

Abstract
Protein phosphatase 2A (PP2A) has been implicated to exert its tumor suppressive function via a small subset of regulatory subunits. In this study, we reported that the specific B regulatory subunits of PP2A B56γ1 and B56γ3 mediate dephosphorylation of p53 at Thr55. Ablation of the B56γ protein by RNAi, which abolishes the Thr55 dephosphorylation in response to DNA damage, reduces p53 stabilization, Bax expression and cell apoptosis. To investigate the molecular mechanisms, we have shown that the endogenous B56γ protein level and association with p53 increase after DNA damage. Finally, we demonstrate that Thr55 dephosphorylation is required for B56γ3-mediated inhibition of cell proliferation and cell transformation. These results suggest a molecular mechanism for B56γ-mediated tumor suppression and provide a potential route for regulation of B56γ-specific PP2A complex function.
Keywords: B56γ subunit, cell proliferation, p53, PP2A, Thr55 dephosphorylation
Introduction
Protein phosphatase 2A (PP2A) is a major cellular serine/threonine phosphatase that plays an important role in balancing phosphorylation signals critical for cell proliferation and differentiation (Virshup, 2000; Schonthal, 2001; Van Hoof and Goris, 2003; Janssens et al, 2005). PP2A consists of a catalytic subunit (C), a scaffolding subunit (A) and a regulatory subunit (B). It exists in mammalian cells as either a core heterodimer (AC) or a heterotrimer (ABC). In general, the catalytic subunit (C) always associates with the scaffolding subunit (A) within the cell. The AC heterodimer can also bind to a B regulatory subunit to form the heterotrimeric holoenzyme. Various B subunits can be categorized into four distinct families on the basis of homology, namely B (B55 or PR55), B′ (B56 or PR61), B″ (PR48/59/72/130) and B′″ (PR93/110). Each of these B families is composed of several isoforms (Sontag, 2001). It is believed that PP2A exercises regulatory flexibility and substrate specificity through the specific association of the core dimer with one of the regulatory B subunits. This characteristic of PP2A contributes to its ability to regulate a multitude of cell functions. The diversity of possible combinations of PP2A subunits that exist as functional holoenzymes also allows this protein to be distinctly regulated.
PP2A functions as a positive and negative regulator of the cell cycle, in the latter capacity potentially acting as a tumor suppressor. The potential tumor suppressive function of PP2A might be achieved by a small subset of PP2A holoenzymes with particular subunit compositions. For example, B56γ3 was found to be involved in cell growth and proliferation control (Chen et al, 2004; Van Hoof and Goris, 2004). The PP2A B56γ3 gene belongs to the PP2A B56 family (also called B' or PR61 subunit), which consists of B56α (PPP2R5A), B56β (PPP2R5B), B56γ (PPP2R5C), B56δ (PPP2R5D) and B56ɛ (PPP2R5E) (Csortos et al, 1996; McCright et al, 1996). The PP2A B56γ gene locus resides at 14q32.2 and encodes four differentially spliced forms, PP2A B56γ1, γ2, γ3 and γ4 (Muneer et al, 2002; Ortega-Lazaro and del Mazo, 2003). As a phosphatase, PP2A was hypothesized to exert its tumor suppressive function by responding to genotoxic stress and acting as a signal transducer to dephosphorylate proteins involved in cell growth and proliferation. In fact, dephosphorylation of p53 by PP2A was first suggested by the finding that treatment of cells with okadaic acid (OA), a PP1/PP2A phosphatase inhibitor, leads to the hyperphosphorylation of cellular p53 protein (Yatsunami et al, 1993; Zhang et al, 1994). More recently, PP2A has been shown to directly dephosphorylate p53 at Ser37 in response to γ irradiation (Dohoney et al, 2004). However, despite the progress in delineating the significance of PP2A in dephosphorylating p53 and in regulating cell growth, the composition, role and regulation of the individual regulatory subunits in these processes remain largely undefined.
p53 functions as a transcriptional factor and induces growth arrest or programmed cell death in response to genotoxic stress (Vogelstein et al, 2000; Sharpless and DePinho, 2002). p53 protein stability and activity are modulated mainly through post-translational modifications. Previous studies in our laboratory showed that p53 is phosphorylated at Thr55 by TATA box binding protein-associated factor 1 (TAF1) under normal cellular growth conditions, and this phosphorylation promotes proteasome-mediated degradation of p53 (Li et al, 2004). In response to DNA damage, Thr55 phosphorylation was found to decrease rapidly (Li et al, 2004), which is associated with p53 protein accumulation. This observation inspired us to explore the role of phosphatase(s) in Thr55 dephosphorylation and activation.
In the present study, we report that the inhibition of PP2A activity by OA blocks Thr55 dephosphorylation in response to DNA damage. Furthermore, we identified the specific B regulatory subunit of PP2A B56γ that is associated with p53 in vivo and responsible for Thr55 dephosphorylation. Overexpression of B56γ3 inhibits cell proliferation and cell transformation in a p53-dependent manner. Consistent with these observations, inhibition of the B56γ protein by RNAi reduces DNA damage-induced p53 stabilization, Bax expression and cell apoptosis. To investigate the molecular mechanisms, we have shown that the endogenous B56γ protein level and association with p53 increase after DNA damage. Collectively, our data present the first evidence that the B56γ-containing complexes of PP2A function as tumor suppressors by, at least in part, dephosphorylating p53 at Thr55 and provide insights into the regulation of B56γ- and B56γ-specific PP2A complexes in response to DNA damage.
Results
PP2A dephosphorylates p53 at Thr55 in response to DNA damage
We previously observed a reduction in Thr55 phosphorylation in response to DNA damage. To investigate whether PP2A is responsible for this dephosphorylation, we tested the effect of the PP2A inhibitor OA on the endogenous Thr55 phosphorylation level. U2OS cells were treated with 10 nM to 1 μM OA and Thr55 phosphorylation was detected using a Thr55 phospho-specific antibody (Ab202; Gatti et al, 2000). As shown in Figure 1A, an increase in Thr55 phosphorylation was clearly detected with 100 nM OA. As controls, the phosphorylation of Ser37, a previously identified dephosphorylation site for PP2A (Dohoney et al, 2004), was also detected under the same conditions, whereas the phosphorylation of Ser15 and Ser20 was unaffected until cells were treated with 1 μM OA (Figure 1A). To confirm the OA treatment results, U2OS cells were also treated with calyculin (CA), another PP2A inhibitor. As shown in Figure 1B, increased Thr55 phosphorylation was detected at all concentrations tested. Together, these results indicate that PP2A may play a role in p53 dephosphorylation at Thr55.
