

Abstract
Autoantibodies have been used as good markers for the prediction of future development of type 1 diabetes mellitus (T1DM), but are not thought to be pathogenic in this disease. The role of B cells that produce autoantibodies in the pathogenesis of human T1DM is largely unknown. In the non-obese diabetic (NOD) mouse model of autoimmune diabetes, it has been shown that B cells may contribute multifariously to the pathogenesis of the disease. Some aspects of deficiencies of B cell tolerance may lead to the circulation of autoreactive B cells. In addition, the antigen-presenting function of autoantigen specific B cells is likely to be particularly important, and autoantibodies are also considered to play a critical role. This review discusses the possible aspects of B cells involved in the development of autoimmune diabetes.
Keywords: type 1 diabetes, NOD mouse, autoimmunity, B cells, costimulation, antigen-presenting cells
Introduction
Type 1 diabetes mellitus (T1DM) is an organ-specific autoimmune disease in which the pancreatic β-cells that produce insulin are the target of immune attack. There is a progressive loss of β-cells and when sufficient numbers of these cells have been destroyed, or their function inhibited, diabetes occurs.
Autoimmune diseases such as T1DM involve the interaction of different subsets of lymphocytes and antigen-presenting cells (APC). These include CD4 and CD8 T lymphocytes, B lymphocytes (B cells), macrophages and dendritic cells which together play an important role in the generation of the autoimmune response. In humans, the process leading to T1DM may take months or years [1]. In animal models such as the non-obese diabetic (NOD) mouse, there is a gradual infiltration of the pancreatic islet β-cells over weeks leading to the development of autoimmune diabetes with some similarities to the human disease from the age of 12 weeks onwards.
An inflammatory infiltrate in islets has been documented in a number of post-mortem studies [2, 3] as well as biopsies of patients with newly-diagnosed diabetes [4]. In the NOD mouse, although there is much evidence that T cells play a major pathogenic role, the infiltrate in the pancreatic β-cells consists not only of CD4 and CD8 T cells, but also of B lymphocytes and other antigen presenting cells. In another model of autoimmune diabetes, the Biobreeding (BB) rat, although not the earliest entrants to the islets, B cells have also been noted to become part of the later infiltrate [5].
There have been many studies documenting the role of T cells as major effectors in the development of diabetes. Nevertheless, over the last few years, there has been increasing interest in the role of B lymphocytes, and studies investigating their role in autoimmune diabetes will be discussed in this review.
B lymphocytes are important cells in immunity
Immunoglobulins (Igs) made up of 2 heavy chains and 2 light chains are the antigen recognition receptors expressed on the surface of B lymphocytes. They serve as a component of the B cell receptor (BCR) together with two other proteins, designated Igα and Igβ, which are important for B cell signaling. In addition, B cells, like T cells, express co-receptors that consist of CD19-CD21-CD81 which enhance the ability of the B cells to respond to antigens. The same Ig molecules that recognize antigen are also produced as antibodies by plasma cells, which are terminally differentiated B cells. Antibodies normally bind to pathogens or toxins in the extracellular spaces. This binding, termed opsonization, neutralizes viruses and toxins and prevents them from entering cells. The antibodies coating the pathogen are recognized by Fc receptors on phagocytic cells and this allows the phagocytes to ingest and destroy bacteria. In addition, natural killer (NK) cells, which do not express antigen-specific receptors, can bind the Fc receptors and this can initiate antibody-dependent cellular cytotoxicity (ADCC). The engagement of Fc receptor activates NK cells which release cytotoxic granules containing perforin and granzymes and cause lysis and apoptosis of the target cell in a manner similar to cytotoxic CD8 T cells. Furthermore, the binding of antibody to antigen forms immune complexes that can activate complement that can directly lyse some organisms.
The BCR binds antigen by the variable regions of the Ig molecule and once bound, an activation signal leads to clonal expansion and specific antibody production. An important feature of the BCR specificity is that antigens recognized by the Ig molecule are endocytosed and are specifically presented on the surface of B cells to T cells, thus concentrating the antigen-presenting ability many fold.
