
Our data demonstrate that under certain conditions in S2 cells, insulin signalling can delay progression through G2/M and reduce cell number. Moreover, partially reducing dTOR signalling by dsRNAi can accelerate progression through G2/M and increase cell number. Such effects have not been reported in other experimental systems, perhaps because sufficiently sensitive analyses have not been performed or because effects on G2/M were masked by coincident effects on G1/S.
We investigated whether inhibiting dTOR signalling in another experimental system, the Drosophila wing, can also promote progression through G2 and M phases, thereby increasing cell number. At the end of larval development, the proliferation of cells in the wing imaginal disc slows and they accumulate in G2 (Neufeld et al, 1998). Two additional mitoses occur during early pupal development, then cells arrest and differentiate into the adult wing. We fed larvae 5 and 10 μM rapamycin, to weakly inhibit dTOR signalling during the last 24 h of larval development and then examined adult wings to see whether extra cell divisions, resulting in increased wing cell number, had occurred. Each wing blade cell possesses a single wing hair; so for each wing, the number of cells within a fixed area was counted and the entire wing area was measured and then the hypothetical number of cells per wing was calculated.
As expected, feeding larvae 10 μM rapamycin to inhibit dTOR signalling reduced wing size (by 13%, P<0.001) wing cell number (by 6%, P<0.001); and wing cell size (by 10%, P<0.001, Figure 6). Importantly, although feeding larvae 5 μM rapamycin slightly reduced wing size (by 4%, P=0.06), these wings contain 6% more cells than control wings (P<0.001) and were composed of cells that were 8% smaller (P<0.001). Thus, very slight reductions in dTOR signalling during wing development, can significantly increase cell number (Figure 6B), supporting the idea that dTOR signalling can also inhibit progression through G2/M in vivo in developing wing imaginal discs.
Figure 6.
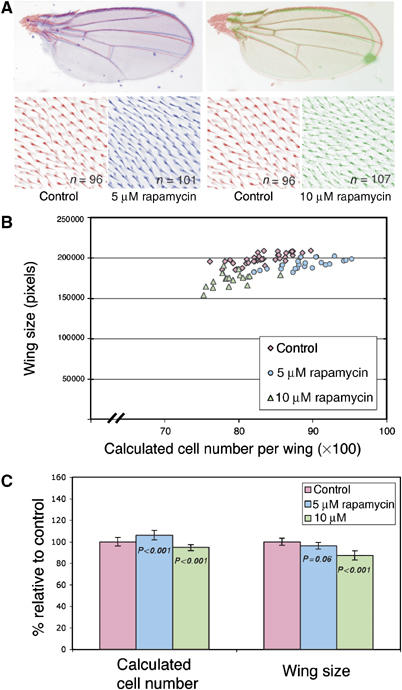
Weak inhibition of dTOR signalling increases Drosophila wing cell number. Drosophila larvae were transferred to food containing 10 or 5 μM rapamycin or ethanol (control) 96 h after egg laying and then the wings of emerging adult males were analysed. (A) Rapamycin at 10 μM (green) reduces wing size and increases wing cell density, whereas 5 μM rapamycin (blue) has less effect on wing size but still increases wing cell density. The hypothetical number of cells per wing was calculated and plotted against wing size (B) or used to calculate the mean wing cell number (C). Data are shown as mean percentage changes±s.d., n=at least 20. Rapamycin at 5 μM increases total wing cell number, indicating that inhibiting dTOR signalling can increase the total number of cell divisions that occur during wing development. Data shown are representative of three independent experiments.
To summarise, by doing short time-scale BrdU pulse-chase experiments on large cell populations, we show that insulin can slow the progression of cultured Drosophila cells from S phase through G2 and M (Figure 2A), and that mild inhibition of dTOR-dRaptor can actually accelerate cell division (Figure 3). Although it is not possible to carry out these types of experiments in vivo, our rapamycin feeding experiments show that, in principle, lowering dTOR signalling can have the same effect during wing development (Figure 6). Thus, our data uncover a new connection between TOR signalling and the cell cycle, and demonstrate that TOR can have dual and opposing effects on cell-cycle progression.