Figure 1.
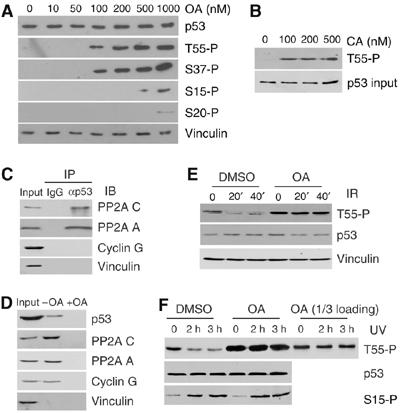
Inhibition of PP2A abolishes DNA damage-induced Thr55 dephosphorylation. (A) U2OS cells were treated with OA for 2 h at different concentrations. p53 protein as well as Thr55, Ser15, Ser37 and Ser20 phosphorylation levels were detected with the indicated antibodies. (B) Thr55 phosphorylation in cells treated with calyculin A (CA). (C) U2OS whole-cell extracts were immunoprecipitated with anti-p53 antibody and immunoblotted with anti-PP2A C, anti-PP2A A, anti-cyclin G and anti-vinculin antibodies. (D) U2OS lysates were incubated with microcystin beads in the absence (−) or presence (+) of 50 nM OA. Proteins associated with beads were analyzed by Western blot analysis with the indicated antibodies. (E, F) U2OS cells were exposed to IR (E) or UV (F) radiation in the absence (DMSO control) or presence of 200 nM okadaic acid (OA). The cells in (E) were treated with OA for 1 h and the cells in (F) were treated for 3 h. To normalize the p53 protein level, cells were treated with MG132 before irradiation. Cells were harvested after DNA damage at the times indicated and Thr55 and Ser15 phosphorylation was assayed.
To provide evidence that PP2A is directly involved in p53 dephosphorylation at Thr55, we determined whether cellular p53 protein could complex with the PP2A core enzyme in vivo. U2OS cell lysates were immunoprecipitated with anti-p53 antibody (DO-1) and the resulting immunocomplexes were analyzed by immunoblotting with anti-PP2A C and A antibodies (IP/IB). As indicated in Figure 1C, immunoprecipitation of p53 resulted in co-precipitation of PP2A. No PP2A was detected when a control antibody (IgG) was used. As cyclin G has been shown to recruit PP2A to Mdm2 that complexes with p53 (Okamoto et al, 2002), we tested whether the interaction of PP2A and p53 observed in our study could be due to the presence of cyclin G/Mdm2. As shown in Figure 1C, no interaction between p53 and cyclin G was detected.
To confirm the interaction, we performed a microcystin-conjugated bead binding assay. Microcystin binds PP1 and PP2A with high affinity (Runnegar et al, 1995; Takai et al, 1995). U2OS cell lysate was incubated with microcystin beads and the proteins that associated with the beads were subjected to immunoblotting with anti-p53, anti-vinculin, anti-PP2A C, anti-PP2A A and anti-cyclin G antibodies. As shown in Figure 1D, a small portion of total cellular p53 was indeed associated with the microcystin beads. As a negative control, microcystin beads were preincubated with 50 nM OA, which specifically competes with microcystin for binding to the PP2A C subunit (+OA; Figure 1D). The presence of cyclin G protein in the microcystin pull-down is consistent with the finding that cyclin G assembles into a quarternary complex with PP2A enzyme (Okamoto et al, 2002). However, cyclin G was not detected in the IP/IB experiments, as shown in Figure 1C, which indicates that p53 does not associate with the cyclin G–PP2A complex. These data suggest that the subset of PP2A complexes involved in p53 dephosphorylation at Thr55 may be distinct from the cyclin G-containing PP2A complex responsible for Mdm2 dephosphorylation.
Our results that PP2A phosphatase activity is involved in Thr55 dephosphorylation of p53 prompted us to test whether PP2A may play a role in Thr55 dephosphorylation in response to DNA damage. As reported previously (Li et al, 2004), a reduction in Thr55 phosphorylation was observed after both IR (Figure 1E) and UV irradiation (Figure 1F). Treating cells with OA, however, completely abolished the reduction (Figure 1E and F). As a control, Ser15 phosphorylation was not affected in the presence of OA (Figure 1F). These data suggest that PP2A plays a specific role in Thr55 dephosphorylation in response to DNA damage. Because the 3 h OA treatment increases the Thr55 phosphorylation levels, lesser amounts of loading (1/3) were also used to exclude the possibility that overloading may mask minor changes in Thr55 phosphorylation after DNA damage.
Specific PP2A regulatory subunits, B56γ1 and B56γ3, mediate Thr55 dephosphorylation
Because the B subunit of PP2A is believed to mediate the substrate specificity of PP2A, we hypothesized that one or more B subunit(s) may mediate the interaction between PP2A and p53. We overexpressed seven HA-tagged B subunits from different B families in U2OS cells and observed interactions between p53 and two isoforms of B56γ, B56γ1 and B56γ3, as well as B55α, a widespread and ubiquitously expressed PP2A B regulatory subunit (Figure 2A and data not shown).
Figure 2.
