

Abstract
The results of trials in which autoantigens have been fed to individuals affected by autoimmune diseases - multiple sclerosis, rheumatoid arthritis and type 1 diabetes - have been disappointing in terms of clinical improvement. This is in striking contrast to the results in experimental rodent models of these diseases. The outcome of the recent DPT-1 trial testing oral insulin in individuals at risk of type 1 diabetes was also disappointing, in contrast to the effects of oral insulin in the non-obese diabetic (NOD) mouse model of type 1 diabetes. However, it is premature to conclude that mucosal tolerance works only in in-bred rodents and not in humans with autoimmune disease. Except for oral insulin in DPT-1, the human trials were performed in individuals with end-stage disease when this form of immune regulation might not be expected to be effective. Importantly, in no trial was an immune response to the autoantigen documented, to demonstrate that the dose was at least bioavailable. Furthermore, mucosal autoantigen administration is a 'double-edged sword' and in rodents can lead not only to regulatory and protective immunity but also to pathogenic, tissue-destructive immunity and exacerbation of autoimmune disease. When suppression of autoimmune disease is observed it may be because autoantigen was administered under conditions which minimize induction of pathogenic immunity. Thus, clinical protocols for mucosal autoantigen administration may need to be modified to favor induction of regulatory immunity. In this short review, we discuss recent studies in autoimmune diabetes-prone NOD mice indicating that with novel modifications mucosal autoantigen administration could be harnessed to prevent type 1 diabetes in humans.
Keywords: type 1 diabetes, immune intervention, mucosal tolerance, DPT-1 trial, CD40, T cells
Mucosal tolerance: definition and mechanisms
Mucosal tolerance refers to the phenomenon of systemic tolerance to challenge with an antigen that has previously been administered via a mucosal route, usually oral or naso-respiratory [1, 2]. Mucosal tolerance was discovered early in the 20th century in models of delayed-type and contact hypersensitivity reactions in guinea pigs, but the mechanisms of tolerance remained ill-defined until the era of modern immunology. The use of cell separation techniques, tests for production of cytokines and transgenic models in which antigen-specific T cells can be tracked in vivo have gradually elucidated mechanisms of mucosal tolerance [3, 4]. It has become evident that antigen administration via mucosal routes can result in distinct types of tolerance depending on the route of administration and dose of antigen. For example, a high dose of oral antigen induces T-cell activation followed by deletion or anergy of responding T cells [5, 6] analogous to parenteral administration of high-dose soluble antigen. This results in extinction of T cells specific to that antigen and unresponsiveness to subsequent antigen challenge, i.e. passive tolerance. In contrast, a low dose of oral antigen does not induce deletion or anergy but, when given repeatedly, induces a distinct type of immune response characterized by the appearance of regulatory-protective T cells that secrete anti-inflammatory cytokines, i.e. active tolerance [7, 8]. These T cells usually belong to the class of CD4 (helper) T cells. Instillation of intact protein antigen onto the nasopharyngeal mucosa also induces T cells that are protective. In this case, both CD4 and CD8 T cells are induced [9]. The ability of nasal instillation to induce regulatory CD8 γδ T cells [10] depends on the absence of degradation of insulin when delivered via this route, in contrast to the oral route, and to the presentation of intact protein by means that are not yet entirely understood. Regulatory T cells induced after oral or intranasal antigen produce anti-inflammatory cytokines such as IL-4, IL-10 and TGF-β. To induce mucosal tolerance, antigen can also be given in the form of aerosol. Administration via these three routes, oral, intranasal and aerosol-inhalation, results in antigen uptake and presentation in different lymphoid compartments in each case. This has been shown by in vivo tracking of antigen-specific T cells labeled with CFSE dye, that allows detection of local T cell proliferation [11, 12]. Accordingly, oral antigen is presented to T cells mostly in mesenteric lymph nodes and to some extent in Peyer's patches, intranasal antigen in deep cervical lymph nodes and inhaled antigen in mediastinal lymph nodes. Repeated exposure to antigen in each case is able to induce regulatory T cells, but the nature of these cells differs, depending on the route and form of antigen. While regulatory cells induced by oral antigen are CD4 T cells [3, 12] and express T cell receptors consisting of αβ heterodimers, in the case of naso-respiratory antigen the regulatory cells belong to a class of CD8 T cells expressing T cell receptors consisting of γδ heterodimers (i.e. γδ T cells). Their CD8 receptor is particular in that it consists of αα homodimers instead of the more conventional αβ-heterodimers [13]. These cells therefore resemble T cells that reside in epithelia of gut and other mucosa. Aerosol insulin induces γδ T cells that protect NOD mice from diabetes. They can produce IL-10 [9] but may also use other mechanisms of suppression that are still incompletely understood.
