

Abstract
Maturation of cytochrome c peroxidase (Ccp1) in mitochondria occurs by the subsequent action of two conserved proteases in the inner membrane: the m-AAA protease, an ATP-dependent protease degrading misfolded proteins and mediating protein processing, and the rhomboid protease Pcp1, an intramembrane cleaving peptidase. Neither the determinants preventing complete proteolysis of certain substrates by the m-AAA protease, nor the obligatory requirement of the m-AAA protease for rhomboid cleavage is currently understood. Here, we describe an intimate and unexpected functional interplay of both proteases. The m-AAA protease mediates the ATP-dependent membrane dislocation of Ccp1 independent of its proteolytic activity. It thereby ensures the correct positioning of Ccp1 within the membrane bilayer allowing intramembrane cleavage by rhomboid. Decreasing the hydrophobicity of the Ccp1 transmembrane segment facilitates its dislocation from the membrane and renders rhomboid cleavage m-AAA protease-independent. These findings reveal for the first time a non-proteolytic function of the m-AAA protease during mitochondrial biogenesis and rationalise the requirement of a preceding step for intramembrane cleavage by rhomboid.
Keywords: AAA protease, cytochrome c peroxidase, intramembrane cleaving peptidase, mitochondria, rhomboid
Introduction
An increasing number of cellular pathways are recognised to be regulated by proteolytic processing. Biologically active protein fragments of membrane proteins can be generated by intramembrane cleaving peptidases, including rhomboids, site-2 proteases, signal peptide peptidases and presenilins (Freeman, 2004; Wolfe and Kopan, 2004). These peptidases possess conserved catalytic residues within their hydrophobic transmembrane domains and generate soluble, biologically active protein fragments by cleaving membrane-anchored proteins within or in close proximity to their transmembrane segments. ATP-dependent proteases, including 26S proteasomes, comprise another group of peptidases that activate various regulatory proteins controlling crucial cellular processes by proteolytic processing (Rape and Jentsch, 2004). At the same time, they conduct the quality surveillance of cellular proteins and degrade misfolded proteins to peptides (Sauer et al, 2004; Ciechanover, 2005; Hanson and Whiteheart, 2005). Increasing evidence suggests that the folding state determines the fate of substrate proteins bound by energy-dependent proteolytic complexes (Lee et al, 2001; Kenniston et al, 2005; Tian et al, 2005; Hoyt et al, 2006). ATP-dependent unfolding of substrates allows substrate entry into barrel-like proteolytic chambers and results in complete degradation (Sauer et al, 2004). Low-complexity regions preceding a tightly folded domain, however, can limit the unfolding capacity of the proteases, halt degradation and lead to the release of proteolytic fragments.
AAA proteases build up conserved membrane-embedded and ATP-dependent proteolytic complexes in mitochondria and also function in both protein quality control and proteolytic activation (Ito and Akiyama, 2005; Nolden et al, 2006). Inactivation of AAA proteases results in pleiotropic cellular defects including the loss of respiratory competence in yeast and axonal degeneration in mammals (Nolden et al, 2006; Rugarli and Langer, 2006). The respiratory deficiency of yeast cells lacking the matrix-exposed m-AAA protease was recently explained by the impaired proteolytic activation of a ribosomal protein, MrpL32 (Nolden et al, 2005). Processing of MrpL32 by the m-AAA protease is required for ribosome assembly and the synthesis of essential respiratory chain subunits within mitochondria (Nolden et al, 2005).
Cytochrome c peroxidase (Ccp1), a haeme-binding ROS scavenger in the intermembrane space, represents another substrate whose maturation depends on the m-AAA protease (Esser et al, 2002). Ccp1 is nuclear-encoded and targeted to mitochondria by a bipartite presequence that is cleaved off in an intriguing process involving both an ATP-dependent and an intramembrane cleaving peptidase (Esser et al, 2002): after m-AAA protease cleavage, the mature form of Ccp1 is generated by the rhomboid-like protease Pcp1 (Rbd1) in the inner membrane. Notably, Ccp1 accumulates in the precursor form in m-AAA protease-deficient mitochondria indicating that m-AAA protease activity is essential for maturation by Pcp1. The m-AAA protease dependence of Ccp1 maturation may reflect proteolytic activation of the rhomboid protease Pcp1 itself. However, this possibility can be excluded as proteolytic cleavage by Pcp1 of another substrate, the dynamin-related GTPase Mgm1 involved in mitochondrial fusion, occurs also in the absence of the m-AAA protease (Herlan et al, 2003; McQuibban et al, 2003). We have therefore investigated in the present paper the interplay of m-AAA and rhomboid proteases during Ccp1 processing in more detail.
Results
Ccp1 processing is not affected by the folding state of the haeme-binding domain
In order to elucidate the determinants of Ccp1 cleavage by the m-AAA protease, we first examined the role of Ccp1 folding for proteolytic processing. Ccp1 contains a stably folded haeme-binding peroxidase domain, which may preclude complete degradation by the m-AAA protease. We therefore replaced histidine 242 by proline, as this mutation has been demonstrated to impair haeme binding (Kaput et al, 1989). Moreover, histidine 248 and asparagine 251, both directly interacting with carbonyl groups of haeme, were mutated to glutamate and aspartate, respectively. Ccp1 and variants thereof, all carrying C-terminal dodecahistidine peptides, were expressed in Δccp1 cells. Whereas the resistance of Δccp1 cells towards hydrogen peroxide was restored upon expression of Ccp1his, Ccp1H242Phis and Ccp1H248EN251Dhis were functionally inactive, indicating that the mutations abolish the peroxidase activity of Ccp1 in vivo (Figure 1A).
Figure 1.
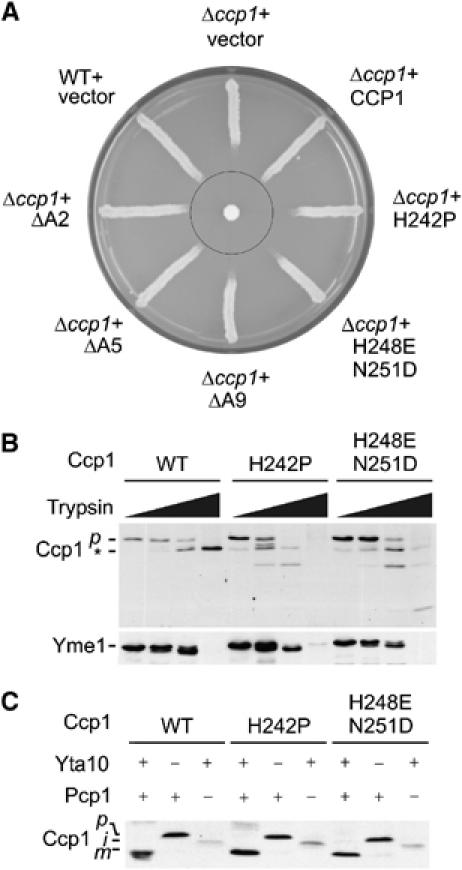
Impaired folding of the haeme-binding domain does not affect processing of Ccp1 in vivo. (A) Complementation analysis. Δccp1 (YTT266) cells harbouring pYX142 (vector) or expressing Ccp1 (CCP1), Ccp1H242P (H242P), Ccp1H248EN251D (H248E N251D), Ccp1ΔA2 (ΔA2), Ccp1ΔA5 (ΔA5) or Ccp1ΔA9 (ΔA9) were grown on selective medium containing glucose. For control, wild-type cells harbouring pYX142 vector (WT+vector) were analysed in parallel. A filter paper containing 30% hydrogen peroxide (2 μl) was placed in the centre of the plate, which was then incubated for 2 days at 30°C. (B) Trypsin resistance of Ccp1 and variants carrying mutations in the haeme-binding domain. After isolation from Δyta10Δccp1 (YTT269) cells expressing Ccp1 or mutant variants thereof, mitochondria were resuspended at a final protein concentration of 1 mg/ml in SHKCl buffer supplemented with 0.05% (v/v) Triton X-100 and 0, 2, 10 or 100 μg/ml trypsin and incubated for 30 min at 4°C. Proteins were TCA-precipitated and analysed by SDS–PAGE and Western blotting using Ccp1- and Yme1-specific antisera. The asterisk indicates Ccp1 fragments generated upon trypsin treatment. (C) Processing of Ccp1 and mutant variants thereof. Extracts of Δccp1 (YTT266), Δyta10Δccp1 (YTT269) or Δpcp1Δccp1 (YTT271) cells expressing Ccp1-his or their mutant variants were analysed by SDS–PAGE and Western blotting using Ccp1-specific antiserum. p, precursor form; i, intermediate form; m, mature form.