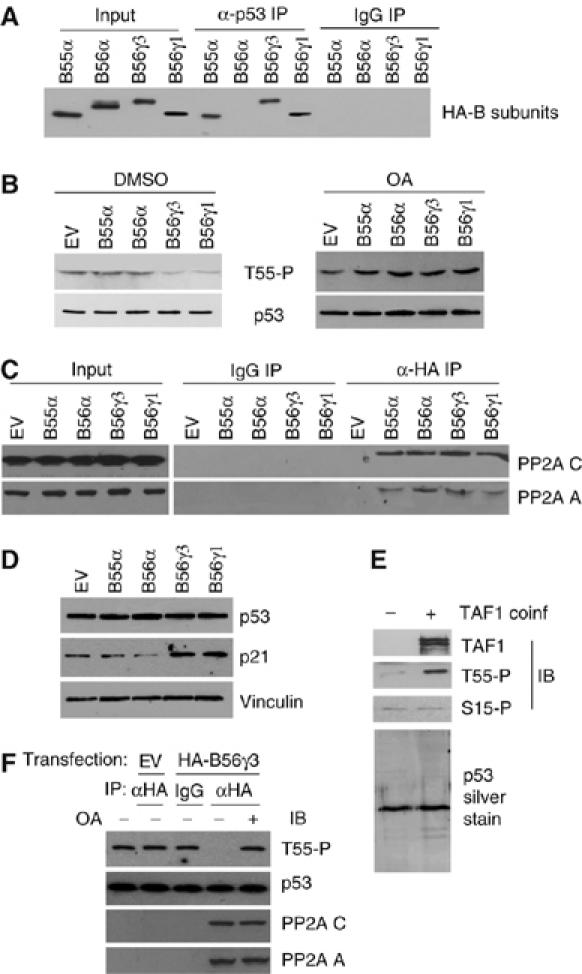
Specific B regulatory subunits B56γ1 and B56γ3 interact with p53 and are responsible for Thr55 dephosphorylation. (A) HA-tagged B55α, B56α, B56γ1 and B56γ3 were overexpressed in U2OS cells. Cell lysates were immunoprecipitated with anti-p53 antibody and immunoblotted with anti-HA antibody. (B) Thr55 phosphorylation levels were analyzed by phospho-specific antibody for Thr55 under B55α, B56α, B56γ1 or B56γ3 overexpression conditions in the absence (DMSO) or presence of OA. (C) HA-tagged B55α-, B56α-, B56γ1- or B56γ3-overexpressed cell lysates were immunoprecipitated with anti-HA antibody and immunoblotted with anti-PP2A A and C antibodies. (D) p53 and p21 protein levels were tested in the absence of MG132. (E) Affinity-purified baculovirus-expressed p53 that is enriched for phosphorylation at Thr55 via TAF1 coinfection. (F) The Thr55 phosphorylated p53 protein was dephosphorylated by immunoprecipitated B56γ3-containing complexes of PP2A in the absence (−) or presence (+) of OA.
Next, we determined whether overexpression of the B subunits that interact with p53 promotes Thr55 dephosphorylation by PP2A. As shown in Figure 2B, overexpression of the B56γ1 and B56γ3 subunits markedly decreased Thr55 phosphorylation, whereas overexpression of B56α, which does not interact with p53 (Figure 2A), had little effect on Thr55 phosphorylation. Interestingly, although B55α was shown to interact with p53 in the co-immunoprecipitation assay (Figure 2A), its overexpression did not affect Thr55 phosphorylation, which suggests that this subunit does not mediate p53 dephosphorylation at Thr55. To ensure that the HA-tagged B subunits were overexpressed as functional regulatory proteins, their interactions with the A and C subunits of PP2A were confirmed in co-immunoprecipitation assays (Figure 2C). Furthermore, the OA treatment completely blocked the dephosphorylation by B56γ1 and B56γ3 (Figure 2B, right), which indicates that these two B56γ subunits specifically mediate p53 dephosphorylation by PP2A at Thr55.
Because previous studies in our laboratory indicated that Thr55 phosphorylation promotes p53 protein degradation, our finding that overexpression of the B56γ1 and B56γ3 subunits decreases Thr55 phosphorylation prompted us to investigate a subsequent stabilization of p53. To our surprise, however, we found that compared with cells transfected with B56α and B55α, which had no effect on Thr55 phosphorylation, the endogenous p53 protein level in cells transfected with the B56γ1 and B56γ3 subunits was largely unaffected (Figure 2D). As B56γ3 has been shown to mediate Mdm2 dephosphorylation and therefore enhance Mdm2-mediated p53 degradation (Okamoto et al, 2002), the unchanged p53 protein level may be due to the combinatory effects of Hdm2 (the human homologue of Mdm2) dephopshorylation, which leads to Hdm2 activation and subsequent p53 degradation, and Thr55 dephosphorylation, which leads to p53 stabilization. However, Thr55 dephosphorylation may activate p53 function without changing its protein levels. To test this possibility, we showed that the p21 protein levels were indeed increased under the B56γ3 and B56γ1 overexpression conditions (Figure 2D), which suggests that B56γ-mediated Thr55 dephosphorylation contributes to p53 activation.
To provide direct evidence that the B56γ3-containing PP2A complexes dephosphorylate p53 at Thr55, we performed an in vitro dephosphorylation assay using immunoprecipitated B56γ3-containing PP2A complexes and baculovirus-expressed and purified p53 that is enriched for phosphorylation at Thr55 by coinfection with TAF1 (Figure 2E). Thr55 phosphorylation levels in the reaction mixture were assayed with Thr55 phospho-specific antibody. As shown in Figure 2F, the addition of immunoprecipitated B56γ3-containing PP2A complexes to the Thr55 phosphorylated p53 protein resulted in a marked reduction in Thr55 phosphorylation, whereas control immunoprecipitations had no effect on dephosphorylation. Furthermore, the OA treatment completely blocked dephosphorylation by immunoprecipitated B56γ3-containing PP2A complexes (Figure 2F). Collectively, our data support the hypothesis that B56γ3-specific PP2A directly dephosphorylates p53 at Thr55.
To further explore the role of B56γ in regulating Thr55 dephosphorylation and p53 function, we designed specific siRNA oligos to knock down endogenous B56γ proteins. As shown in Figure 3A, the cellular B56γ3 protein was successfully ablated by the specific siRNA oligos, whereas the protein level of B56α and B56β, two other B56 family isoforms, remained unchanged. However, the endogenous Thr55 phosphorylation level was largely unaffected by B56γ ablation (Figure 3A). Because PP2A plays a role in Thr55 dephosphorylation in response to DNA damage (Figure 1), we reasoned that B56γ might specifically function after DNA damage. To test this possibility, we assayed Thr55 dephosphorylation after DNA damage under the B56γ ablation condition. Compared with controls, the knockdown of endogenous B56γ proteins in U2OS cells indeed abolishes Thr55 dephosphorylation in response to both UV and IR treatments (Figure 3B). In contrast, the knockdown of B56γ has no effect on Ser37 phosphorylation (Figure 3B). These data suggest that B56γ specifically dephosphorylates p53 at Thr55 in response to DNA damage. Significantly, a decrease in p53 stabilization (4.5-fold versus 6-fold) and Bax induction (3.7-fold versus 2.2-fold) was observed under B56γ ablation conditions in the absence of the proteosome inhibitor MG132 (Figure 3C), which indicates that B56γ plays a role in p53 activation in response to DNA damage. To further investigate the functional link between B56γ-mediated Thr55 dephosphorylation and p53 activation, we determined whether B56γ-mediated Thr55 dephosphorylation could play a role in cell apoptosis in response to DNA damage. As shown in Figure 3D, UV treatment causes 55.1% of cells to undergo apoptosis, whereas under the B56γ ablation condition, the same treatment only results in 19.2% of cells to undergo apoptosis. As the antibody specific for PP2A B56γ used in these experiments recognizes only the B56γ2 and B56γ3 splice variants, we were not able to determine whether B56γ1 and B56γ4 were also ablated. However, as B56γ1, B56γ2, B56γ3 and B56γ4 share exons 1–11, the introduction of the specific siRNA oligos should target all four B56γ isoforms. In light of the previously demonstrated inhibition of Thr55 phosphorylation by overexpression of B56γ1 and B56γ3 (Figure 2), these results suggest that at least two of the B56γ isoforms, γ1 and γ3, function to mediate dephosphorylation of p53 at Thr55 in response to DNA damage.