Why should the same signalling pathway both accelerate progression through G1/S and delay progression from S phase through G2/M? It is already known that various cells can maintain their cell-cycle time and accommodate alterations in the rates of progression through one cell-cycle transition (for example, G1/S) by inducing a compensatory change in the rate of progression through another cell-cycle transition (for example G2/M: Reis and Edgar, 2004). Feedback mechanisms involving components of the cell-cycle machinery are responsible for at least some of this ‘cell-cycle compensation'. Our data show that cells that have been through a normal G1/S-phase transition, then are labelled in S phase with BrdU and treated with insulin still go from S phase through G2/M more slowly. Thus, the ability of insulin to slow progression through G2/M is independent of its ability to accelerate progression through G1/S, and so is not solely due to a feedback mechanism involving the cell-cycle machinery.
Perhaps the dual and opposing effects of this signalling pathway on cell-cycle progression have evolved as a mechanism to prevent ‘runaway cell division' under high nutrient conditions. This response may also have evolved to allow starving cells to pass rapidly through mitosis, an energy consuming process, before their full response to starvation can begin. Interestingly, when Schizosaccharomyces pombe are starved, they increase their cell division rate within 1 h and divide at a smaller size (Fantes and Nurse, 1977, 1978). Although this observation has been taken to support the existence of a G2/M cell size checkpoint that is under nutrient control, it might also indicate that it is beneficial for cells to divide as rapidly as possible once they are starved. Indeed, the induction of autophagy, a cellular response to nutrient deprivation that provides a source of amino acids to allow cell survival, is inhibited in mitotic cells until they have exited telophase (Eskelinen et al, 2002). If autophagy cannot be induced until cells have reentered the G1 phase of the cell cycle, the rapid passage of starved cells through G2/M would ensure that minimal time, energy and resources are wasted, and that autophagy can begin more quickly. Intriguingly, inhibiting TOR can also induce autophagy (Noda and Ohsumi, 1998; Cutler et al, 1999; Abeliovich et al, 2000; Kamada et al, 2000; Gutierrez et al, 2004; Ravikumar et al, 2004; Guertin and Sabatini, 2005); so lowered TOR signalling may have dual roles that aid cell survival during starvation. The inhibition of TOR signalling may both promote the rapid progression of cells in S phase, through G2/M and into G1, and, once the cells are in G1, induce autophagy to provide an alternative supply of nutrients.
Further experiments are required to characterise the molecular connections responsible for the ability of reduced levels of nutrients and TOR signalling to induce cell division. Intriguingly, in Xenopus eggs, rapamycin treatment rapidly increases the translation of the CDK phosphatase, Cdc25A, which promotes cell division (Schwab et al, 1999). Indeed, Thomas speculated that inhibiting TOR signalling might reduce cell size by inducing cell division at a smaller than normal size, because of this increased translation of Cdc25A (Thomas, 2000). Such a molecular mechanism should also result in accelerated cell division, and thus allow cells to begin autophagy more rapidly upon starvation.
Materials and methods
Cell culture
Kc167 and S2 cells were cultured at 23°C in Schneider's medium (Gibco) supplemented with 10% foetal bovine serum (FBS), 50 U/ml penicillin and 50 μg/ml streptomycin (Invitrogen, complete medium). For experiments, cells were seeded at a concentration of 1 × 106 cells/ml in 75 cm2 flasks (30 ml per flask) or in six-well plates at 3 ml per well). Bovine insulin (Sigma-Aldrich) was used at a concentration of 1 μM. For rapamycin treatment, 1 or 100 nM rapamycin (Sigma-Aldrich) was added to the culture medium 1 h before insulin stimulation. For experiments with diluted Schneider's medium, cells were seeded in PBSA-diluted Schneider's medium supplemented with 10% FBS for 24 h before insulin treatment.
Cell volume, cell count and cell-cycle analysis
Kc167 and S2 cells were cultured as described above in six-well plates and triplicate wells were used for each time point. A total of 300 μl from each well was diluted in 15 ml of Isoton (Beckman–Coulter) and analysed for cell volume and cell count using a Z2 Coulter Counter (Multisizer II, Beckman–Coulter). The remaining cells were collected by centrifugation, fixed by vortexing and incubating in cold 70% ethanol at 4°C for at least 30 min, then washed twice in PBSA and resuspended in 500 μl of 20 μg/ml RNaseA (Sigma-Aldrich) and 40 μg/ml propidium iodide in PBSA (Sigma-Aldrich). Propidium iodide-stained cells were analysed on a FACScalibur flow cytometer using CellQuest (Becton Dickinson).
BrdU pulse-chase experiments
S2 cells were cultured as described above in six-well plates for 12 h, 15 μM BrdU (Sigma-Aldrich) was added for 15 min and then the cells were washed with PBSA. After three washes, BrdU-free medium plus or minus 1 μM insulin was added to wells. In experiments where rapamycin was used, medium plus or minus 100 nM rapamycin was added to cells after the PBSA washes. At 1 h after rapamycin incubation, 1 μM insulin was added to medium.