Similar to Ccp1, both mutant variants accumulated in the precursor form when expressed in Δyta10Δccp1 cells lacking the m-AAA protease subunit Yta10 (Figure 1B). To assess the folding state of the haeme-binding domain, we isolated mitochondria from Δyta10Δccp1 cells expressing the Ccp1 variants. Mitochondrial membranes were lysed with Triton X-100 and extracts were incubated with trypsin (Figure 1B). A protease-resistant fragment corresponding in size to the peroxidase domain of Ccp1 was generated by trypsin treatment of the precursor form, demonstrating stable folding in vivo (Figure 1B). In contrast, Ccp1H242P and Ccp1H248EN251D were trypsin sensitive, indicating that the mutations destabilised the peroxidase domain of Ccp1 (Figure 1B).
The processing of Ccp1 variants harbouring a destabilised haeme-binding domain was analysed by immunoblotting in Δccp1 cells, which lack the rhomboid protease Pcp1 or Yta10, a subunit of the m-AAA protease (Figure 1C). Mature, precursor and intermediate forms of Ccp1 were detected in Δccp1, Δyta10Δccp1 and Δpcp1Δccp1 cells, respectively (Figure 1C). Strikingly, Ccp1H242P and Ccp1H248EN251D also accumulated in intermediate forms in Δpcp1Δccp1 cells and in mature forms in Δccp1 cells (Figure 1C). Thus, Ccp1 variants carrying mutations in the haeme-binding domain are processed normally, demonstrating that Ccp1 cleavage by the m-AAA protease does not depend on the global folding of the peroxidase domain.
We noticed that Ccp1, independent of its folding state, accumulated at significantly reduced levels in Δpcp1Δccp1 mitochondria when compared to Δccp1 mitochondria and Δyta10Δccp1 mitochondria (Figure 1C). It is conceivable that Ccp1 is degraded if it cannot be cleaved by the rhomboid protease. Interestingly, similar levels of Ccp1 were observed in Δccp1, Δyta10Δccp1 and Δyta10Δpcp1Δccp1 mitochondria (see Figure 2D), indicating that turnover of the intermediate form of Ccp1 is dependent on the m-AAA protease.
Figure 2.
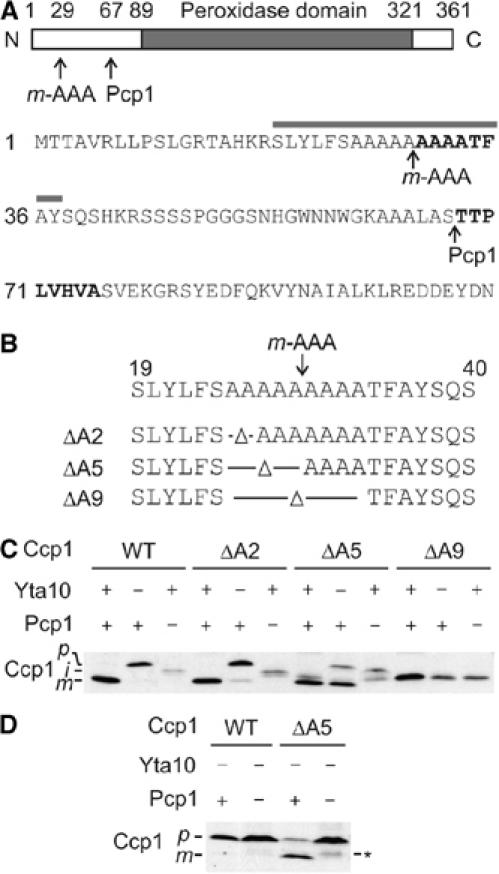
An alanine stretch within the Ccp1 presequence determines the m-AAA dependence of rhomboid cleavage. (A) Upper panel: Domain structure of Ccp1. Processing sites of the m-AAA protease and Pcp1 within Ccp1 and the peroxidase domain of Ccp1 are indicated. Lower panel: Amino-acid sequence of the N-terminal region of the Ccp1 precursor protein. N-terminal amino-acid residues determined by Edman sequencing of intermediate and mature forms of Ccp1 are shown in bold. The grey bar indicates the 19-amino-acid window used in Figure 3D. (B) Schematic representation of Ccp1 mutants lacking two (ΔA2), five (ΔA5) or nine (ΔA9) alanine residues surrounding the processing site of the m-AAA protease. (C) In vivo processing of Ccp1 variants harbouring mutations in the alanine stretch. Lysates of Δccp1 (YTT266), Δyta10Δccp1 (YTT269) or Δpcp1Δccp1 (YTT271) cells expressing Ccp1 or mutant variants were analysed by SDS–PAGE and Western blotting using Ccp1-specific antiserum. p, precursor form; i, intermediate form; m, mature form. (D) In vivo processing of Ccp1ΔA5. Lysates of Δyta10Δpcp1Δccp1 (YTT273) cells expressing Ccp1 or Ccp1ΔA5 (ΔA5) were analysed as in (C). p, precursor form; m, mature form. The asterisk indicates a form of Ccp1ΔA5 generated by an unknown peptidase.
m-AAA protease cleavage within the alanine stretch of Ccp1
To further characterise the functional interaction of both proteases, we first determined the cleavage site of the m-AAA protease within Ccp1. A variant of Ccp1 harbouring a C-terminal histidine tag was expressed in Δpcp1 cells and the intermediate form accumulating in these cells was purified by metal chelating chromatography. The N-terminal sequence of the Ccp1 intermediate was determined by Edman degradation (Figure 2A). This analysis revealed that Ccp1 is cleaved by the m-AAA protease between alanine residues 29 and 30, that is, within a stretch of nine subsequent alanine residues in the presequence of Ccp1 (Figure 2A). Similar experiments performed in Δccp1 cells identified threonine 68 as the N-terminal amino-acid residue of mature Ccp1, confirming previous reports (Kaput et al, 1982).
The length of alanine stretch determines the fate of Ccp1 in vivo
To examine the role of the alanine stretch for Ccp1 biogenesis, Ccp1 variants lacking two (ΔA2), five (ΔA5) or nine (ΔA9) of the alanine residues were generated and expressed in Δccp1 cells (Figure 2B). Mitochondria isolated from these cells were fractionated with increasing concentrations of digitonin to assess the submitochondrial localisation of the variants (Supplementary Figure S1). In agreement with localisation in the intermembrane space, Ccp1 was released to the supernatant at digitonin concentrations at which Yme1, a marker for the intermembrane space, became accessible to externally added trypsin (Supplementary Figure S1A). Similarly, the Ccp1 variants ΔA2 and ΔA5 were localised to the intermembrane space (data not shown). In contrast, ΔA9 was released in concert with degradation of the matrix protein Mge1 by trypsin, indicating that ΔA9 is mislocalised to the matrix space (Supplementary Figure S1B). Consistently, expression of ΔA2 or ΔA5 but not ΔA9 suppressed the oxidant-sensitive phenotype of Δccp1 cells (see Figure 1A). Thus, the alanine stretch is required for sorting of Ccp1 to the intermembrane space, a prerequisite for its maturation.