Figure 3.
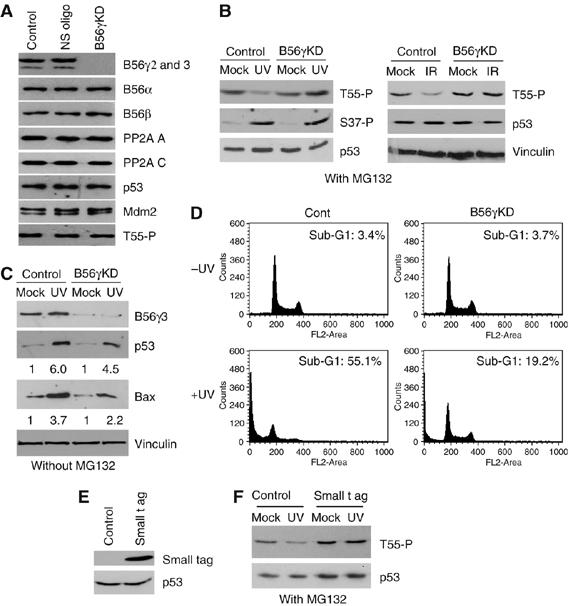
B56γ mediates Thr55 dephosphorylation in response to DNA damage. (A) U2OS cells were transfected with siRNA oligos targeted to B56γ or a control nonspecific oligo (NS). The protein levels of B56γ, B56α, B56β, p53, Mdm2, vinculin and Thr55 phosphorylation of p53 were analyzed. (B) U2OS cells were subjected to UV or IR radiation with (B56γ KD) or without (control) B56γ protein ablation. Phosphorylation levels of p53 at Thr55 and Ser37 were tested in the presence of MG132. The p53 and Bax protein levels were also assayed in the absence of MG132 (C). (D) Reduction of the DNA damage-induced cell apoptotic response in U2OS cells by RNAi-mediated B56γ ablation. (E) SV40 t ag was overexpressed in U2OS cells. (F) U2OS cells were subjected to UV radiation with (small t ag) or without (control) SV40 small t antigen overexpression. Thr55 phosphorylation of p53 was assayed in the absence (mock) or presence of UV radiation.
A recent study has indicated that SV40 small t antigen (t ag) displaces the B56γ3 subunit from the PP2A trimeric holoenzyme, leading to cell transformation (Chen et al, 2004). To confirm the B56γ ablation results, t ag was overexpressed in U2OS cells (Figure 3E) and Thr55 phosphorylation levels were assayed under normal growth and DNA damage conditions. Compared with cells transfected with the empty CMV vector, overexpression of t ag in U2OS cells abolished Thr55 dephosphorylation under DNA damage but not normal growth conditions (Figure 3F). These results support the notion that the specific PP2A regulatory subunits, B56γ1 and B56γ3, mediate Thr55 dephosphorylation in response to DNA damage.
B56γ is upregulated in response to DNA damage
The observation that B56γ mediates dephosphorylation of p53 at Thr55 under DNA damage but not normal growth conditions prompted us to investigate the mechanisms involved in the DNA damage-induced dephosphorylation. U2OS cells were exposed to γ-irradiation and B56γ protein level was assayed by immunoblotting. Interestingly, the B56γ2 and B56γ3 protein levels increased rapidly after irradiation (Figure 4A). The protein level of the B55α subunit, which we found to be associated with p53, as well as that of the PP2A C and A subunits remained constant after DNA damage. These results are consistent with the finding that overexpression of the B56γ subunits decreases Thr55 phosphorylation (Figure 2) and suggest that the DNA damage increases the B56γ protein level, which plays a role in recruiting the PP2A C subunit to p53 and sequential Thr55 dephosphorylation. To test this hypothesis, we compared the interaction between endogenous B56γ and p53 before and after DNA damage. As shown in Figure 4B, a significant increase in B56γ–PP2A C association was detected after DNA damage by microcystin bead pull-down assay. In contrast, the association of B55α–PP2A C remained unchanged after DNA damage. Likewise, increased association of the PP2A C, A and B56γ subunits with p53 was also detected after DNA damage with IP/IB (Figure 4C), suggesting an enhanced interaction of p53 and B56γ-specific PP2A complexes. Collectively, these results indicate that DNA damage increases the B56γ protein level and enhances the formation of B56γ-specific PP2A complexes, which, in turn, lead to dephosphorylation of p53 at Thr55.
Figure 4.
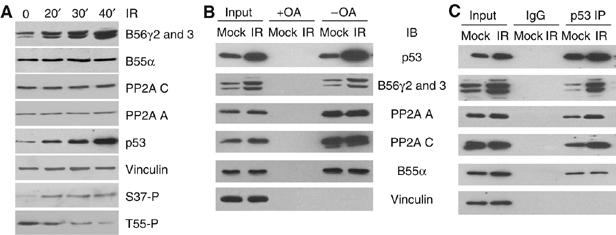
Increases in B56γ protein levels and in the association with p53 correlate with Thr55 dephosphorylation. (A) Whole-cell extracts of γ-irradiated U2OS cells were subjected to immunoblotting with anti-B56γ, anti-B55α, anti-PP2A C, anti-PP2A A, anti-p53, anti-phospho-Ser37 and anti-vinculin antibodies. Thr55 phosphorylation was assayed with anti-phospho-p53 antibody as described. (B) U2OS cells were exposed to IR and association of p53 and B56γ3 with PP2A C was detected by microcystin bead pull-down assay. (C) Association of p53 with B56γ3-containing PP2A complex after DNA damage was detected by p53 immunoprecipitation (IP) and PP2A A, PP2A C and B56γ immunoblotting (IB).