Mucosal antigen-induced regulatory T cells and the prevention of diabetes in NOD mice
The two most important cytokines made by mucosal antigen-induced, regulatory T cells [13], TGF-β and IL-10, counter-regulate two important aspects of the immune response, namely activation of antigen-presenting dendritic cells into potent stimulators of antigen-specific effector T cells and commitment of responding naïve T cells to effector Th1 or Th2 cells [14-18]. In type 1 diabetes and in NOD mice, T cells that destroy islet β-cells are mostly Th1 effector CD4 T cells and cytotoxic CD8 T cells [19]. Therefore, if regulatory cytokines were produced locally in islets, islet destruction should be suppressed. Indeed, local production of TGF-β in islets (in transgenic mice expressing TGF-β under the rat insulin promoter) protects NOD mice from developing spontaneous diabetes [20], and co-transfer of islet-specific T cell lines transduced with a gene-construct driving IL-10 production prevents adoptive transfer of diabetes in NOD mice [21]. Thus, if regulatory T cells that produce these cytokines can be directed to home to the appropriate tissue and respond to their antigen there, they should be able to suppress inflammation locally.
Mucosal administration of islet autoantigens is able to induce regulatory T cells that prevent adoptive transfer of diabetes in NOD mice. The anatomical localization of these regulatory cells has seldom been followed due to difficulties in their tracking. Homann and co-workers [8] used CFSE dye to label regulatory T cells induced by oral insulin and showed that these T cells homed in pancreatic lymph nodes and proliferated there. However, it is possible that in other cases regulatory cells also need to act locally in islets to suppress islet-destructive immunity. Autoantigens administered include insulin as a whole protein given orally or as aerosol inhalation [10, 22], a CD4 T-cell specific peptide of insulin (B-chain amino acids 9-23) given intranasally [23, 24], GAD65 as a whole protein (produced transgenetically in plants) given orally [25] or a mixture of CD4 T-cell specific GAD peptides given intranasally [26]. Reported rates of protection afforded by these regulatory cells vary a little, but typically diabetes incidence has reduced from 80%-90% to 20%-40%. However, in humans at risk of developing type 1 diabetes we can hardly envisage preventive measures based on purification and transfer of regulatory cells induced in another individual. To obtain more pertinent information, the above-mentioned rates of protection should be compared to rates of protection from spontaneous diabetes occurrence in these studies. Protection from spontaneous diabetes was determined in five out of six of these studies, but two of these lacked data on co-transfer. In one out of the other three, the incidence of spontaneous diabetes (49%) in the control group was much lower than in the co-transfer experiment (92%) [22], making direct comparisons difficult. In the two remaining studies, the spontaneous incidence of diabetes dropped from 79% to 49% [10] and from 90% to 40% [26]; in comparison, in co-transfer experiments with purified regulatory cells the incidence dropped from 82% to 19% and 90% to 20%, respectively. This suggests that the beneficial effect of mucosal autoantigen administration can be accentuated by purifying the relevant regulatory T-cell population and that the immune response to mucosal antigen may not be solely tolerance-promoting, but perhaps includes (an)other component(s) that might undermine the beneficial effects of induced regulatory T cells.
Antigen administration into mucosal surfaces can exacerbate autoimmunity
It is well known that several factors influence the immune response to mucosal antigen administration. First of all, antigen dose is critical; the tolerance-promoting effect of oral insulin, collagen and MBP has been reported to be lost above a critical dose [22, 27, 28]. Age also influences the outcome; tolerance is classically induced only after infancy [2, 29] and autoantigen administration in the neonatal period may exacerbate disease [30]. It is noteworthy that oral and intranasal autoantigen administration during clinical disease may also exacerbate disease [31, 32]. Thus, at least three factors - dose, age and disease stage - can determine the outcome of mucosal antigen administration. One treatment that is effective in a given disease model or laboratory strain can fail in another model or strain, reflecting the importance of genetic makeup of the individual. In one model, arthritis was induced in healthy DBA mice simply by repeated feeding with type II collagen [33]. In transgenic C57BL/6 mice expressing ovalbumin as a model antigen in pancreatic β-cells, adoptively transferred T-cells were activated and diabetes induced by either oral, nasal or aerosol ovalbumin [11, 34]. These studies indicate the potential of mucosal antigen to induce a pathogenic systemic immune response.
Induction of cytotoxic T lymphocytes following mucosal administration of antigen
Cytotoxic T lymphocytes (CTL) are essential for our immune defense because they kill virally-infected and transformed, potentially-neoplastic cells. Herpes simplex virus can cause a blistering disease in the mouth and genitourinary tract. In a study evaluating the strength of CTL response and duration of CTL memory in mucosal tissues, it was found that intranasal immunization with Herpes simplex glycoprotein (expressed in a recombinant adenovirus) was clearly more effective than systemic immunization [35]. Intranasal immunization with CTL epitope peptides of HIV virus, or with ovalbumin together with LPS as an adjuvant, induced potent CTL responses systemically [36]. Thus, intranasal administration of antigen seems to be a good way of inducing CTL-mediated immunity. We observed that intranasal ovalbumin protein alone without adjuvant frequently induces a strong systemic CTL response, while oral and to a lesser extent aerosol ovalbumin do also [11]. In the transgenic mouse model referred to above [34] (mice expressing ovalbumin in islet β-cells, receiving a small number of T cells specific to ovalbumin), ovalbumin administration via any of these routes can result in the development of diabetes [11]. This indicates that mucosal autoantigen administration bears the potential hazard of inducing antigen-specific CTL with tissue-destructive potential.