Whereas processing of ΔA9 was impaired, ΔA2 and ΔA5 accumulated in the mature form in Δccp1 cells (Figure 2C). To further assess processing of ΔA2 and ΔA5, Ccp1 variants were also expressed in Δpcp1Δccp1 and Δyta10Δccp1 cells. Intermediate forms of both ΔA2 and ΔA5 were detected in Δpcp1Δccp1 cells, indicating cleavage by the rhomboid protease in Δccp1 cells (Figure 2C). Strikingly, a small portion of ΔA2 and the majority of ΔA5 accumulated in the mature form in Δyta10Δccp1 cells lacking the m-AAA protease (Figure 2C). This is not due to a mislocalisation of ΔA5 within Δyta10Δccp1 mitochondria. ΔA5 was localised to the intermembrane space by digitonin fractionation of both Δccp1 and Δyta10Δccp1 mitochondria (Supplementary Figure S1C).
Another peptidase may mediate maturation of Ccp1 variants in the absence of the m-AAA protease. However, this possibility can be excluded, as processing was completely inhibited upon expression of ΔA5 in Δyta10Δpcp1Δccp1 cells (Figure 2D). Notably, ΔA5 accumulated in the intermediate form in Δpcp1Δccp1 mitochondria (Figure 2C), indicating that the m-AAA protease can cleave this Ccp1 variant and that maturation is mediated by Pcp1. We therefore conclude that shortening the alanine stretch of Ccp1 alleviates the requirement of the m-AAA protease for Pcp1-mediated maturation.
Facilitated membrane dislocation renders rhomboid cleavage of Ccp1 independent of the m-AAA protease
The effect of deletions within the alanine stretch suggests that adjacent amino-acid residues may affect the dependence of Pcp1-mediated processing on the m-AAA protease. Alanine residues in the presequence of Ccp1 are surrounded by hydrophobic residues (see Figure 2A), suggesting that this segment may form a transmembrane helix. We determined the hydrophobicity of this region using the membrane protein explorer (MPEx) programme, which is based on experimentally derived Wimley–White hydropathy scale (Wimley et al, 1996). The transfer free energy of bilayer-to-water partitioning (ΔG) of the 19-amino-acid residue window S19–Y37 was calculated to be +1.17 kcal/mol, indicating that this segment containing the alanine stretch can be stably inserted into the hydrophobic membrane (Figure 3A, black line). Consistently, the precursor form of Ccp1 accumulating in Δyta10 mitochondria was resistant against alkaline extraction, a characteristic feature of integral membrane proteins (Supplementary Figure S2A; Michaelis et al, 2005). It should be noted, however, that the hydropathy of this region is close to the threshold to form a stable transmembrane segment and that the analysed region is surrounded by amino-acid residues that do not favour the formation of transmembrane helices (see Figure 2A). Therefore, a 19-amino-acid residue window of this region lacking five alanines is not predicted to form a stable transmembrane helix, although alanine residues themselves are almost neutral in hydrophobicity (Figure 3A, grey line). Accordingly, the precursor form of ΔA5 accumulating in Δyta10Δpcp1Δccp1 mitochondria was extracted from the membrane at alkaline pH (Supplementary Figure S2B).
Figure 3.
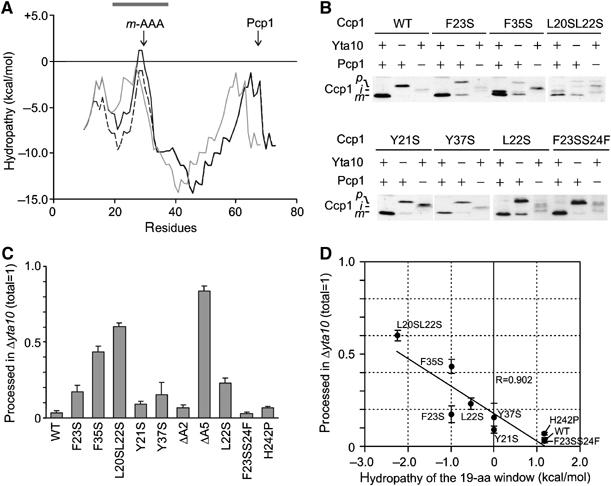
The hydrophobicity of the Ccp1 sorting signal determines the m-AAA protease dependence of Ccp1 processing. (A) Hydropathy plot of the N-terminal 80 amino acids of Ccp1 (black line), Ccp1ΔA5 (grey line) and Ccp1F23S (dotted line) according to MPEx. The grey bar indicates the 19-amino-acid window used in (D). (B) In vivo processing of Ccp1 (WT) and mutant variants. Samples were analysed as in Figure 1C. (C) Quantification of m-AAA protease-independent processing of Ccp1 mutants. Precursor and mature forms of Ccp1 or their variants were detected in Δyta10Δccp1 (YTT269) cells using an infrared scanner (Odyssey). (D) Correlation of processing efficiencies in Δyta10Δccp1 with the hydrophobicity of the sorting signal. Amounts of mature Ccp1 (total was set to 1) were plotted against the hydropathy of the 19-amino-acid window (centre=A29) of Ccp1 variants. Deletion mutants in the sorting signal were not considered for this analysis as the deletion causes a shift of the window.
These findings suggest that the hydropathy of this region determines whether Ccp1 maturation depends on the m-AAA protease. We reasoned that mutations that decrease the hydrophobicity of amino-acid residues adjacent to the alanine stretch should have similar effects on the dependence of Ccp1 maturation on the m-AAA protease as deletions within the alanine stretch. Replacement of phenylalanine 23 by serine decreases the calculated ΔG of the segment S19–Y37 by −1.0 kcal/mol, indicating that the transmembrane helix in Ccp1F23S is destabilised to a similar extent as in ΔA5 (Figure 3A, dotted line). Indeed, Ccp1F23S was recovered from the supernatant fraction upon alkaline extraction of mitochondrial membranes (Supplementary Figure S2C). Expression of Ccp1F23S in Δyta10Δccp1 and Δpcp1Δccp1 cells revealed that ∼20% of Ccp1F23S was matured by Pcp1 independent of the m-AAA protease (Figure 3B and C). This effect was almost completely reversed if the ΔG value of the segment was increased (+1.17 kcal/mol) to a similar level as in wild-type Ccp1 by mutating serine 24 to phenylalanine within Ccp1F23S (Figure 3B and C).
To substantiate the role of the hydropathy of the Ccp1 sorting sequence for processing, we analysed the m-AAA protease dependence of a series of Ccp1 variants that carried substitutions at hydrophobic residues flanking the alanine stretch (Figure 3B and C). A significant correlation was observed between the ΔG of the 19-amino-acid window S19–Y37 and the requirement of the m-AAA protease for Pcp1-mediated maturation (Figure 3D). Substitutions of tyrosine residues, generally less favoured within transmembrane helices, were less effective in alleviating the m-AAA protease dependence of maturation (Figure 3B and C). Taken together, decreased hydrophobicity and, concomitantly, destabilization of the transmembrane helix and facilitated dislocation from the lipid bilayer render Ccp1 maturation by Pcp1 independent of the m-AAA protease.
Proteolytic activity of the m-AAA protease is dispensable for maturation of Ccp1 in vivo
How can these findings be reconciled with proteolytic processing of Ccp1 by the m-AAA protease? To address this question, Ccp1 maturation was examined in cells expressing proteolytically inactive m-AAA protease variants. Glutamate residues within the consensus metal-binding sites HExxH of the proteolytic centre of both subunits of the m-AAA protease, Yta10 and Yta12, were replaced by glutamine (Yta10E559Q, Yta12E614Q). Moreover, aspartate 634 and 689 of Yta10 and Yta12, respectively, were substituted by alanine (Yta10D634A, Yta12D689A). Crystal structures of homologous bacterial AAA proteases identified these amino-acid residues as essential for Zn2+ binding and for proteolytic activity (Bieniossek et al, 2006; Suno et al, 2006).