B56γ3 inhibits cell proliferation and transformation in a p53-dependent manner
As B56γ3 has been reported to inhibit cell proliferation in lung cancer cells (Chen et al, 2004), we proceeded to test whether B56γ3 could function through dephosphorylation of p53. We considered the possibility that different cell lines might differ in ways other than p53 status, and therefore chose the HCT116 p53−/− and p53+/+ human colon cancer cells (Bunz et al, 1998). Compared with cells transfected with the empty CMV vector, overexpression of B56γ3 in HCT116 p53+/+ cells reduced cell proliferation (Figure 5A, left). In contrast, overexpression of B56γ3 in HCT116 p53−/− cells had a much less effect on cell proliferation (Figure 5A, right). The presence of overexpressed B56γ3 during the analysis was confirmed by immunoblotting analysis (Figure 5C, left). Notably, significant increases in the p21 protein levels were also observed in HCT116 p53+/+, but not in p53−/−, cells under the B56γ3 overexpression conditions (Figure 5C, left). Cell doubling times from three independent cell growth experiments are shown in Figure 5D. These results indicate that B56γ3 inhibits cell proliferation, at least in part, through p53.
Figure 5.
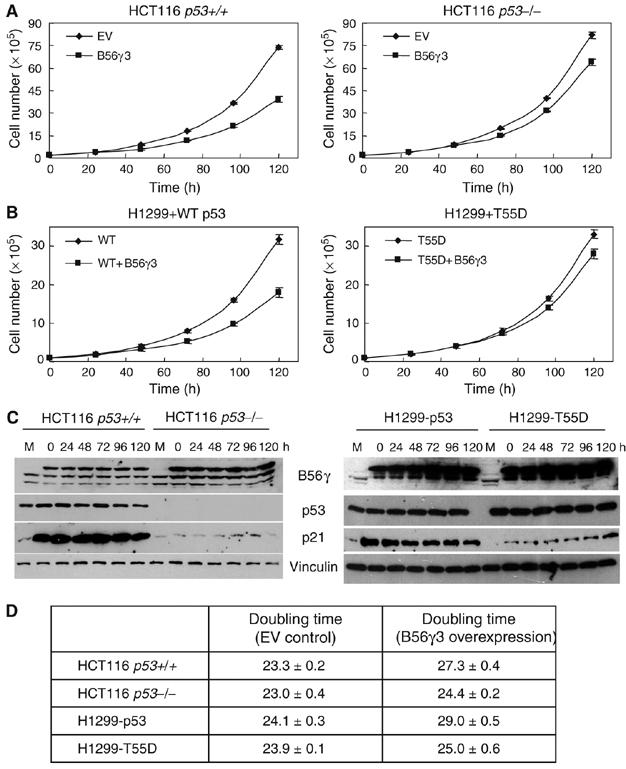
B56γ3 inhibits cell proliferation in a p53-dependent manner. Representatives of cell proliferation of the HCT116 human colon cancer cell lines (A) or the H1299 human lung cancer cell line (B) transfected with either B56γ3 or a control CMV empty vector. H1299+p53: H1299 transfected with wild-type p53; H1299+T55D: H1299 transfected with T55D. Error bars show average±s.d. from triplicate plates in one representative experiment. (C) Immunoblotting of the transfected HA-B56γ3, endogenous B56γ3/B56γ2, p53 and p21 proteins in HCT116 and H1299 cells. M: empty vector-transfected cell lysate at seeding. (D) Cell doubling times show average±s.d. from three independent experiments. EV: empty vector.
To assay the role of p53 dephosphorylation in B56γ3-mediated inhibition of cell proliferation, we reintroduced wild-type p53 and a p53 dephosphorylation mutant (alteration of Thr to Asp at position 55, T55D) into H1299 human lung cancer cell line, a p53−/− cancer cell line, and assayed the effect of B56γ3 on cell proliferation rate. As in HCT116−/− cells, overexpression of B56γ3 alone had a moderate effect on cell proliferation rate in H1299 cells (data not shown). Overexpression of B56γ3 together with wild-type p53, however, significantly inhibits cell proliferation (Figure 5B), supporting the notion that B56γ3 inhibits cell proliferation in a p53-dependent manner. By comparison, introduction of B56γ3 together with T55D failed to significantly affect the proliferation rate. The presence of overexpressed B56γ3 and p53 during the analysis was confirmed by immunoblotting analysis (Figure 5C, right). Cell doubling times from three independent cell growth experiments are shown in Figure 5D. These results suggest that p53 dephosphorylation contributes to B56γ3-mediated inhibition of cell proliferation.
The observation that B56γ3 inhibits cell proliferation in a p53-dependent manner prompted us to investigate whether B56γ3 affects p53-induced cell G1 arrest. As shown in Figure 6A, overexpression of p53 resulted in more cells in the G1 phase (from 52 to 60%) and fewer in the S phase (from 19 to 14%) in H1299 cells and more importantly, the p53-induced cell G1 arrest is significantly enhanced by B56γ3. Overexpression of T55D also induces cell G1 arrest. However, B56γ3 has no effect on the T55D-induced cell G1 arrest (Figure 6A). In light of the previous demonstration that B56γ3 significantly inhibits cell proliferation in the presence of wild-type p53, but not T55D (Figure 5), these results suggest that B56γ3 induces cell G1 arrest in a p53 dephosphorylation-dependent manner.
Figure 6.

B56γ3 functions in cell G1 arrest and transformation via Thr55 dephosphorylation. (A) H1299 cells were transfected with B56γ3 together with wild-type p53 or T55D. The cell-cycle profile was analyzed 60 h after transfection. (B) Anchorage-independent growth of HCT116 cells in the presence or absence of B56γ3 as indicated. EV: empty vector. (C) Anchorage-independent growth of H1299 cells in the presence or absence of p53, T55D and B56γ3 as indicated.
Because B56γ3 suppression has been linked to cell transformation and tumorigenicity (Chen et al, 2004), we reasoned that B56γ3 might function through dephosphorylation of p53 at Thr55. To test this, we assayed whether p53 is required for inhibition of anchorage-independent cell growth by B56γ3. As shown in Figure 6B, the HCT116 p53+/+ human colon cancer cells transfected with B56γ3 formed much less colonies in soft agar than those formed by the cells transfected with a control vector. In contrast, the HCT116 p53−/− human colon cancer cells transfected with B56γ3 formed only slightly less colonies in soft agar than those formed with a control vector. These data suggest that B56γ3 inhibits anchorage-independent cell growth in a p53-dependent manner.
To assay the role of Thr55 dephosphorylation in B56γ3-mediated inhibition of anchorage-independent cell growth, we reintroduced wild-type p53 and T55D into H1299 human lung cancer cell line and assayed the effect of B56γ3 on colony formation in soft agar. As shown in Figure 6C, overexpression of B56γ3 together with wild-type p53 significantly reduced colony formation in soft agar, whereas introduction of B56γ3 together with T55D had only a moderate effect on anchorage-independent cell growth (Figure 6C). These data support the notion that B56γ3 inhibits anchorage-independent cell growth through dephosphorylation of p53 at Thr55. Collectively, our data provide a molecular basis for B56γ3-mediated tumor suppression.