The DPT-1 trial: failure of oral insulin to protect from diabetes development
Encouraging results in NOD mice paved the way to clinical trials with oral insulin for the prevention of type 1 diabetes. A multicenter clinical trial in the United States called the Diabetes Prevention Trial-1 (DPT-1) was launched in 1995 to test if insulin administered either intravenously and subcutaneously or, alternatively, orally could prevent the development of diabetes in healthy, islet antibody-positive individuals assessed to have a high risk of developing type 1 diabetes [37]. The parenteral insulin trial was an open study that showed no effect to delay diabetes development. The oral insulin trial, in islet autoantibody-positive type 1 relatives with a 25-50% risk of diabetes over 5 years, was randomized and placebo-controlled. 372 individuals were enrolled and given a daily capsule of either insulin crystals (7.5 mg) or a placebo, starting in 1996. Unfortunately, in the primary analysis the rate of diabetes development was similar in both groups. For further details, visit http://www.bsc.gwu.edu/trialnet/en/public/odpt.html.
Why did DPT-1 fail to show a protective effect of oral insulin? With the number of subjects included and the quality standards employed, it is clear that even a modest effect would have been detected. Thus, either the dose used had no effect to induce insulin-specific regulatory immunity or induced regulatory immunity that had no clinical effect. In all probability, the dose used, 7.5 mg, was unlikely to have been sufficient and bioavailable; if extrapolated to a mouse this dose equates to only a few micrograms, whereas milligram amounts of oral insulin were required to exert a protective effect in the mouse [22]. Ideally, antigen-specific T cells, in particular regulatory T cells, should be monitored as mechanistic markers of mucosal antigen-induced tolerance, but T-cell assays are not yet in routine practice. The emerging application of HLA tetramers to identify antigen-specific T cells [38, 39] could provide a solution for monitoring the frequencies of antigen-specific T cells in blood samples, but would require detailed knowledge of the relevant HLA-restricted T-cell epitopes. If tracking insulin-specific T cells was possible, analysis of their surface phenotype could be used to reveal their possible mucosal origin and predicted regulatory function. The best characterized phenotypic markers of gut-derived T cells are α4β7-integrin and chemokine receptor CCR9 [40].
Enhancing the immunoprotective effect of mucosal autoantigen
Induction of CTL to ovalbumin [11, 34], and the existence of insulin-specific, diabetogenic CTL in NOD mice [41] suggested that induction of CTL in response to oral or intranasal insulin is an obvious concern. This concern proved to be realistic, as it has recently been shown that intranasal proinsulin induces CTL [42]. The possibility of concomitant induction of CTL by mucosal insulin has led us to consider potential modifications to the standard protocols. We describe here two modifications to mucosal tolerance protocols that avoid the concomitant generation of CTL, and one potential adjunct therapy that could help regulatory T cells induced by oral antigen to better colonize the inflamed islets. These are by no means the only possibilities; e.g. see cytokine co-administration studies [43, 44].
Blockade of CD40L-CD40 signaling during immune response to oral insulin
CD40L (CD154) is a surface receptor that belongs to the TNF-ligand family and is expressed mainly on activated T cells [45]. It binds CD40, an inducible member of the TNF receptor family, which can be expressed on a variety of cell types, most importantly on B lymphocytes, monocytes, macrophages and dendritic cells. Ligation of CD40 induces activation of NF-κB via TRAF-2 [46] and thereby activates gene expression and various functions of the cells. One way by which CD4 helper T cells "help" CD8 T cells to become cytotoxic T cells (CTL) is to activate dendritic cells (which present antigen to a CD8 T cell) via CD40L-CD40 interactions [47]. This turns the dendritic cell into a potent activator of CD8 T cell; conversely, in the absence of CD40L-CD40 signaling the dendritic cell may not be able to activate a CD8 T cell to become a competent CTL. From previous work [34], it was known that CTL induction in response to oral antigen requires CD4 T-cell help This prompted us to ask if this help is, at least in part, mediated by CD40L-CD40 signaling. Therefore, we tested the effect of CD40L-blockade on CTL induction in response to oral ovalbumin in C57BL/6 mice. We found that generation of systemic CTL immunity was greatly reduced when CD40L was blocked with an antibody. Importantly, we found that the mice had also become tolerant to subsequent challenge with ovalbumin, indicating that oral tolerance was not compromised and that CD40L is not necessary for induction of oral tolerance [48]. Kweon et al. [49] had earlier addressed the same question by using gene-targeted mice and found that oral tolerance was compromised in CD40L-deficient mice. This suggests that CD40L is, in fact, necessary. The reason of this discrepancy is not completely clear. However, it is known that CD40L-deficient mice lack germinal centers in their lymphoid organs, cannot mount antibody responses to protein antigens and have low serum levels of immunoglobulins other than IgM [50]. Although B cells and immunoglobulins apparently do not play a direct role in the induction of T-cell tolerance in response to oral antigen, CD40L-deficient mice have numerous other immune abnormalities [51] which could influence tolerance induction. Furthermore, the effect of transient blockade with antibody may be different from gene targeting. Finally, the dose and administration protocols were different in our study (multiple small doses, i.e. 0.5 mg x 5) and that of Kweon et al. (one large 25 mg dose) [49].