Yta10 and Yta12 variants were expressed in different combinations in Δyta10Δyta12 cells and the activity of mutant m-AAA proteases was assessed in vivo, monitoring maturation of Ccp1 and the ribosomal protein MrpL32 (Figure 4). Expression of wild-type complexes or complexes carrying a mutation only in one subunit in Δyta10Δyta12 cells allowed maturation (Figure 4A, lanes 1, 3, 4, 6 and 7). Mutations of aspartate residues involved in Zn2+ binding impaired processing more severely than mutations within the consensus metal-binding motif. Nevertheless, MrpL32 maturation was observed and respiratory growth was maintained in the presence of either Yta10D634A or Yta12D689A (Figure 4A and D; data not shown). However, MrpL32 processing and respiratory growth was completely inhibited in the presence of mutant variants of both Yta10 and Yta12 (Figure 4A, lanes 5 and 8; data not shown). This is in agreement with previous findings (Nolden et al, 2005) and demonstrates that only concomitant mutations in the proteolytic domains of both Yta10 and Yta12 inactivate the m-AAA protease. In striking contrast to MrpL32, ∼20% of Ccp1 accumulated in the mature form in mitochondria harbouring Yta10E559Q and Yta12E614Q (Figure 4A, lane 5 and C). Deletion of PCP1 in these cells inhibited maturation, indicating that the mature form of Ccp1 is generated by Pcp1 in the presence of proteolytically inactive m-AAA protease (Figure 4B). We therefore conclude that proteolysis by the m-AAA protease is not obligatory for Ccp1 maturation by rhomboid.
Figure 4.
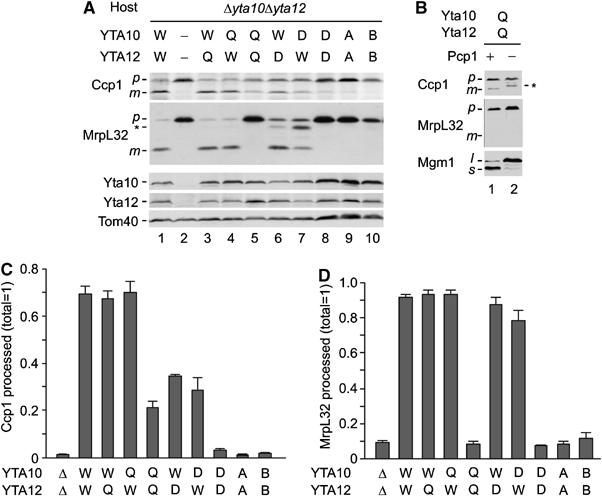
The ATPase but not the protease activity of m-AAA protease is essential for Ccp1 processing. (A) In vivo processing of Ccp1 by mutant m-AAA proteases. Yta10, Yta12 and various mutants were expressed in Δyta10Δyta12 (YKO200) cells from centromeric plasmids under the control of the ADH1 promoter. m-AAA protease subunits carrying mutations in the proteolytic domain (Q, Yta10E559Q or Yta12E614Q; D, Yta10D634A or Yta12D689A) or in Walker A or B motifs of the AAA domains (A, Yta10K334A or Yta12K394A; B, Yta10E388Q or Yta12E448Q) were expressed, as indicated. Cell lysates were analysed by SDS–PAGE and Western blotting using antisera directed against Ccp1, MrpL32, Yta10, Yta12 and Tom40. p, precursor form; m, mature form; asterisk, intermediate form of MrpL32. (B) Pcp1-dependent maturation of Ccp1 in cells harbouring proteolytically inactive m-AAA protease. Yta10E559Q and Yta12E614Q were expressed in Δyta10Δyta12 (YKO200; lane 1) or Δyta10Δpcp1Δccp1 (YTT273; lane 2) and cell lysates were analysed by SDS–PAGE and Western blotting using antisera directed against Ccp1, MrpL32 and Mgm1. p, precursor form; m, mature form; l, long form; s, short form; asterisk, a form generated by an unknown peptidase. (C) Quantification of Ccp1 processing. Precursor and mature forms of Ccp1 detected by immunoblotting were quantified using an infrared scanner (Odyssey). Standard deviations are indicated (n=3). (D) Quantification of MrpL32 processing. Precursor and mature forms of MrpL32 were quantified as in (C).
Coexpression of Yta10D634A and Yta12D689A did not allow significant processing of Ccp1 or MrpL32, pointing to differences in the activity of m-AAA proteases carrying mutations in their proteolytic domains (Figure 4A, lane 8 and C). Notably, Ccp1 and MrpL32 maturation was also inhibited in the presence of mutant m-AAA proteases harbouring mutations in conserved sequence motifs of the AAA domains of both Yta10 and Yta12 (Yta10K334A/Yta12K394A Walker A motif; Yta10E388Q/Yta12E448Q Walker B motif; Figure 4A, lanes 9 and 10 and C). It is therefore conceivable that the ATPase rather than the proteolytic activity of the m-AAA protease is required to ensure maturation of Ccp1 in vivo.
The efficiency of Ccp1 maturation correlates with ATPase activities of m-AAA protease variants
We established an in vitro assay allowing determination of ATPase activities of the m-AAA protease variants. Mitochondria were isolated from Δyta10Δyta12 cells expressing Yta12 and Yta10 modified with a C-terminal hexahistidine tag. After lysis with the non-ionic detergent NP-40, m-AAA protease complexes were purified to near homogeneity by metal chelating chromatography (Figure 5A). ATPase activities of wild-type and mutant m-AAA proteases were measured in vitro (Figure 5B and C). An eluate fraction derived from Δyta10Δyta12 mitochondria did not show significant ATPase activity, excluding contaminations by other mitochondrial ATPases (data not shown). Consistently, no ATPase activity was observed for m-AAA protease variants harbouring mutations in the Walker A- or Walker B-motifs of both Yta10 and Yta12 (Figure 5B and C). m-AAA protease complexes hydrolysed ∼2.9±0.5 μmol ATP/min/mg. Mutations in the proteolytic centre of either (Yta10E559Q) or Yta12 (Yta12E614Q) reduced the ATPase activity of the m-AAA protease to ∼70% (Figure 5B and C). m-AAA protease complexes built up of Yta10E559Q and Yta12E614Q subunits exerted only ∼10% of wild-type activity (Figure 5B and C). The reduced ATPase activity of m-AAA protease variants harbouring mutations within proteolytic domains indicates a functional interdependence of both catalytic activities, which has also been observed for bacterial FtsH (Karata et al, 1999).
Figure 5.