Cells were harvested at the time points specified by pipetting, collected by centrifugation, fixed in cold 70% ethanol and stored at 4°C. Subsequent steps were performed at room temperature. Fixed cells were washed twice in PBSA, permeabilised by incubating in 2 M HCl for 30 min and then washed twice in PBSA and once in PBS-BT (PBSA+0.1% BSA+0.2% Tween 20). A 2 μl portion of monoclonal mouse anti-BrdU (Becton Dickinson) was added directly to the cell pellet. This was incubated for 20 min in the dark and then cells were washed twice in PBS-BT and incubated in 50 μl of FITC-conjugated rabbit anti-mouse F(ab′)2 fragments (DAKO) diluted 1:10 in PBS-BT for 20 min. Cells were washed in PBSA, treated with 50 μl 100 μg/ml RNaseA (Sigma-Aldrich) for 15 min and 250 μl 40 μg/ml propidium iodide for a further 30 min and then analysed by flow cytometry.
dsRNAi
DNA templates for dsRNA synthesis were PCR amplified from fly genomic DNA using primers that contained 5′T7 RNA polymerase-binding sites preceded by a GAA overhang and followed by sense or antisense sequences. The following primers were used: dTOR: sense primer 5′-CGAACCTTATGCTGGATCGT-3′, antisense primer 5′-TGCCTGTTAGCTTGCACTTG-3′; dS6K: sense primer 5′-CTAACA TATCGCGGCAAAT-3′, antisense primer 5′-AGCGCAATATACTC GAGGC-3′; dRictor: sense primer 5′-AGGCCTGCAAAACGATAGG-3′, antisense primer 5′-ACGATTGCTGTTTCCTGTGG-3′; dRaptor: sense primer 5′-GCACCAGCTCCATTAACGAT-3′, antisense primer 5′-CGTATGTGCGTACCGAATTG-3′; DRONC: sense primer 5′-ATG CAGCCGCCGGAGCTCGAG-3′, antisense primer 5′- CGAGGAGGT CACCATTGTCAG-3′; DIAP1: sense primer 5′-ATGGCATCTGTTG TAGCTGAT-3′, antisense primer 5′- GCGTGCTGTTCCCAGGGCTCG-3′. dsRNA was synthesised using the T7 MEGAscript Transcription Kit (Ambion) as directed. 2 × 106 cells were seeded in 1 ml of complete medium per well of a six-well plate, 25 μg of dsRNA was added, cells were incubated for 1 h and then a further 2 ml complete medium was added.
Immunofluorescence
S2 cells were seeded at 1 × 106 cells/ml on glass coverslips coated with polylysine. The cells were allowed to adhere for 30 min, then 100 nM rapamycin or DMSO (control) was added to the medium for 1 h, followed by 1 μM insulin and the plates were incubated at 23°C for a further 9 h. Cells were fixed in PLP (periodate lysine paraformaldehyde=0.2 M lysine buffer adjusted to pH 7.4 with 0.2 M NaH2PO4 and 0.2 M Na2HPO4 with 2% final paraformaldehyde concentration added just before fixation) at room temperature for 20 min. Coverslips were washed three times in PBS+0.3% Tween 20, incubated for 1 h in blocking buffer (PBS+1% Tween 20+10% goat serum) and then incubated overnight in blocking buffer containing polyclonal anti-phospho-histone H3 (Ser10) diluted 1/500 (Upstate). Coverslips were washed four times in blocking buffer and incubated for 1 h in Alexa Fluor 488 donkey anti-rabbit IgG (Molecular Probes) diluted 1/1000 in blocking buffer. Coverslips were washed three times in PBS+0.3% Tween 20 and once in PBS and then mounted on slides and to visualised on a Nikon Eclipse1000 Fluorescence microscope using a Nikon Plan Apo × 100 objective. Images were acquired and processed using Openlab 4.0.4.