The promising results with transient blockade of CD40 during oral tolerance induction to ovalbumin prompted us to test if oral tolerance to insulin could be improved in NOD mice by the same modification. To start the treatment when considerable autoimmune activity was present in islets and thus, to mimic a relevant clinical situation, we gave oral insulin (1 mg of porcine insulin, Sigma) to female NOD mice at 8 weeks of age, and repeated this treatment at 10 weeks of age. Both oral insulin and control groups were further divided into two subgroups that received either a control (isotype-matched) monoclonal antibody (6C8, anti-human Bcl-2) or an antibody (MR-1) that blocks CD40L binding to CD 40. This was critical because MR-1 antibody alone can prevent the development of diabetes in NOD mice if given at an early age (4 weeks of age) but not later (10 weeks of age) [52]. Thus, starting at 8 weeks of age (as in our protocol) was thought to circumvent this problem, but yet necessitated a control group. The four groups were followed for the development of diabetes until 6 months age, and although none of the groups was effectively protected from developing diabetes, CD40L blockade combined to oral insulin delayed the onset of diabetes (Figure 1). By 20 weeks of age the incidence of diabetes was 20% in this group, compared to 60% in the three other groups (p < 0.05, Fisher exact test, two tailed). This is promising in light of the fact that with our protocol, i.e. just two doses and at relatively late time points (8 and 10 weeks of age), oral insulin alone did not yet show any protective effect. Therefore, it seems possible that transient CD40L blockade can accentuate the tolerance-promoting effects of oral insulin.
Figure 1.
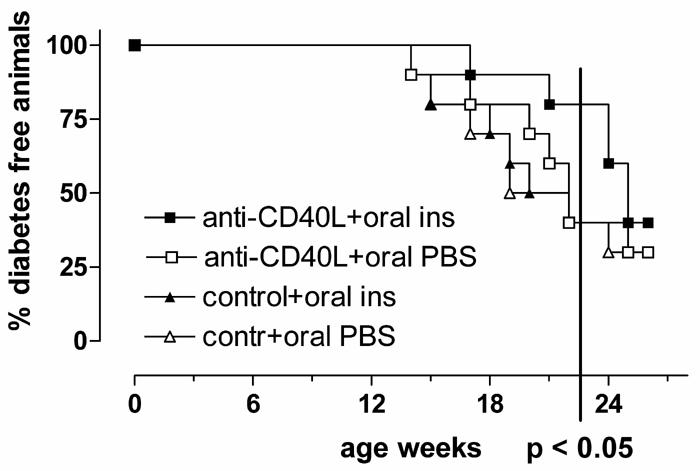
Transient blockade of CD40L facilitates the effect of oral insulin to suppress development of diabetes. NOD mice were fed twice (at 8 and 10 weeks age) with 1 mg of porcine insulin or PBS and simultaneously received a subcutaneous injection of anti-CD40L or control antibody.
Disabling an integral CTL epitope in proinsulin peptide
Insulin is a major autoantigen both in human type 1 diabetes and in NOD mice [53, 54]. It is synthesized in β-cells as a prohormone, proinsulin, comprising the A and B chains present in the mature insulin hormone linked by a connecting C-peptide, which is cleaved after A and B chains are covalently linked by disulfide bonds during post-translational modification of the prohormone. There is sound evidence that proinsulin is also an important autoantigen [55, 56]. In addition to immunodominant epitopes in the B chain (B 9-23 for CD4 T-cells and B15-23 for CD8 T cells), T cells both in human type 1 diabetes patients and in NOD mice recognize a peptide spanning the B chain and C chain (B24-C36) of proinsulin [57, 58]. Intranasal administration of this peptide is able to generate regulatory CD4 T cells that prevent the adoptive transfer of diabetes (i.e. when co-transferred with diabetogenic T cells). However, mice treated with this peptide were not protected themselves [42], suggesting that the immune response to this peptide is more complex than the mere generation of regulatory T cells. Interestingly, this peptide was also discovered to contain a monomer peptide (B25-C34) able to bind to H-2Kd, a class I restriction molecule in the MHC of NOD mice that is able to present antigen to CD8 T cells. Immunization of NOD mice with this peptide generated peptide-specific CTL, and administration of this peptide to pre-weaned NOD mice significantly reduced diabetes incidence indicating its involvement in the development of diabetes [42]. Thus, a peptide that is recognized by CD4 T cells acting as regulatory T cells also contains a shorter peptide which is recognized by CTL. To prevent unwanted generation of CTL after intranasal administration of this peptide, proinsulin peptide B24-C36 was modified. A valine at residue C34 is essential for peptide binding to H-2Kd and thus, for its ability to present antigen to CD8 T cells. Therefore, the peptide was truncated from this end. Truncated B24-C33 administered intranasally induced CD4 T cells that prevented adoptive transfer of diabetes (i.e. regulatory cells) and, importantly, also prevented development of diabetes in the treated mice [42]. Therefore, disabling an integral CTL epitope may be essential to reveal the immunoprotective effect of mucosal antigen.