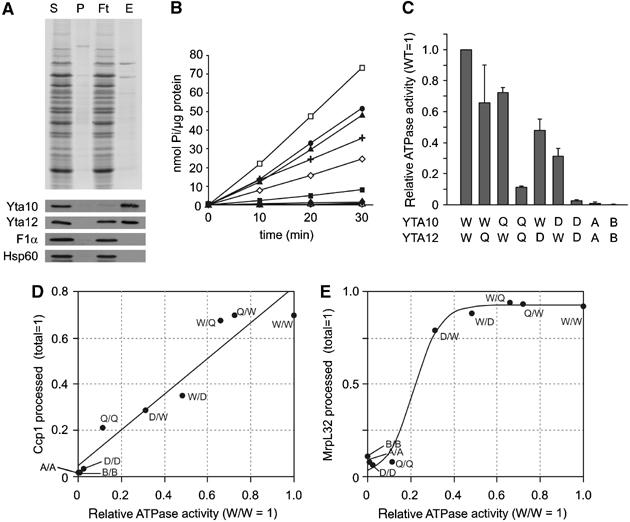
ATPase activities of purified m-AAA protease variants. (A) Purification of m-AAA protease variants by metal chelating chromatography. After expression of Yta10 harbouring a C-terminal histidine tag and Yta12 in Δyta10Δyta12 cells, m-AAA protease complexes were isolated by Ni-NTA chromatography. Upper panel: SDS–PAGE analysis of various fractions followed by Coomassie staining: S, soluble fraction applied to Ni-NTA beads (40 μg); P, insoluble pellet fraction after solubilisation; Ft, flow-through fraction (40 μg); E, eluate fraction (80 ng protein). Bottom panel: Western blot analysis of various fractions using antisera directed against Yta10, Yta12, Hsp60 and the α-subunit of the F1-particle of the F1FO-ATP synthase. (B, C) ATPase activities of m-AAA protease variants in vitro. A kinetic analysis is shown in (B). The ATPase activities of the variants relative to the wild-type m-AAA protease are shown in (C). Standard deviations are indicated (n=3). Open square (W/W), Yta10/Yta12; closed square (Q/Q), Yta10E559Q/Yta12E614Q; closed diamond (D/D), Yta10D634A/Yta12D689A; open circle (A/A), Yta10K334A/Yta12K394A; open triangle (B/B), Yta10E388Q/Yta12E448Q; closed circle (Q/W), Yta10E559Q/Yta12; closed triangle (W/Q), Yta10/Yta12E614Q; open diamond (D/W), Yta10D634A/Yta12; cross (W/D), Yta10/Yta12D689A. (D) Correlation of Ccp1 processing efficiencies in vivo with the in vitro ATPase activity of m-AAA protease variants. Accumulating mature Ccp1 (quantified in Figure 4C) was plotted against relative ATPase activities of m-AAA protease variants. Linear fitting (black line; R=0.962) is shown. (E) Correlation of processing efficiencies of MrpL32 in vivo with the ATPase activity in vitro. Sigmoid fitting is shown (SSE: 0.0181).
An even stronger impairment of ATPase activities was observed upon mutation of aspartate residues involved in Zn2+ binding in Yta10 (Yta10D634A) or Yta12 (Yta12D689A) (Figure 5B and C). The presence of Yta10D634A or Yta12D689A diminished the ATPase activities of m-AAA proteases to ∼30 or 50%, respectively (Figure 5B and C). Mutations in both subunits almost completely abolished the ATPase activity of the complex, similar to mutations in Walker A or Walker B motifs of both Yta10 and Yta12 (Figure 5B and C).
These experiments revealed a striking correlation between the ATPase activities of m-AAA protease variants in vitro and the efficiency of Ccp1 processing in vivo (Figure 5D). Whereas mutant m-AAA proteases lacking significant ATPase activities did not promote Ccp1 processing, intermediate processing efficiencies were observed in the presence of variants exhibiting intermediate ATPase activities.
It is conceivable that the dependence of Ccp1 processing on the ATPase activity of the m-AAA protease reflects solely the general ATP requirement for proteolysis. We therefore examined the ATP dependence of MrpL32 processing by the m-AAA protease. In contrast to Ccp1, we did not observe a linear but rather a sigmoid correlation between MrpL32 processing and the ATPase activities of m-AAA protease variants (Figure 5E). Low ATPase activities, which are not sufficient to promote Ccp1 processing, allow efficient processing of MrpL32. The apparently different ATP requirements for processing of Ccp1 and MrpL32 may reflect differences in substrate handling and suggest that Ccp1 processing depends on additional ATP consuming processes. These findings therefore support our model that rhomboid cleavage requires the ATP-dependent membrane dislocation driven by the m-AAA protease.
Membrane dislocation of Ccp1 by the m-AAA protease
To directly monitor dislocation of Ccp1 by the m-AAA protease, we introduced a tobacco etch virus protease cleavage site (TCS) after serine 45 of Ccp1, which is located between the two processing sites of Ccp1 (Ccp1TCS; Figure 6A). Insertion of TCS did not affect the processing of Ccp1 by the m-AAA protease and rhomboid (Supplementary Figure S3A). To examine whether the TEV cleavage site is exposed to the intermembrane space of mitochondria in the absence of the m-AAA protease, we coexpressed Ccp1TCS with a TEV variant targeted to the intermembrane space (IMS-TEV; Kondo-Okamoto et al, 2003) in Δyta10Δpcp1Δccp1 cells. Targeting of IMS-TEV was verified by the cleavage of a model substrate that resides in this compartment and was coexpressed in the cells for control (Supplementary Figure S3C). Immunoblotting of cellular extracts revealed that the precursor form of Ccp1TCS was converted to a proteolytic fragment upon expression of IMS-TEV, demonstrating the accessibility of TCS in the intermembrane space (Figure 6B). In contrast, the intermediate form accumulating upon exogenous expression of the m-AAA protease in these cells remained resistant to cleavage by IMS-TEV (Figure 6B).
Figure 6.
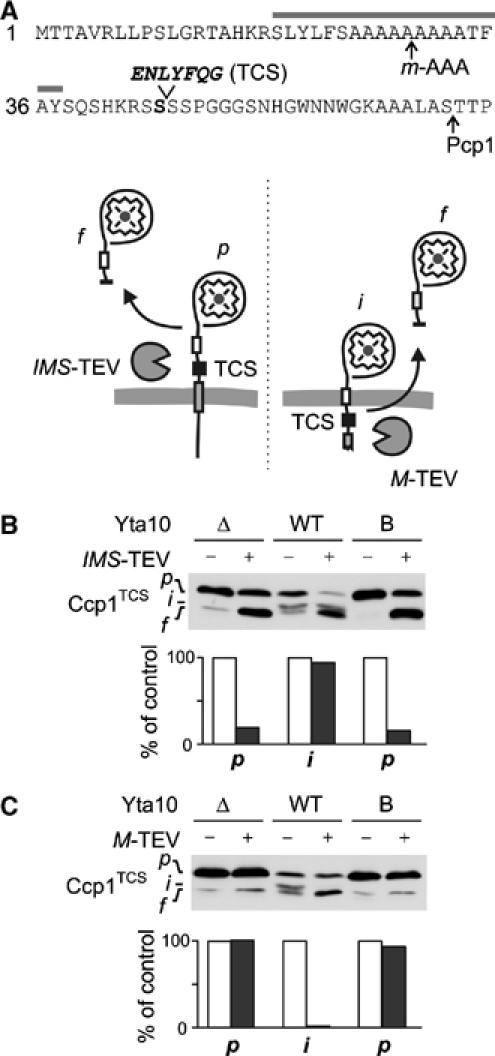
m-AAA protease dislocates the precursor of Ccp1 to the matrix. (A) The presequence of Ccp1TCS harbouring a TEV cleavage site (TCS) after S45 (Ccp1TCS). Grey bar, hydrophobic sorting signal; arrows, processing sites. Lower panel: Potential topologies of Ccp1TCS in the inner membrane and cleavage by TEV proteases located in the intermembrane (IMS-TEV) or matrix space (M-TEV). (B) Cleavage of Ccp1TCS by IMS-TEV in vivo. The precursor of Ccp1TCS accumulates in Δyta10Δpcp1Δccp1 cells (Δ), whereas the intermediate form is generated upon expression of Yta10, which at least partially complemented the processing deficiency (WT). To examine the accessibility of TCS from the intermembrane space, IMS-TEV, Yta10 (WT) and Yta10E388Q (B) were expressed when indicated and Ccp1 cleavage was monitored by immunoblotting. p, precursor form; i, intermediate form; f, proteolytic fragment produced by TEV cleavage. We observed the accumulation of a similar-sized proteolytic fragment of Ccp1TCS in some cells even in the absence of TEV, which might result from the degradation of Ccp1 molecules mislocalised to the matrix. To avoid any interference with our interpretation, we only quantified the stability of precursor (Δ, B) and intermediate forms (WT) in the presence or absence of TEV in the lower panel. Values were expressed as a per cent of precursor or intermediate form in the absence of TEV. (C) Cleavage of Ccp1TCS by M-TEV in vivo. M-TEV and Ccp1TCS were coexpressed in Δyta10Δpcp1Δccp1 cells (Δ) containing Yta10 (WT) or Yta10E388Q (B) when indicated. Ccp1 cleavage was analysed and quantified as in (B).