Discussion
In this study, we show that PP2A dephosphorylates the tumor suppressor p53 at Thr55 in response to DNA damage. Importantly, we identified a specific B regulatory subunit of PP2A, B56γ, which mediates Thr55 dephosphorylation by PP2A and showed that B56γ3 inhibits cell proliferation and transformation in a p53-dependent manner. Furthermore, we report an induction in B56γ protein level and in the association with p53 in response to DNA damage and provide insights into regulation of B56γ- and B56γ-specific PP2A complexes. The implications of our findings in relation to the tumor suppressive function of PP2A are discussed as follows.
B56γ3 as a tumor suppressor and implication in human cancers
Several lines of evidence have linked B56γ isoforms to PP2A's tumor suppressive function. For example, a melanoma cell line with a truncated B56γ1 isoform showed increased invasiveness and enhanced malignant phenotypes as compared with its parental melanoma cell line (Ito et al, 2003). Moreover, B56γ was absent in all 10 human lung cancer cell lines investigated, whereas the PP2A C, A and B levels were within the normal range (Chen et al, 2004). Interestingly, Chen et al (2004) also found that suppression of B56γ expression increased cell proliferation and permitted anchorage-independent growth, similar to the functional consequences of small t overexpression. These results suggest that B56γ is involved in cell growth and proliferation control. Because the individual B regulatory subunit is believed to specifically recruit substrates to the PP2A holoenzyme, the most likely molecular mechanisms for the action of B56γ tumor suppression should be related to the functions of the proteins that B56γ targets for dephosphorylation by PP2A. Indeed, B56γ3-containing PP2A complexes were recruited by cyclin G to Mdm2, which leads to dephosphorylation of Mdm2 at Thr216. The resulting hypophosphorylated Mdm2, however, is more efficient for p53 degradation (Okamoto et al, 2002). Although these studies provide evidence that the B56γ3-containing PP2A complexes affect p53 stability, the function of the dephosphorylation (which leads to p53 degradation) corresponding to the role of B56γ3 as a potential tumor suppressor remains unclear.
In the present study, we identified B56γ as the B regulatory subunit responsible for Thr55 dephosphorylation of p53, thus providing a molecular basis for B56γ's function as a potential tumor suppressor. Owing to the fact that mouse Mdm2 is not present in our system, we were unable to investigate the effect of Thr216 dephosphorylation on Thr55 dephosphorylation and p53 stability. It is possible that, under our assay conditions, cyclin G recruits B56γ3-containing PP2A complexes to dephosphorylate Hdm2. The fact that B56γ overexpression, which leads to Thr55 dephosphorylation (Figure 2B), did not change the p53 protein levels (Figure 2D) supports this view. The combinatory effects of Hdm2 dephosphorylation, which leads to Hdm2 activation and subsequent p53 degradation, and Thr55 dephosphorylation, which leads to p53 stabilization, would lead to unchanged p53 protein level.
Finally, emerging evidence has begun to indicate that PP2A plays an important role in cell transformation, particularly in human cancer. Interestingly, a recent study suggests that perturbation of two signaling pathways, involving p53 and Raf, suffices for the tumorigenic conversion of normal murine fibroblasts, whereas perturbation of six pathways, involving p53, pRb, PP2A, telomerase, Raf and Ral-GEFs, is needed for human fibroblasts, which indicates the importance of PP2A in human cancer progression (Rangarajan et al, 2004). Because Thr55 is absent in mouse p53, it is tempting to speculate that our discovery may also provide a molecular basis for this human-specific tumor suppressive function of PP2A. Ultimately, it will be interesting to test this hypothesis in human cancer samples to further understand the roles of PP2A and Thr55 dephosphorylation in human cancer.
Mechanisms for activation of B56γ3-specific PP2A complexes
Although the B56γ-containing PP2A complex has been shown to function as a tumor suppressor, little is known about its regulation in response to DNA damage. Our studies establish, for the first time, that B56γ is upregulated after DNA damage and thus reveal a mechanism for understanding the potential tumor suppressive function of PP2A. This finding has two implications. First, although PP2A is considered a single enzyme, it actually represents a family of specifically modulated enzymes that regulate many different pathways within the cell. For this reason, tight regulation of each PP2A enzyme is very important. Much research has been carried out on the regulatory B subunits of PP2A, but the precise regulatory mechanisms that control PP2A B subunit composition are still poorly understood. The increase in B56γ3 protein level may thus provide a general mechanism for regulation of specific PP2A function.
Second, our observation of the increase in B56γ3 protein level and p53 association after DNA damage may illustrate a potential route for regulation of PP2A tumor suppressor function in response to DNA damage. Although the underlying molecular mechanisms are unclear, the fact that B56γ3 protein levels increased in response to DNA damage in a manner similar to p53 suggests that similar mechanisms may apply to the regulation of B56γ3. Clearly, more studies are needed to further understand the mechanisms of B56γ3 regulation.
The dynamics of Thr55 phosphorylation
We previously showed phosphorylation of p53 at Thr55 by TAF1 under cell growth conditions, which leads to Mdm2-mediated protein degradation and subsequent cell growth (Li et al, 2004). In response to DNA damage, however, p53 was dissociated from TAF1, resulting in reduced Thr55 phosphorylation (Li et al, 2004). In the present study, we show that, in addition to p53 dissociation from TAF1, association of p53 and B56γ-containing PP2A complexes also plays a role in Thr55 dephosphorylation. Although the precise mechanisms involved in TAF1 dissociation and B56γ-containing PP2A complex association remain unclear, these studies imply that balanced Thr55 phosphorylation may be important in controlling cell growth and death (Figure 7). Just as keeping p53 protein levels low is important for cell growth, keeping the p53 protein prepared for a rapid and finely regulated response to genotoxic stress is crucial in maintaining genomic stability and preventing proliferation of damaged cells. Thus, by dephosphorylating p53 at Thr55, B56γ-containing PP2A complexes shift the balance of Thr55 toward dephosphorylation in response to DNA damage, thereby functioning as a tumor suppressor.
Figure 7.