Taking advantage of the differential migration behavior of mucosa-derived vs. diabetogenic T cells
It is known that T cells acquire tissue-selective homing properties when they are activated. The particular property acquired depends on the lymphoid compartment where activation occurs [59]. Accordingly, T cells that are activated in gut-associated lymphoid tissues up-regulate expression of the α4β7 integrin homing receptor, while T cells that are activated in skin-draining lymph nodes instead acquire the capacity to bind P-selectin [40]. The P-selectin ligand is expressed on naïve T cells in a form that is not able to bind P-selectin, but during activation can be modified with carbohydrates to mediate binding to P-selectin [60]. Earlier work [61] and unpublished results from one of our laboratories (A.H.) indicate that T cells responding to oral antigen can home into islets by using LPAM-1 (α4β7 integrin), and that T cells responding to islet-derived antigen in pancreas-draining lymph nodes home using VLA-4 (α4β1 integrin), not LPAM-1. Therefore, by blocking the function of VLA-4 it may be possible to inhibit the homing of diabetogenic T cells (activated in pancreas-draining lymph nodes) without compromising the homing of regulatory T cells induced by oral insulin (employing LPAM-1 for their homing). This hypothesis awaits further testing. The possible ways of modifying the response to intranasal or oral insulin in favor of regulatory T cell induction are shown in Figure 2.
Figure 2.
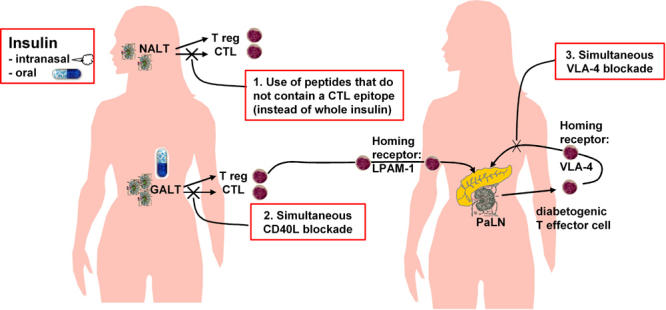
Potential ways of modifying the response to intranasal or oral insulin in favor of regulatory T-cell induction and their homing to pancreas. Left: Insulin administration nasally or orally could induce both regulatory T cells (Treg) and cytotoxic T cells (CTL) in nasal- (NALT) or gut-associated (GALT) lymphoid tissue. Right: These T cells home to the pancreas using LPAM-1 (α4β7-integrin). Diabetogenic effector T cells are generated in pancreatic lymph nodes (PaLN) and use VLA-4 (α4β1-integrin) to home to the pancreas. Induction of harmful CTL in response to intranasal or oral insulin could be prevented by use of peptides devoid of CTL epitopes (1. red box) or simultaneous blockade of CD40L (2. red box). Homing of Treg to islets could be favored by simultaneously blocking VLA-4 (3. red box) which mediates homing of diabetogenic effector T cells to pancreas.
Conclusions and future prospects
Antigen administered via mucosal routes (oral, intranasal, aerosol inhalation) induces regulatory T cells that can suppress systemic responses to the same antigen (mucosal tolerance), but simultaneously can also induce CTL. This 'double-edged sword' is a potential hazard when attempting to prevent an autoimmune disease such as type 1 diabetes by mucosal delivery of an autoantigen. There is now evidence that intranasal (pro)insulin peptide administration induces pathogenic CTL [42]. The possibility of inducing pathogenic CTL should therefore be considered in ongoing trials with intranasal insulin, although a pilot study in children receiving intranasal insulin found a decline in humoral and proliferative T-cell responses to insulin [62], thus suggesting that intranasal insulin induces mucosal tolerance. We propose that in intranasal protocols, peptides rather than protein should be administered. Preferentially, a peptide should be designed for administration after possible CTL epitopes have been disabled. In oral protocols, adjunct therapies such as transient blockade of CD40L-CD40 costimulatory pathway and/or selective blockade of T-cell homing targeting α4β1-integrin could be useful.
We find it premature to conclude that administration of autoantigens such as insulin or GAD via mucosal routes is not a feasible strategy to prevent type 1 diabetes. However, the selection of antigen and the administration protocol should be carefully considered. In the design of new trials, studies should be prioritized in NOD mice, in which spontaneous disease, as opposed to an adoptive transfer experiment, is the readout of "clinical" effect [63]. Furthermore, it would be important to extend findings in NOD mice to other disease models, such as the BB rat, in which NK cells are important [64], and the LCMV model, in which autoimmune diabetes is induced by virus infection [65]. Results from a variety of animal models could better reflect the human situation, where genetic heterogeneity and several pathogenic mechanisms may be involved, and may increase significance and safety. Hopefully, growing knowledge on the diverse effects of mucosal autoantigen administration will help us to design new clinical trials with appropriate modifications to more effectively target the autoimmune pathology.
Acknowledgments
A.H. is supported by the Finnish Diabetes Research Foundation and, by Finnish Academy, Sigrid Juselius Foundation and Juvenile Diabetes Research Foundation International as a participant in a national consortium funded by these three.