Western blotting
Cells were lysed in 50 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 1% (v/v) Triton X-100, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 0.27 M sucrose, 0.1% (v/v) 2-mercaptoethanol and Complete™ Protease Inhibitor mixture (Roche). Then the extracts were cleared of nuclei and debris by centrifugation for 10 min at 13 000 r.p.m. A 75 μg portion of protein per lane was resolved on NuPage™ 4–12% gradient SDS–PAGE gels (Invitrogen), transferred onto Immobilon-P membranes (Millipore) and analysed by immunoblotting as described (Lizcano et al, 2003). Primary antibodies used include affinity-purified polyclonal rabbit anti-CTdAKT (Lizcano et al, 2003) and anti-phospho-dAKT (Ser505) (Lizcano et al, 2003) sheep anti-dS6K (Lizcano et al, 2003), and the polyclonal rabbit anti-phospho Drosophila p70 S6K (Thr389, Cell Signalling Technology). Secondary antibodies were conjugated to Alexa Fluor 680 (Molecular Probes) or IRDye™ 800 (Rockland Immunochemicals), and signals were detected using the linear Odyssey Infrared Imaging System (LI-COR Biosciences).
Feeding larvae rapamycin
First instar yw larvae were picked from 1 h egg lays, seeded at a density of 70 larvae per vial and maintained at 25°C. At 96 h after egg laying, larvae were transferred to vials containing food plus 5 μM rapamycin, 10 μM rapamycin or ethanol (control). Male wings were mounted and analysed as described (Marygold et al, 2005).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figures Legend
Acknowledgments
We thank the CRUK London Research Institute FACS laboratory, Equipment Park and Fly Facility for technical support and Pascal Meier for S2 cells expressing p35 and DRONC and DIAP1 primers and cDNAs. We are grateful to Steven Marygold, Nicolas Tapon, Buzz Baum and Pascal Meier for many helpful discussions, and Grant Otto and Michael Howell for critical reading of the manuscript. This study was supported by Cancer Research UK.
References
- Abeliovich H, Dunn WA Jr, Kim J, Klionsky DJ (2000) Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 151: 1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikopoulos G, Ceddia RB, Sweeney G, Hilliker AJ (2004) Insulin reduces apoptosis and increases DNA synthesis and cell size via distinct signalling pathways in Drosophila Kc cells. Cell Prolif 37: 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E (1999) Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell 97: 865–875 [DOI] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E (2001) An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol 11: 213–221 [DOI] [PubMed] [Google Scholar]
- Cutler NS, Heitman J, Cardenas ME (1999) TOR kinase homologs function in a signal transduction pathway that is conserved from yeast to mammals. Mol Cell Endocrinol 155: 135–142 [DOI] [PubMed] [Google Scholar]
- Dann SG, Thomas G (2006) The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett 580: 2821–2829 [DOI] [PubMed] [Google Scholar]
- Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, Backer JM, Lucocq JM (2002) Inhibition of autophagy in mitotic animal cells. Traffic 3: 878–893 [DOI] [PubMed] [Google Scholar]
- Fantes P, Nurse P (1977) Control of cell size at division in fission yeast by a growth modulated size control over nuclear division. Exp Cell Res 107: 377–386 [DOI] [PubMed] [Google Scholar]
- Fantes PA, Nurse P (1978) Control of the timing of cell division in fission yeast. Cell size mutants reveal a second control pathway. Exp Cell Res 115: 317–329 [DOI] [PubMed] [Google Scholar]
- Gao X, Neufeld TP, Pan D (2000) Drosophila PTEN regulates cell growth and proliferation through PI3K-dependent and -independent pathways. Dev Biol 221: 404–418 [DOI] [PubMed] [Google Scholar]
- Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C (1999) Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev 13: 3244–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM (2005) An expanding role for mTOR in cancer. Trends Mol Med 11: 353–361 [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766 [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189 [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Lamb RF (2005) Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci 30: 35–42 [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD (1997) Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106: 348–360 [DOI] [PubMed] [Google Scholar]
- Huang H, Potter CJ, Tao W, Li DM, Brogiolo W, Hafen E, Sun H, Xu T (1999) PTEN affects cell size, cell proliferation and apoptosis during Drosophila eye development. Development 126: 5365–5372 [DOI] [PubMed] [Google Scholar]
- Igaki T, Yamamoto-Goto Y, Tokushige N, Kanda H, Miura M (2002) Down-regulation of DIAP1 triggers a novel Drosophila cell death pathway mediated by Dark and DRONC. J Biol Chem 277: 23103–23106 [DOI] [PubMed] [Google Scholar]
- Ikeya T, Galic M, Belawat P, Nairz K, Hafen E (2002) Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr Biol 12: 1293–1300 [DOI] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150: 1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiessling S, Green DR (2006) Cell survival and proliferation in Drosophila S2 cells following apoptotic stress in the absence of the APAF-1 homolog, ARK, or downstream caspases. Apoptosis 11: 497–507 [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163–175 [DOI] [PubMed] [Google Scholar]