Naïve antigen-specific B cells do not, in general become activated by antigen alone. The activation of B cells and their subsequent differentiation into antibody-producing plasma cells usually requires CD4+ T helper (Th) cells (Figure 1). B cells obtain help from activated helper T cells in the form of CD154-CD40 and other costimulatory interactions. Once B cells become activated, they increase expression of costimulatory molecules such as B7.1 and B7.2 on the surface. These activated B cells can then stimulate antigen-specific CD4 Th cells (Figure 1). These T cells (both Th1 and Th2) produce cytokines which further aid in B cell differentiation, expansion and isotype switching leading to antibody (including autoantibody) production.
Figure 1. B cell and CD4 T cell interaction.
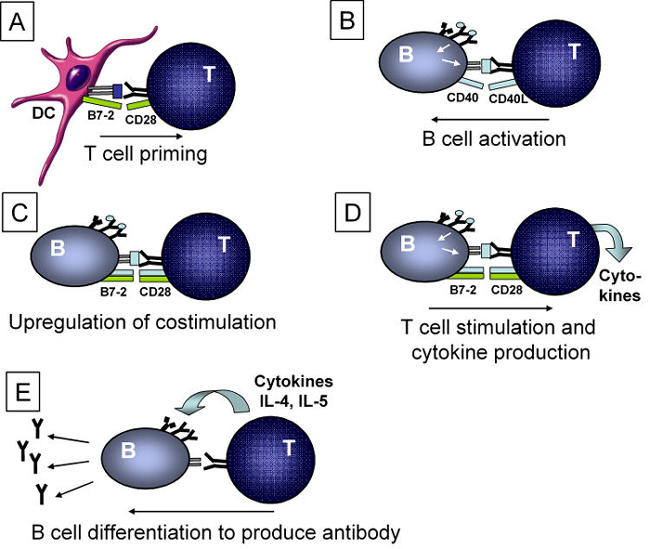
A. Naïve T cells are generally primed by antigen-presenting cells such as dendritic cells. B. Antigen-specific T cells recognizing antigens presented by B cells stimulate the B cells through CD40 ligand (CD40L) interactions with CD40. C. Activated B cells upregulate costimulatory molecules such as B7.2 and B7.1 which are then able to stimulate T cells. D. T cells activated by specific antigen-recognition together with costimulation produce cytokines. E. T cell-derived cytokines stimulate B cells to switch isotypes and differentiate to produce antibodies.
Autoantibodies in diabetes
Autoantibodies have long been associated with T1DM. Indeed, the recognition that the immune system plays a predominant role in T1DM was based on the detection of autoantibodies which are present in pre-diabetic and newly diagnosed patients with diabetes. The earliest type of autoantibody was termed islet cell antibody (ICA) and was measured by binding to frozen human pancreas. In recent years a number of methods and types of tissue section for this measurement have been used but, in general, standardization is difficult [6]. More defined autoantibodies to islet autoantigens include those to insulin [7], glutamic acid decarboxylase (GAD) [8, 9] and the tyrosine phosphatase IA-2 [10-13]. ICA are made up of antibodies mainly to GAD and IA-2 [14, 15], but also to other undefined autoantigens. Currently, according to standardization workshops, it is possible to measure these autoantibodies, particularly in combination, by radioimmunoassay to a high sensitivity and with a low false positive rate [6]. The presence of autoantibodies is currently the best marker for ongoing autoreactivity and is a major predictive tool for the future development of type 1 diabetes. In family studies with long-term follow up, combinations of autoantibodies are known to be highly predictive of future development of disease when positive in relatives of patients with type 1 diabetes. This applies particularly if they also have other susceptibility genes, such as specific human leukocyte antigen (HLA) alleles [16]. The type of autoantibodies is also important and recent work suggests that it is possible to distinguish between insulin autoantibodies that are associated with disease and those that are not, using phage display technology [17].