TCS may be translocated across the inner membrane in Δpcp1 cells harbouring the m-AAA protease and therefore may not be accessible from the intermembrane space. To examine this possibility, we coexpressed in Δpcp1 cells Ccp1TCS with a matrix-targeted TEV variant (M-TEV; Kondo-Okamoto et al, 2003) whose correct localisation was experimentally verified in control experiments (Supplementary Figure S3C). In striking contrast to cells expressing IMS-TEV, the intermediate form of Ccp1 was cleaved in cells expressing M-TEV and m-AAA protease exogenously, whereas the Ccp1 precursor form accumulating in the absence of the m-AAA protease was resistant towards TEV cleavage (Figure 6C). We conclude that TCS, located in between the two processing sites of Ccp1, is only exposed to the matrix space in the intermediate form, which is generated by the m-AAA protease. Moreover, TCS was not accessible from the matrix in the presence of an m-AAA protease variant that carried a mutation in the AAA domain of Yta10 (Yta10E388Q; Figure 6C) and which did not support respiratory growth (data not shown). Conversely, TCS introduced into the Ccp1 variant Ccp1F23S harbouring a sorting signal with decreased hydrophobicity is exposed to the matrix and cleaved by M-TEV even in the absence of the m-AAA protease (data not shown).
Taken together, these experiments reveal that the conversion of the precursor form of Ccp1 to the mature form is accompanied by a partial membrane dislocation of Ccp1, which depends on the ATPase activity of the m-AAA protease.
Conserved residues in the pore loop are essential for Ccp1 processing
Most AAA+ proteins form hexameric ring structures that allow substrates to enter the central channel (Sauer et al, 2004; Hanson and Whiteheart, 2005). To examine whether Ccp1 extracted from the membrane by the m-AAA protease is inserted into the predicted central pore of the protease, we mutated conserved amino-acid residues within a conserved loop, which is exposed to the central channel in the crystal structure of eubacterial AAA proteases (Bieniossek et al, 2006; Suno et al, 2006). This loop contains an aromatic-hydrophobic-glycine motif (FVG in Yta10 and Yta12), which is conserved within AAA+ proteins (Figure 7A) and has been linked to substrate translocation in other AAA proteins (Sauer et al, 2004; Hanson and Whiteheart, 2005). We mutated concomitantly amino acids within and neighbouring this loop in Yta10 and Yta12, coexpressed the variants in Δyta10Δyta12 cells and assessed Ccp1 processing in vivo. Substitution of F361 in Yta10 and F421 in Yta12 by serine, alanine or glutamic acid abolished cleavage of Ccp1 (Figure 7B, lanes 3–5). Ccp1 processing was also inhibited upon expression of Yta10G363A and Yta12G423A in Δyta10Δyta12 cells (Figure 7B, lane 6), or significantly reduced upon replacement of methionine residues neighbouring the pore motif by lysine (Figure 7B, lane 7).
Figure 7.
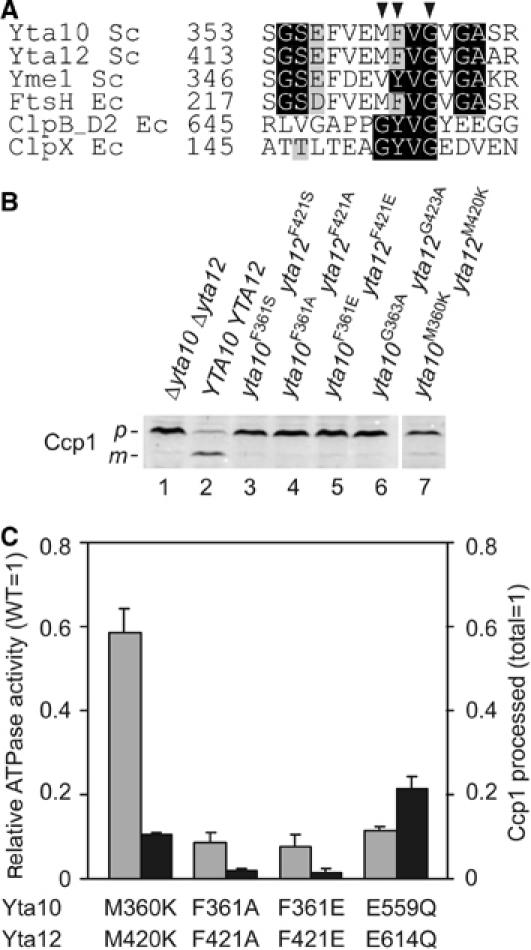
Conserved amino acids of central pore loops of the m-AAA protease are essential for Ccp1 processing. (A) Sequence alignment of central pore loop motifs of AAA+ proteins. Arrowheads indicate amino acids mutated in Yta10 and Yta12. (B) Role of conserved amino acids of the central pore motif for Ccp1 processing. Δyta10Δyta12 cells expressing Yta10 and Yta12 (lane 2) or m-AAA protease variants with mutations in methionine (Yta10M360KYta12M420K; lane 7), phenylalanine (Yta10F361SYta12F421S; Yta10F361AYta12F421A; Yta10F361EYta12F421E; lanes 3–5) or glycine (Yta10G363AYta12G423A; lane 6) within the central pore loop were analysed by SDS–PAGE and Western blotting using antiserum directed against Ccp1. p, precursor form; m, mature form. (C) Comparison of Ccp1 processing in vivo with ATPase activities of m-AAA protease variants in vitro. ATPase activities (grey bars) are given relative to wild-type m-AAA protease and compared with the efficiency of Ccp1 processing (black bars). For comparison, corresponding values for the m-AAA protease variant harbouring point mutations within proteolytic centres (Yta10E559QYta12E614Q) are indicated.
To scrutinise potential effects of these mutations on the ATPase activity of the m-AAA protease, we purified mutant variants carrying mutations within the pore loop motif and determined their ATPase activities in vitro. We observed a strongly impaired ATPase activity of some m-AAA protease variants harbouring mutant pore loops (Figure 7C). Mutations in conserved phenylalanine residues F361 and F421 of Yta10 and Yta12, respectively, inhibited the ATPase by ∼90%, that is, to a similar extent as mutations in the proteolytic sites of the m-AAA protease (see Figure 5C). However, an m-AAA protease variant harbouring mutations in methionine residues next to the pore loop motif of Yta10 and Yta12 (Yta10M360KYta12M420K) retained ∼60% ATPase activity. This is remarkable as Ccp1 processing was reduced to ∼10% in these mutant cells (Figure 7B, lane 7). Thus, mutations in the pore loop motif had a severe effect on Ccp1 processing, which cannot be solely explained by a reduced ATPase activity of the mutant proteases. It therefore appears that maturation of Ccp1 depends critically on the central pore loop and the insertion of Ccp1 into the central channel of the m-AAA protease.
Discussion
Our findings reveal an unexpected role of the m-AAA protease for Ccp1 maturation by rhomboid (Figure 8A). Mutations in the proteolytic domains of both m-AAA protease subunits impair respiratory growth of yeast cells and processing of MrpL32 (Arlt et al, 1998; Nolden et al, 2005). In contrast, the maturation of Ccp1 depends only on the ATPase but not the proteolytic activity of the m-AAA protease and therefore represents the first known non-proteolytic function of the m-AAA protease within mitochondrial biogenesis. Monitoring the accessibility of a TEV protease cleavage site inserted into the Ccp1 precursor, we provide in vivo evidence for an ATP-dependent vectorial membrane dislocation of the Ccp1 precursor, which depends on the m-AAA protease. Consistently, a decrease in the hydrophobicity of the Ccp1 sorting signal facilitates a passive dislocation of the Ccp1 precursor and alleviates the dependence of rhomboid processing on the m-AAA protease (Figure 8B).