References
- 1.Weiner HL, Friedman A, Miller A, Khoury SJ, al-Sabbagh A, Santos L, Sayegh M, Nussenblatt RB, Trentham DE, Hafler DA. Oral tolerance: immunologic mechanisms and treatment of animal and human organ-specific autoimmune diseases by oral administration of autoantigens. Annu Rev Immunol. 1994;12:809–837. doi: 10.1146/annurev.iy.12.040194.004113. [DOI] [PubMed] [Google Scholar]
- 2.Strobel S, Mowat AM. Immune responses to dietary antigens: oral tolerance. Immunol Today. 1998;19:173–181. doi: 10.1016/s0167-5699(97)01239-5. [DOI] [PubMed] [Google Scholar]
- 3.Garside P, Mowat AM. Mechanisms of oral tolerance. Crit Rev Immunol. 1997;17:119–137. doi: 10.1615/critrevimmunol.v17.i2.10. [DOI] [PubMed] [Google Scholar]
- 4.Fowler E, Weiner HL. Oral tolerance: elucidation of mechanisms and application to treatment of autoimmune diseases. Biopolymers. 1997;43:323–335. doi: 10.1002/(SICI)1097-0282(1997)43:4<323::AID-BIP5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Inobe J, Marks R, Gonnella P, Kuchroo VK, Weiner HL. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature. 1995;376:177–180. doi: 10.1038/376177a0. [DOI] [PubMed] [Google Scholar]
- 6.Benson JM, Whitacre CC. The role of clonal deletion and anergy in oral tolerance. Res Immunol. 1997;148:533–541. doi: 10.1016/s0923-2494(98)80147-8. [DOI] [PubMed] [Google Scholar]
- 7.von Herrath MG. Bystander suppression induced by oral tolerance. Res Immunol. 1997;148:541–554. doi: 10.1016/s0923-2494(98)80148-x. [DOI] [PubMed] [Google Scholar]
- 8.Homann D, Holz A, Bot A, Coon B, Wolfe T, Petersen J, Dyrberg TP, Grusby MJ, von Herrath MG. Autoreactive CD4+ T cells protect from autoimmune diabetes via bystander suppression using the IL-4/Stat6 pathway. Immunity. 1999;11:463–472. doi: 10.1016/s1074-7613(00)80121-1. [DOI] [PubMed] [Google Scholar]
- 9.Hanninen A, Harrison LC. Gamma delta T cells as mediators of mucosal tolerance: the autoimmune diabetes model. Immunol Rev. 2000;173:109–119. doi: 10.1034/j.1600-065x.2000.917303.x. [DOI] [PubMed] [Google Scholar]
- 10.Harrison LC, Dempsey-Collier M, Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J Exp Med. 1996;184:2167–2174. doi: 10.1084/jem.184.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanninen A, Braakhuis A, Heath WR, Harrison LC. Mucosal antigen primes diabetogenic cytotoxic T-lymphocytes regardless of dose or delivery route. Diabetes. 2001;50:771–775. doi: 10.2337/diabetes.50.4.771. [DOI] [PubMed] [Google Scholar]
- 12.Faria AM, Weiner HL. Oral tolerance: mechanisms and therapeutic applications. Adv Immunol. 1999;73:153–264. doi: 10.1016/s0065-2776(08)60787-7. [DOI] [PubMed] [Google Scholar]
- 13.von Herrath MG, Harrison LC. Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol. 2003;3:223–232. doi: 10.1038/nri1029. [DOI] [PubMed] [Google Scholar]
- 14.Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499–1505. doi: 10.1084/jem.20012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill N, Sarvetnick N. Cytokines: promoters and dampeners of autoimmunity. Curr Opin Immunol. 2002;14:791–797. doi: 10.1016/s0952-7915(02)00403-x. [DOI] [PubMed] [Google Scholar]
- 16.Jonuleit H, Schmitt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol. 2001;22:394–400. doi: 10.1016/s1471-4906(01)01952-4. [DOI] [PubMed] [Google Scholar]
- 17.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 18.Sato K, Yamashita N, Matsuyama T. Human peripheral blood monocyte-derived interleukin-10-induced semi-mature dendritic cells induce anergic CD4(+) and CD8(+) T cells via presentation of the internalized soluble antigen and cross-presentation of the phagocytosed necrotic cellular fragments. Cell Immunol. 2002;215:186–194. doi: 10.1016/s0008-8749(02)00021-7. [DOI] [PubMed] [Google Scholar]
- 19.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 20.King C, Davies J, Mueller R, Lee MS, Krahl T, Yeung B, O'Connor E, Sarvetnick N. TGF-beta1 alters APC preference, polarizing islet antigen responses toward a Th2 phenotype. Immunity. 1998;8:601–613. doi: 10.1016/s1074-7613(00)80565-8. [DOI] [PubMed] [Google Scholar]
- 21.Moritani M, Yoshimoto K, Ii S, Kondo M, Iwahana H, Yamaoka T, Sano T, Nakano N, Kikutani H, Itakura M. Prevention of adoptively transferred diabetes in nonobese diabetic mice with IL-10-transduced islet-specific Th1 lymphocytes. A gene therapy model for autoimmune diabetes. J Clin Invest. 1996;98:1851–1859. doi: 10.1172/JCI118986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang ZJ, Davidson L, Eisenbarth G, Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci U S A. 1991;88:10252–10256. doi: 10.1073/pnas.88.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniel D, Wegmann DR. Intranasal administration of insulin peptide B:9-23 protects NOD mice from diabetes. Ann N Y Acad Sci. 1996;778:371–372. doi: 10.1111/j.1749-6632.1996.tb21146.x. [DOI] [PubMed] [Google Scholar]