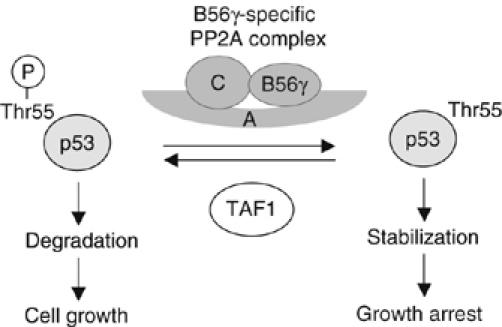
A proposed model for the dynamics of Thr55 phosphorylation by TAF1 and PP2A.
We by no means imply that the regulation of p53 by other modifications is not important in controlling p53 protein levels and transcriptional activity. In fact, our data that p53 is associated with B56γ-containing PP2A complexes in response to DNA damage imply that other modifications may occur on p53 before Thr55 dephosphorylation. Likewise, B56γ-containing PP2A complexes may also regulate p53 through other pathways. For example, PP2A has been shown to play a role in ARF signaling to p53 pathway (Moule et al 2004). Thus, it will be interesting to investigate the interplay between different signaling pathways and Thr55 phosphorylation and to further elucidate under what conditions the B56γ-containing PP2A complexes exhibit tumor suppressive function.
Materials and methods
Cell culture and plasmids
U2OS cells were cultured in McCoy's 5A medium supplemented with 10% fetal calf serum. To induce DNA damage, the cells were subjected to UV radiation (10 J/m2) or γ-radiation (6 Gy) and harvested at the time points indicated. In PP2A inhibition experiments, cells were treated with 0.01–1 μM OA, 0.1–0.5 μM CA or DMSO control for 2 h or as indicated.
B56α and B56γ3 plasmids were gifts from Dr D Virshup. B55α was amplified from the human B-cell cDNA library, a gift from Dr J Bachant, and cloned into the pCMV5-HA vector in KpnI/BamHI sites. SV40 t ag was amplified from rpSVP1A, a gift from Dr A Berk at UCLA, and cloned into the pcDNA3.1 vector in HindIII/BamHI sites. Both insertions were confirmed by DNA sequencing.
Western blot, immunoprecipitation and microcystin bead binding assays
Whole-cell extract was prepared by lysing the cells in a buffer containing 50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 0.5% NP-40, 1 mM DTT, 2 μg/ml aprotinin and 2 μg/ml leupeptin. Cell lysates were subjected to SDS–PAGE, followed by Western blot analysis with anti-p53 (DO1, Santa Cruz Biotechnology), anti-phospho-Ser15 (Cell Signaling Technology), anti-phospho-Ser20 (Cell Signaling Technology), anti-phospho-Ser37 (Cell Signaling Technology), cyclin G (Santa Cruz Biotechnology), anti-PP2A A subunit (Upstate), anti-PP2A C subunit (1D6, Upstate), anti-PP2A B subunit (Upstate), anti-PP2A B56α (Santa Cruz Biotechnology), anti-PP2A B56β (Santa Cruz Biotechnology), anti-PP2A B56γ3 (a gift from Drs C Kamibayashi and M Mumby, or generated by ourselves against full-length B56γ3), anti-Mdm2 (N-20, Santa Cruz Biotechnology) or anti-vinculin (VIN-11-5, Sigma) antibodies. For Thr55 dephosphorylation, the cell lysate was incubated for 6 h at 4°C with 1.2 μl of phospho-specific antibody for Thr55 (Ab202) and 15 μl of Protein A agarose beads (Pierce Biotechnology). Beads were washed three times with lysis buffer and proteins were eluted with Laemmli sample buffer and subjected to immunoblotting. For interaction of p53 with transfected B subunits, 1 × 107 U2OS cells were transfected with 2 μg HA-tagged B subunits using Lipofectamine (Invitrogen) and lysed 28 h after transfection. Immunoprecipitation was performed using 0.5 μg of anti-p53 polyclonal antibody (FL393, Santa Cruz) and 15 μl of Protein A agarose beads. The amounts of B subunits in the immunoprecipitates were determined by Western blot analysis with anti-HA antibody. For interaction of p53 with endogenous B56γ3, 1 ml of U2OS cell lysate from 3 × 107 cells was incubated with 0.5 μg of FL393 antibody. For microcystin bead binding assay, the U2OS cell lysate (from 4 × 107 cells) was incubated with 20 μl of microcystin beads (Upstate) for 2 h at 4°C. The beads were washed and the protein was dissociated from the beads using Laemmli buffer and then subjected to immunoblotting.
In vitro PP2A dephosphorylation assay
Baculovirus-expressed p53 that is enriched for phosphorylation at Thr55 via TAF1 coinfection was affinity-purified as described (Piluso et al, 2005). To obtain B56γ3-containing PP2A complex, immunoprecipitation was performed with HA-B56γ3-transfected U2OS cell lysate (2 × 107 cells per IP) and 10 μl of anti-HA antibody-conjugated Protein A beads. The dephosphorylation assay was performed in dephosphorylation buffer (20 mM HEPES, pH 7.0, 1 mM DTT, 1 mM MnCl2, 100 μg/ml BSA and 50 μM leupeptin) (Scheidtmann et al, 2003) with immunoprecipitated B56γ3-containing PP2A complex at 30°C for 30 min in the presence or absence of OA.
Inhibition of endogenous B56γ3 by siRNA
Two 21-nt siRNA duplexes with 3′dTdT overhangs corresponding to the B56γ3 cDNA sequences 305 (5′-UCCUACGGGAGCGGAAUUU-3′) and 1045 (5′-UCAGUGACAACGCAGCGAA-3′) were synthesized. Twenty picomoles of either B56γ3 siRNA or a non-silencing control oligo (Qiagen) was transfected into a 60-mm plate of U2OS cells using Lipofectamine 2000 reagent according to the manufacturer's protocol (Invitrogen).
Cell apoptosis and cell cycle profile analysis
For apoptosis assay, U2OS cells were transfected with B56γ-siRNA or a non-silencing control siRNA (NS) and collected 20 h after UV treatment, fixed, stained with propidium iodide and analyzed by FACScan flow cytometry (Becton Dickinson) for apoptotic cells (sub-G1) according to DNA content.
For cell cycle analysis, H1299 cells were transfected with B56γ3 together with either wild-type p53 or T55D. Cells were harvested 60 h after transfection, fixed in paraformaldehyde and stained with propidium iodide. Cell cycle phase distributions of GFP-positive cells were determined by FACScan flow cytometry.
Cell proliferation and anchorage-independent growth assays
To generate proliferation curves for HCT116 cells, cells were transfected with either B56γ3 or a control CMV empty vector using Fugene (Roche), seeded in triplicate and counted at 0, 24, 48, 72, 96 and 120 h post seeding. To generate proliferation curves for H1299 cells, cells were cotransfected with wild-type p53 or T55D and B56γ3 or a control CMV empty vector, seeded in triplicate and counted at 0, 24, 48, 72, 96 and 120 h post seeding.