Figure 8.
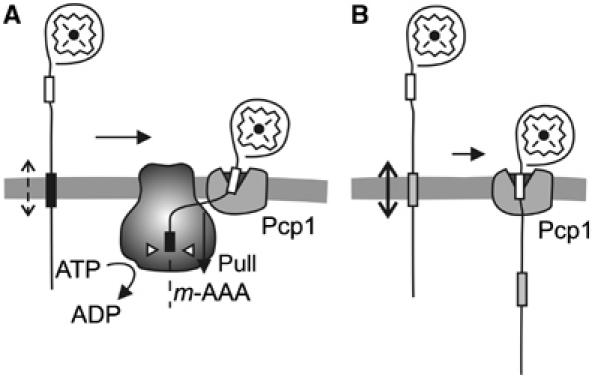
The functional interplay of m-AAA and rhomboid protease during Ccp1 processing. (A) The hydrophobic sorting signal (black box) triggers insertion of newly imported Ccp1 into the mitochondrial inner membrane. The m-AAA protease mediates the ATP-dependent dislocation of the hydrophobic segment from the membrane, making the second processing site (white box) accessible for the rhomboid protease Pcp1. (B) A reduced hydrophobicity of the sorting signal of mutant Ccp1 facilitates the passive dislocation of Ccp1 preproteins from the membrane and allows m-AAA protease-independent maturation by rhomboid.
Several lines of evidence suggest that the ability to mediate membrane dislocation of substrate proteins is conserved among AAA proteases. Similar to Ccp1, misfolded or non-assembled membrane proteins are extracted from the membrane bilayer for proteolysis by AAA proteases (Kihara et al, 1999; Leonhard et al, 2000). Short protein segments protruding from the membrane surface are sufficient to allow m-AAA protease-dependent degradation of large domains exposed to the opposite membrane side (Leonhard et al, 2000). It is therefore conceivable that this general activity of the m-AAA protease is exploited during Ccp1 biogenesis to ensure maturation by the rhomboid protease Pcp1. Moreover, mammalian m-AAA proteases can functionally substitute for the yeast homologue in protein processing (Nolden et al, 2005; Koppen et al, 2007), suggesting that the ability for membrane dislocation is also conserved in mammalian orthologues. Interestingly, the i-AAA protease Yme1 was recently found to be required for the import of mammalian PNPase into the intermembrane space of yeast mitochondria, consistent with a function for membrane translocation (Rainey et al, 2006).
The degradation of misfolded membrane proteins as well as protein processing by the m-AAA protease is ATP-dependent and abolished by mutations in the AAA domains of its subunits. We observed, however, a strikingly different dependence on the ATPase activity of the m-AAA protease for maturation of Ccp1 and MrpL32. Whereas MrpL32 is efficiently cleaved by protease variants with low ATPase activities, we observed a linear correlation between Ccp1 processing and the ATPase activity of the m-AAA protease. As Ccp1 folding does not interfere with its processing, differences in protein stability of Ccp1 and MrpL32 are unlikely to account for the apparently higher ATP demand for Ccp1 maturation. Our experiments rather suggest that the membrane dislocation of Ccp1 imposes additional ATP requirements for its maturation. Consistently, the translocation of the TEV cleavage site inserted into the Ccp1 precursor was inhibited by mutations in the AAA domains of the m-AAA protease and facilitated by decreasing the hydrophobicity of the Ccp1 sorting signal. Evidence for an additional energy requirement for the extraction of membrane-embedded protein segments was recently provided also for ER proteins that are dislocated to the cytosol for degradation by 26S proteasomes (Carlson et al, 2006). Although dispensable for the degradation of protein domains protruding from the membrane, the AAA protein p97/cdc48 is required for the dislocation of transmembrane segments of an ER membrane protein. It thus appears that a number of AAA proteins acting on different cellular membranes share the ability to drive vectorial protein dislocation.
The role of the m-AAA protease for Ccp1 processing is reminiscent of the function of the mtHsp70 motor for the cleavage of the dynamin-like GTPase Mgm1 by rhomboid (Herlan et al, 2004). In contrast to Ccp1, rhomboid cleavage of Mgm1 does not depend on the m-AAA protease but on a pulling force exerted by mtHsp70. It is tempting to speculate that the different nature of the sorting signals of both proteins may account for the different dependence on the m-AAA protease. Ccp1 harbours a rather short matrix-targeting segment at the N-terminus followed by a hydrophobic sorting signal that lacks proline or positively charged amino-acid residues. These residues, present within the sorting signal of Mgm1, reduce the efficiency of lateral sorting to the inner membrane (Meier et al, 2005) and thereby allow the mtHsp70 motor to further translocate newly imported Mgm1 into the matrix space. Efficient lateral sorting of Ccp1, on the other hand, may result in the release of newly imported Ccp1 from the TIM23 translocase. This could make the recruitment of mtHsp70 to short protein segments protruding into the matrix more difficult, as docking sites for the mtHsp70 motor at the inner membrane surface are no longer available. Thus, a delicate balance appears to exist between the efficiencies of lateral sorting and of vectorial membrane dislocation, which ensures the sorting of Ccp1 to the intermembrane space. Of note, proteolytic processing of the Mgm1 homologue OPA1 in mammalian cells has recently been linked to an m-AAA protease (Ishihara et al, 2006). It remains to be analysed whether OPA1 processing also involves membrane dislocation of OPA1 driven by an m-AAA protease.
The observation that membrane dislocation rather than processing allows Ccp1 maturation by mitochondrial rhomboid is in agreement with other rhomboid-like proteases whose activity does not depend on a preceding processing step (Freeman, 2004). This contrasts other intramembrane cleaving peptidases whose substrates are first cleaved, most likely to loosen steric restrictions and facilitate a stochastic vectorial movement of substrates relative to the membrane bilayer (Ye et al, 2000; Lemberg and Martoglio, 2002). It is therefore conceivable that the recruitment of accessory factors mediating membrane dislocation is of general relevance for cleavage by rhomboid proteases and alleviates the dependence on a preceding proteolytic event.
Materials and methods
Determination of the processing site of Ccp1
Mitochondria harbouring Ccp1-his (10 mg) were solubilised in 1 ml of buffer A (50 mM potassium phosphate buffer pH 8.0, 300 mM NaCl, 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM), 1 mM phenylmethylsulphonylfluoride (PMSF), EDTA-free protease inhibitor cocktail (Roche)). The suspension was incubated at 4°C for 30 min under vigorous shaking. Insoluble material was removed by centrifugation (30 000 g, 30 min) and the supernatant was loaded on Protino Ni-NTA column (Macherey-Nagel). To increase the binding efficiency, the flow-through fraction was reloaded twice onto the column. After washing twice with 320 μl of buffer B (50 mM potassium phosphate buffer pH 8.0, 300 mM NaCl, 0.1% (w/v) DDM, 1 mM PMSF, EDTA-free protease inhibitor cocktail (Roche)), bound proteins were eluted with 3 × 240 μl buffer C (50 mM potassium phosphate buffer pH 8.0, 300 mM NaCl, 250 mM imidazole) and subjected to SDS–PAGE. Intermediate and mature forms of Ccp1 were analysed by Edman sequencing using an ABI Procise 491 sequencer.