- 24.Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23) Proc Natl Acad Sci U S A. 1996;93:956–960. doi: 10.1073/pnas.93.2.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma SW, Zhao DL, Yin ZQ, Mukherjee R, Singh B, Qin HY, Stiller CR, Jevnikar AM. Transgenic plants expressing autoantigens fed to mice to induce oral immune tolerance. Nat Med. 1997;3:793–796. doi: 10.1038/nm0797-793. [DOI] [PubMed] [Google Scholar]
- 26.Tian J, Atkinson MA, Clare-Salzler M, Herschenfeld A, Forsthuber T, Lehmann PV, Kaufman DL. Nasal administration of glutamate decarboxylase (GAD65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J Exp Med. 1996;183:1561–1567. doi: 10.1084/jem.183.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang ZY, Lee CS, Lider O, Weiner HL. Suppression of adjuvant arthritis in Lewis rats by oral administration of type II collagen. J Immunol. 1990;145:2489–2493. [PubMed] [Google Scholar]
- 28.Nagler-Anderson C, Bober LA, Robinson ME, Siskind GW, Thorbecke GJ. Suppression of type II collagen-induced arthritis by intragastric administration of soluble type II collagen. Proc Natl Acad Sci U S A. 1986;83:7443–7446. doi: 10.1073/pnas.83.19.7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strobel S. Immunity induced after a feed of antigen during early life: oral tolerance v. sensitisation. Proc Nutr Soc. 2001;60:437–442. doi: 10.1079/pns2001119. [DOI] [PubMed] [Google Scholar]
- 30.Miller A, Lider O, Abramsky O, Weiner HL. Orally administered myelin basic protein in neonates primes for immune responses and enhances experimental autoimmune encephalomyelitis in adult animals. Eur J Immunol. 1994;24:1026–1032. doi: 10.1002/eji.1830240503. [DOI] [PubMed] [Google Scholar]
- 31.Bai XF, Li HL, Shi FD, Liu JQ, Xiao BG, Van der Meide PH, Link H. Complexities of applying nasal tolerance induction as a therapy for ongoing relapsing experimental autoimmune encephalomyelitis (EAE) in DA rats. Clin Exp Immunol. 1998;111:205–210. doi: 10.1046/j.1365-2249.1998.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer AL, Benson JM, Gienapp IE, Cox KL, Whitacre CC. Suppression of murine chronic relapsing experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. J Immunol. 1996;157:4230–4238. [PubMed] [Google Scholar]
- 33.Terato K, Ye XJ, Miyahara H, Cremer MA, Griffiths MM. Induction by chronic autoimmune arthritis in DBA/1 mice by oral administration of type II collagen and Escherichia coli lipopolysaccharide. Br J Rheumatol. 1996;35:828–838. doi: 10.1093/rheumatology/35.9.828. [DOI] [PubMed] [Google Scholar]
- 34.Blanas E, Carbone FR, Allison J, Miller JF, Heath WR. Induction of autoimmune diabetes by oral administration of autoantigen. Science. 1996;274:1707–1709. doi: 10.1126/science.274.5293.1707. [DOI] [PubMed] [Google Scholar]
- 35.Gallichan WS, Rosenthal KL. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med. 1996;184:1879–1890. doi: 10.1084/jem.184.5.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porgador A, Staats HF, Faiola B, Gilboa E, Palker TJ. Intranasal immunization with CTL epitope peptides from HIV-1 or ovalbumin and the mucosal adjuvant cholera toxin induces peptide-specific CTLs and protection against tumor development in vivo. J Immunol. 1997;158:834–841. [PubMed] [Google Scholar]
- 37.Schatz DA, Bingley PJ. Update on major trials for the prevention of type 1 diabetes mellitus: the American Diabetes Prevention Trial (DPT-1) and the European Nicotinamide Diabetes Intervention Trial (ENDIT) J Pediatr Endocrinol Metab. 2001;14(Suppl 1):619–622. doi: 10.1515/jpem.2001.14.s1.619. [DOI] [PubMed] [Google Scholar]
- 38.Reijonen H, Kwok WW, Nepom GT. Detection of CD4+ autoreactive T cells in T1D using HLA class II tetramers. Ann N Y Acad Sci. 2003;1005:82–87. doi: 10.1196/annals.1288.009. [DOI] [PubMed] [Google Scholar]
- 39.Mallone R, Nepom GT. MHC Class II tetramers and the pursuit of antigen-specific T cells: define, deviate, delete. Clin Immunol. 2004;110:232–242. doi: 10.1016/j.clim.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med. 2002;195:135–141. doi: 10.1084/jem.20011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong FS, Karttunen J, Dumont C, Wen L, Visintin I, Pilip IM, Shastri N, Pamer EG, Janeway CA Jr. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat Med. 1999;5:1026–1031. doi: 10.1038/12465. [DOI] [PubMed] [Google Scholar]
- 42.Martinez NR, Augstein P, Moustakas AK, Papadopoulos GK, Gregori S, Adorini L, Jackson DC, Harrison LC. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin Invest. 2003;111:1365–1371. doi: 10.1172/JCI17166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiner HL, Gonnella PA, Slavin A, Maron R. Oral tolerance: cytokine milieu in the gut and modulation of tolerance by cytokines. Res Immunol. 1997;148:528–533. doi: 10.1016/s0923-2494(98)80146-6. [DOI] [PubMed] [Google Scholar]