For anchorage-independent growth assays of HCT116 cells, cells were transfected with either B56γ3 or a control CMV empty vector, seeded in triplicate in 0.35% Noble Agar (Fisher) and counted 4 weeks post seeding. For anchorage-independent growth assays of H1299 cells, cells were cotransfected with wild-type p53 or T55D and B56γ3 or a control CMV empty vector, seeded in triplicate in 0.35% Noble Agar and counted 4 weeks post seeding.
Acknowledgments
We are very grateful to Drs C Kamibayashi and M Mumby for providing anti-B56γ3 antibody, to Dr DM Virshup for providing PP2A B56 expression constructs, to Dr AJ Berk for providing SV40 plasmid and to Dr J Bachant for providing a human B cell cDNA library. We thank Dr JA Traugh and all members of our laboratory, particularly AG Li, for many helpful discussions. This work was supported by NIH grant CA75180 from the National Institute of Cancer.
References
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC (2004) Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5: 127–136 [DOI] [PubMed] [Google Scholar]
- Csortos C, Zolnierowicz S, Bako E, Durbin SD, DePaoli-Roach AA (1996) High complexity in the expression of the B′ subunit of protein phosphatase 2A: evidence for the existence of at least seven novel isoforms. J Biol Chem 271: 2578–2588 [DOI] [PubMed] [Google Scholar]
- Dohoney KM, Guillerm C, Whiteford C, Elbi C, Lambert PF, Hager GL, Brady JN (2004) Phosphorylation of p53 at serine 37 is important for transcriptional activity and regulation in response to DNA damage. Oncogene 23: 49–57 [DOI] [PubMed] [Google Scholar]
- Gatti A, Li H-H, Traugh JA, Liu X (2000) Phosphorylation of human p53 on Thr-55. Biochemistry 39: 9837–9842 [DOI] [PubMed] [Google Scholar]
- Ito A, Koma Y, Watabe K, Nagano T, Endo Y, Nojima H, Kitamura Y (2003) A truncated isoform of the protein phosphatase 2A B56γ regulatory subunit may promote genetic instability and cause tumor progression. Am J Pathol 162: 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J, Van Hoof C (2005) PP2A: the expected tumor suppressor. Curr Opin Genet Dev 15: 34–41 [DOI] [PubMed] [Google Scholar]
- Li H-H, Li AG, Sheppard HM, Liu X (2004) Phosphorylation on Thr55 by TAF1 mediates degradation of p53: a role for TAF1 in cell G1 progression. Mol Cell 13: 867–878 [DOI] [PubMed] [Google Scholar]
- McCright B, Brothman AR, Virshup DM (1996) The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. Genomics 36: 168–170 [DOI] [PubMed] [Google Scholar]
- Moule MG, Collins CH, McCormick F, Fried M (2004) Role for PP2A in ARF signaling to p53. Proc Natl Acad Sci USA 91: 8974–8978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muneer S, Ramalingam V, Wyatt R, Schultz RA, Minna JD, Kamibayashi C (2002) Genomic organization and mapping of the gene encoding the PP2A B56gamma regulatory subunit. Genomics 79: 344–348 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Li H, Jensen MR, Zhang T, Taya Y, Thorgeirsson SS, Prives C (2002) Cyclin G recruits PP2A to dephosphorylate Mdm2. Mol Cell 9: 761–771 [DOI] [PubMed] [Google Scholar]
- Ortega-Lazaro JC, del Mazo J (2003) Expression of the B56delta subunit of protein phosphatase 2A and Mea1 in mouse spermatogenesis: identification of a new B56gamma subunit (B56gamma4) specifically expressed in testis. Cytogenet Genome Res 103: 345–351 [DOI] [PubMed] [Google Scholar]
- Piluso LG, Wei G, Li AG, Liu X (2005) Purification of acetyl-p53 using p300 co-infection and the baculovirus expression system. Protein Expr Purif 40: 370–378 [DOI] [PubMed] [Google Scholar]
- Rangarajan A, Hong SJ, Gifford A, Weinberg RA (2004) Species- and cell type-specific requirements for cellular transformation. Cancer Cell 6: 171. [DOI] [PubMed] [Google Scholar]
- Runnegar M, Berndt N, Kong SM, Lee EY, Zhang L (1995) In vivo and in vitro binding of microcystin to protein phosphatases 1 and 2A. Biochem Biophys Res Commun 216: 162–169 [DOI] [PubMed] [Google Scholar]
- Scheidtmann KH, Mumby MC, Rundell K, Walter G (2003) Dephosphorylation of simian virus 40 large-T antigen and p53 protein by protein phosphatase 2A: inhibition by small-t antigen. Mol Cell Biol 11: 1996–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonthal AH (2001) Role of serine/threonine protein phosphatase 2A in cancer. Cancer Lett 170: 1–13 [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA (2002) p53: good cop/bad cop. Cell 110: 9–12 [DOI] [PubMed] [Google Scholar]
- Sontag E (2001) Protein phosphatase 2A: the Trojan horse of cellular signaling. Cell Signal 13: 7–16 [DOI] [PubMed] [Google Scholar]
- Takai A, Sasaki K, Nagai H, Mieskes G, Isobe M, Isono K, Yasumoto T (1995) Inhibition of specific binding of okadaic acid to protein phosphatase 2A by microcystin-LR, calyculin-A and tautomycin: method of analysis of interactions of tight-binding ligands with target protein. Biochem J 306: 657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoof C, Goris J (2003) Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochimica Biophysica Acta 1640: 97–104 [DOI] [PubMed] [Google Scholar]
- Van Hoof C, Goris J (2004) PP2A fulfills its promises as tumor suppressor: which subunits are important? Cancer Cell 5: 105–106 [DOI] [PubMed] [Google Scholar]
- Virshup DM (2000) Protein phosphatase 2A: a panoply of enzymes. Curr Opin Cell Biol 12: 180–185 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Yatsunami J, Komori A, Ohta T, Suganuma M, Fujiki H (1993) Hyperphosphorylation of retinoblastoma protein and p53 by okadaic acid, a tumor promoter. Cancer Res 53: 239–241 [PubMed] [Google Scholar]
- Zhang W, McClain C, Gau JP, Guo XY, Deisseroth AB (1994) Hyperphosphorylation of p53 induced by okadaic acid attenuates its transcriptional activation function. Cancer Res 54: 4448–4453 [PubMed] [Google Scholar]