Purification of the m-AAA protease from mitochondria
Δyta10Δyta12 (YKO200) cells carrying plasmids for expression of Yta10 and Yta12 (or their mutant variants) were grown on selective SC-glucose medium at 30°C. The culture was diluted into YP medium supplemented with 1.75% (w/v) galactose, 0.5% (w/v) lactate and 0.25% (w/v) glucose and further incubated for 14 h at 30°C. Cells were collected and mitochondria were isolated by differential centrifugation as described previously (Tatsuta and Langer, 2007). Mitochondria were resupended (4 mg/ml) in buffer D (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM PMSF, EDTA-free protease inhibitor cocktail (Roche), 10% (v/v) glycerol, 50 mM imidazole, 1% (w/v) NP-40) and incubated for 20 min at 4°C. Insoluble material was removed by centrifugation at 30 000 g for 20 min. The supernatant was mixed with His-trap beads (GE Healthcare) for 1 h at 4°C. Beads were washed three times with buffer D containing 70 mM imidazole and 0.2% (w/v) NP-40. Bound proteins were eluted with buffer E (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 10% (v/v) glycerol, 300 mM imidazole, 0.2% (w/v) NP-40). After SDS–PAGE analysis, protein amounts were determined densitometrically using the Deep Purple fluorescent dye and the Typhoon TRIO scanner (GE Healthcare) and phosphorylase B (Sigma) as standard.
TEV cleavage assay in mitochondria
The assay was performed according to Kondo-Okamoto et al (2003). Briefly, cells expressing Ccp1TCS (pYX142-ccp1S45TCS) or GFPTCS (pYX142-Cytb2(1–220)-3HA-TCS-GFP or pYX142-Su9(1–69)-3HA-TCS-GFP) and TEV protease (p416-PGAL1-Cytb2(1–220)-TEV (IMS-TEV) or p416-PGAL1-Su9(1–69)-TEV (Matrix-TEV)) were grown in SC-Ura/Leu supplemented with 0.1% glucose and 2% raffinose. Logarithmically growing cells (OD600∼1.5) were collected and inoculated in SC-Ura/Leu supplemented with 2% galactose and 2% raffinose to induce TEV. After 4 h, cell extracts were analysed by SDS–PAGE and immunoblotting using Ccp1- and GFP-specific antiserum. Ccp1 without insertion of a TCS was not cleaved by TEV (Supplementary Figure S3B).
Miscellaneous
Yeast strains and growth used in this study are summarised in Supplementary data. Moreover, the following procedures are described in Supplementary data: cloning and mutagenesis procedures; determination of ATPase activities in vitro; hydropathy analysis.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We thank K Okamoto (University of Utah) for the mitochondrial TEV system, D Tils for expert technical assistance and M Graef for critical reading of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 635), the European Union (6th framework programme) and the German-Israeli-Project (DIP grant F.5.1) to TL.
References
- Arlt H, Steglich G, Perryman R, Guiard B, Neupert W, Langer T (1998) The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. EMBO J 17: 4837–4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniossek C, Schalch T, Bumann M, Meister M, Meier R, Baumann U (2006) The molecular architecture of the metalloprotease FtsH. Proc Natl Acad Sci USA 103: 3066–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson EJ, Pitonzo D, Skach WR (2006) p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J 25: 4557–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 6: 79–87 [DOI] [PubMed] [Google Scholar]
- Esser K, Tursun B, Ingenhoven M, Michaelis G, Pratje E (2002) A novel two-step mechanism for removal of a mitochondrial signal sequence involves the m-AAA complex and the putative rhomboid protease Pcp1. J Mol Biol 323: 835–843 [DOI] [PubMed] [Google Scholar]
- Freeman M (2004) Proteolysis within the membrane: rhomboids revealed. Nat Rev Mol Cell Biol 5: 188–197 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW (2005) AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6: 519–529 [DOI] [PubMed] [Google Scholar]
- Herlan M, Bornhovd C, Hell K, Neupert W, Reichert AS (2004) Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol 165: 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlan M, Vogel F, Bornhövd C, Neupert W, Reichert AS (2003) Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem 278: 27781–27788 [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Zich J, Takeuchi J, Zhang M, Govaerts C, Coffino P (2006) Glycine–alanine repeats impair proper substrate unfolding by the proteasome. EMBO J 25: 1720–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25: 2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Akiyama Y (2005) Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol 59: 211–231 [DOI] [PubMed] [Google Scholar]
- Kaput J, Brandriss MC, Prussak-Wiechowska T (1989) In vitro import of cytochrome c peroxidase into the intermembrane space: release of the processed form by intact mitochondria. J Cell Biol 109: 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaput J, Goltz S, Blobel G (1982) Nucleotide sequence of the yeast nuclear gene for cytochrome c peroxidase precursor. Functional implications of the pre sequence for protein transport into mitochondria. J Biol Chem 257: 15054–15058 [PubMed] [Google Scholar]
- Karata K, Inagawa T, Wilkinson AJ, Tatsuta T, Ogura T (1999) Dissecting the role of a conserved motif (the second region of homology) in the AAA family of ATPases. Site-directed mutagenesis of the ATP-dependent protease FtsH. J Biol Chem 274: 26225–26232 [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Sauer RT (2005) Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA 102: 1390–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Akiyama Y, Ito K (1999) Dislocation of membrane proteins in FtsH-mediated proteolysis. EMBO J 18: 2970–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo-Okamoto N, Shaw JM, Okamoto K (2003) Mmm1p spans both the outer and inner mitochondrial membranes and contains distinct domains for targeting and foci formation. J Biol Chem 278: 48997–49005 [DOI] [PubMed] [Google Scholar]
- Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T (2007) Variable, tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A (2001) ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell 7: 627–637 [DOI] [PubMed] [Google Scholar]
- Lemberg MK, Martoglio B (2002) Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol Cell 10: 735–744 [DOI] [PubMed] [Google Scholar]
- Leonhard K, Guiard B, Pellechia G, Tzagoloff A, Neupert W, Langer T (2000) Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol Cell 5: 629–638 [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Saurya S, Freeman M (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423: 537–541 [DOI] [PubMed] [Google Scholar]
- Meier S, Neupert W, Herrmann JM (2005) Proline residues of transmembrane domains determine the sorting of inner membrane proteins in mitochondria. J Cell Biol 170: 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis G, Esser K, Tursun B, Stohn JP, Hanson S, Pratje E (2005) Mitochondrial signal peptidases of yeast: the rhomboid peptidase Pcp1 and its substrate cytochrome c peroxidase. Gene 354: 58–63 [DOI] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T (2005) The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123: 277–289 [DOI] [PubMed] [Google Scholar]
- Nolden M, Kisters-Woike B, Langer T, Graef M (2006) Quality control of proteins in the mitochondrion. Top Curr Genet 16: 119–147 [Google Scholar]
- Rainey RN, Glavin JD, Chen HW, French SW, Teitell MA, Koehler CM (2006) A new function in translocation for the mitochondrial i-AAA protease Yme1: import of polynucleotide phosphorylase into the intermembrane space. Mol Cell Biol 26: 8488–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Jentsch S (2004) Productive rupture: activation of transcription factors by proteasomal processing. Biochim Biophys Acta 1695: 209–213 [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Langer T (2006) Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia. Trends Mol Med 12: 262–269 [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA (2004) Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119: 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suno R, Niwa H, Tsuchiya D, Zhang X, Yoshida M, Morikawa K (2006) Structure of the whole cytosolic region of ATP-dependent protease FtsH. Mol Cell 22: 575–585 [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Langer T (2007) Studying proteolysis within mitochondria. Methods Mol Biol 372: 343–362 [DOI] [PubMed] [Google Scholar]
- Tian L, Holmgren RA, Matouschek A (2005) A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol 12: 1045–1053 [DOI] [PubMed] [Google Scholar]
- Wimley WC, Creamer TP, White SH (1996) Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry 35: 5109–5124 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Kopan R (2004) Intramembrane proteolysis: theme and variations. Science 305: 1119–1123 [DOI] [PubMed] [Google Scholar]
- Ye J, Dave UP, Grishin NV, Goldstein JL, Brown MS (2000) Asparagine–proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc Natl Acad Sci USA 97: 5123–5128 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3