- Kim SE, Cho JY, Kim KS, Lee SJ, Lee KH, Choi KY (2004) Drosophila PI3 kinase and Akt involved in insulin-stimulated proliferation and ERK pathway activation in Schneider cells. Cell Signal 16: 1309–1317 [DOI] [PubMed] [Google Scholar]
- Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M (2006) DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol 26: 7258–7268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Kim SH, Kim SE, Jang IH, Ahn Y, Lee WJ, Choi KY (2002) Drosophila extracellular signal-regulated kinase involves the insulin-mediated proliferation of Schneider cells. J Biol Chem 277: 14853–14858 [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Hafen E (2004) Growth Regulation by Insulin and TOR Signaling in Drosophila. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD (1996) The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J 15: 6584–6594 [PMC free article] [PubMed] [Google Scholar]
- Leulier F, Ribeiro PS, Palmer E, Tenev T, Takahashi K, Robertson D, Zachariou A, Pichaud F, Ueda R, Meier P (2006) Systematic in vivo RNAi analysis of putative components of the Drosophila cell death machinery. Cell Death Differ 13: 1663–1674 [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Alrubaie S, Kieloch A, Deak M, Leevers SJ, Alessi DR (2003) Insulin-induced Drosophila S6 kinase activation requires phosphoinositide 3-kinase and protein kinase B. Biochem J 374: 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10: 457–468 [DOI] [PubMed] [Google Scholar]
- Marygold SJ, Coelho CM, Leevers SJ (2005) Genetic analysis of RpL38 and RpL5, two minute genes located in the centric heterochromatin of chromosome 2 of Drosophila melanogaster. Genetics 169: 683–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron M, Lasko P, Sonenberg N (2003) Signaling from Akt to FRAP/TOR targets both 4E-BP and S6K in Drosophila melanogaster. Mol Cell Biol 23: 9117–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G (1999) Drosophila S6 kinase: a regulator of cell size. Science 285: 2126–2129 [DOI] [PubMed] [Google Scholar]
- Muro I, Berry D, Huh J, Chen C, Huang H, Yoo S, Guo M, Baehrecke E, Hay B (2006) The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development 133: 3305–3315 [DOI] [PubMed] [Google Scholar]
- Muro I, Hay BA, Clem RJ (2002) The Drosophila DIAP1 protein is required to prevent accumulation of a continuously generated, processed form of the apical caspase DRONC. J Biol Chem 277: 49644–49650 [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA (1998) Coordination of growth and cell division in the Drosophila wing. Cell 93: 1183–1193 [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273: 3963–3966 [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E (2000) Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 14: 2689–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Rintelen F, Stocker H, van der Kaay J, Downes CP, Hafen E, Thomas G (2002) dS6K-regulated cell growth is dPKB/dPI(3)K-independent, but requires dPDK1. Nat Cell Biol 4: 251–255 [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36: 585–595 [DOI] [PubMed] [Google Scholar]
- Reis T, Edgar BA (2004) Negative regulation of dE2F1 by cyclin-dependent kinases controls cell cycle timing. Cell 117: 253–264 [DOI] [PubMed] [Google Scholar]
- Sarbassov D, Ali S, SEngupta S, Sheen J, Hsu P, Bagley A, Markhard A, Sabatini D (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22: 159–168 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- Schwab MS, Kim SH, Terada N, Edfjall C, Kozma SC, Thomas G, Maller JL (1999) p70(S6K) controls selective mRNA translation during oocyte maturation and early embryogenesis in Xenopus laevis. Mol Cell Biol 19: 2485–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, Glatz K (2006) Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am J Surg Pathol 30: 83–89 [DOI] [PubMed] [Google Scholar]
- Thomas G (2000) An encore for ribosome biogenesis in the control of cell proliferation. Nat Cell Biol 2: E71–E72 [DOI] [PubMed] [Google Scholar]
- Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ (1999) Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol 1: 500–506 [DOI] [PubMed] [Google Scholar]
- Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ (1999) Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr Biol 9: 1019–1029 [DOI] [PubMed] [Google Scholar]
- Xu D, Wang Y, Willecke R, Chen Z, Ding T, Bergmann A (2006) The effector caspases drICE and dcp-1 have partially overlapping functions in the apoptotic pathway in Drosophila. Cell Death Differ 13: 1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP (2000) Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 14: 2712–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann KC, Ricci JE, Droin NM, Green DR (2002) The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol 156: 1077–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figures Legend