- 44.Ma S, Huang Y, Yin Z, Menassa R, Brandle JE, Jevnikar AM. Induction of oral tolerance to prevent diabetes with transgenic plants requires glutamic acid decarboxylase (GAD) and IL-4. Proc Natl Acad Sci U S A. 2004;101:5680–5685. doi: 10.1073/pnas.0307420101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 46.He L, Grammer AC, Wu X, Lipsky PE. TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-kappaB activation. J Biol Chem. 2004 doi: 10.1074/jbc.M407284200. forthcoming. [DOI] [PubMed] [Google Scholar]
- 47.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 48.Hanninen A, Martinez NR, Davey GM, Heath WR, Harrison LC. Transient blockade of CD40 ligand dissociates pathogenic from protective mucosal immunity. J Clin Invest. 2002;109:261–267. doi: 10.1172/JCI13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kweon MN, Fujihashi K, Wakatsuki Y, Koga T, Yamamoto M, McGhee JR, Kiyono H. Mucosally induced systemic T cell unresponsiveness to ovalbumin requires CD40 ligand-CD40 interactions. J Immunol. 1999;162:1904–1909. [PubMed] [Google Scholar]
- 50.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 51.Grewal IS, Flavell RA. The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev. 1996;153:85–106. doi: 10.1111/j.1600-065x.1996.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 52.Balasa B, Krahl T, Patstone G, Lee J, Tisch R, McDevitt HO, Sarvetnick N. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J Immunol. 1997;159:4620–4627. [PubMed] [Google Scholar]
- 53.Eisenbarth GS, Moriyama H, Robles DT, Liu E, Yu L, Babu S, Redondo M, Gottlieb P, Wegmann D, Rewers M. Insulin autoimmunity: prediction/precipitation/prevention type 1A diabetes. Autoimmun Rev. 2002;1:139–145. doi: 10.1016/s1568-9972(02)00035-6. [DOI] [PubMed] [Google Scholar]
- 54.Narendran P, Mannering SI, Harrison LC. Proinsulin-a pathogenic autoantigen in type 1 diabetes. Autoimmun Rev. 2003;2:204–210. doi: 10.1016/s1568-9972(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 55.French MB, Allison J, Cram DS, Thomas HE, Dempsey-Collier M, Silva A, Georgiou HM, Kay TW, Harrison LC, Lew AM. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes. 1997;46:34–39. doi: 10.2337/diab.46.1.34. [DOI] [PubMed] [Google Scholar]
- 56.Steptoe RJ, Ritchie JM, Harrison LC. Transfer of hematopoietic stem cells encoding autoantigen prevents autoimmune diabetes. J Clin Invest. 2003;111:1357–1363. doi: 10.1172/JCI15995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen W, Bergerot I, Elliott JF, Harrison LC, Abiru N, Eisenbarth GS, Delovitch TL. Evidence that a peptide spanning the B-C junction of proinsulin is an early Autoantigen epitope in the pathogenesis of type 1 diabetes. J Immunol. 2001;167:4926–4935. doi: 10.4049/jimmunol.167.9.4926. [DOI] [PubMed] [Google Scholar]
- 58.Rudy G, Stone N, Harrison LC, Colman PG, McNair P, Brusic V, French MB, Honeyman MC, Tait B, Lew AM. Similar peptides from two beta cell autoantigens, proinsulin and glutamic acid decarboxylase, stimulate T cells of individuals at risk for insulin-dependent diabetes. Mol Med. 1995;1:625–633. [PMC free article] [PubMed] [Google Scholar]
- 59.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 60.Fuhlbrigge RC, Kieffer JD, Armerding D, Kupper TS. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature. 1997;389:978–981. doi: 10.1038/40166. [DOI] [PubMed] [Google Scholar]
- 61.Hanninen A, Salmi M, Simell O, Jalkanen S. Mucosa-associated (beta 7-integrinhigh) lymphocytes accumulate early in the pancreas of NOD mice and show aberrant recirculation behavior. Diabetes. 1996;45:1173–1180. doi: 10.2337/diab.45.9.1173. [DOI] [PubMed] [Google Scholar]
- 62.Harrison LC, Honeyman MC, Steele CE, Stone NL, Sarugeri E, Bonifacio E, Couper JJ, Colman PG. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care. 2004;27:2348–2355. doi: 10.2337/diacare.27.10.2348. [DOI] [PubMed] [Google Scholar]
- 63.Hanninen A, Hamilton-Williams E, Kurts C. Development of new strategies to prevent type 1 diabetes: the role of animal models. Ann Med. 2003;35:546–563. doi: 10.1080/07853890310014597. [DOI] [PubMed] [Google Scholar]
- 64.Nakamura N, Woda BA, Tafuri A, Greiner DL, Reynolds CW, Ortaldo J, Chick W, Handler ES, Mordes JP, Rossini AA. Intrinsic cytotoxicity of natural killer cells to pancreatic islets in vitro. Diabetes. 1990;39:836–843. doi: 10.2337/diab.39.7.836. [DOI] [PubMed] [Google Scholar]
- 65.von Herrath MG, Dockter J, Oldstone MB. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity. 1994;1:231–242. doi: 10.1016/1074-7613(94)90101-5. [DOI] [PubMed] [Google Scholar]