Autoantibodies have also been found in animal models of diabetes. Insulin autoantibodies are present in NOD mice and early expression of these autoantibodies predicts earlier onset of the disease [18]. However, neither anti-GAD nor I-A2 antibodies were consistently found in all colonies of mice although the efficiency of measurement is variable [19]. Autoantibodies associated with other manifestations of autoimmune disease were also detected in NOD mice, including anti-thyroid antibodies and thyroiditis [20] and autoimmune hemolytic anemia [21]. In addition to production of islet cell autoantibodies in the serum, Verdaguer and colleagues have recently identified B cells infiltrating the pancreas as secreting antibodies in situ and reacting to pancreatic nervous system elements [22]. These studies complemented earlier investigations showing that, in addition to GAD, other components of cells of the nervous system are a target for attack by autoreactive T cells in diabetes [23].
There are many reports describing the measurement of autoantibodies in diabetes and their use in prediction of disease, and thus, this topic will not be further reviewed in detail here. In T1DM, antibodies themselves are not thought to be directly pathogenic. They do not react against cell surface or matrix antigens such as in autoimmune thrombocytopenic purpura or autoimmune hemolytic anemia. Moreover, they do not cause disease by forming immune complexes such as in systemic autoimmune diseases including systemic lupus erythematosus (SLE). However, there are other roles of autoantibodies in disease development and these will be discussed in later sections. Although a very useful tool, the assumed non-pathogenic nature of autoantibodies has meant that B cells were largely ignored in terms of their role in the pathogenesis of diabetes. Indeed, it has been shown that it is possible to develop T1DM in the absence of B cells, as seen in a patient who had X-linked agammaglobulinemia. This individual had a mutation in Bruton's tyrosine kinase gene (BTK) and hence, severe B cell deficiency, but still developed T1DM [24]. Although it is possible that B cells are not absolutely necessary for the disease onset, such as in this rare case, this does not rule out the role(s) of these cells in the development of diabetes in non-immune deficient patients. It has become increasingly clear that B cells do play an important role in disease development, certainly in the animal models of autoimmune diabetes, and the evidence for this will be discussed in the next sections.
B cells in autoimmune diabetes in the NOD mouse
IgM is an important molecule for B cell ontogeny. Gene targeting of IgM (μMT-/-) leads to B cell developmental arrest, and thus, no mature B cells in the periphery in μMT-/- mice. [25]. The development of diabetes was severely impaired when μMT-/- mice were crossed to NOD mice [26-29], although an early publication with small numbers of mice had shown less protection [30]. Similarly, B cell development was inhibited in mice by anti-mouse Ig treatment [31] or administration of anti-IgM antibody and the antibody-treated mice did not develop diabetes compared with mice treated with a control antibody [32]. It was not possible to reconstitute NOD.μMT-/- mice with B cells alone, due to CD8 T cell attack on the B cells [33]. This CD8 T cell attack occurred because these T cells developing in mice that are deficient in B cells recognized the reconstituting B cells as foreign. However, when the NOD.μMT-/- mice were irradiated and reconstituted with syngeneic NOD bone marrow, to provide T cell precursors that do not recognize B cells as foreign, together with mature peripheral B cells, the incidence of diabetes was considerably increased [33].
Although diabetes is much reduced, the B cell-deficient mice do develop insulitis and the onset of insulitis may relate to the number of backcross generations to the NOD genetic background. Studies in μMT-/- mice that had been backcrossed 8 times [27] or 10 times to NOD mice [34] had shown very little insulitis at 12 weeks [27, 34] whereas insulitis occurred as early as 8 weeks in NOD.μMT-/- backcrossed 14 generations to NOD mice [35], even though overall, insulitis is reduced. This indicates that B cells may not be absolutely required for the initiation stage of the autoimmune process but they can traffic to the islets and facilitate the development of diabetes. It is possible that their role is more prominent later in the expansion of the diabetogenic repertoire.
Chiu and colleagues [36] performed an elegant experiment where they transplanted NOD embryonic 15-day-old fetal thymi (FT) into 3-4 week-old NOD.scid mice under the kidney capsule to generate T cells that had developed in an environment devoid of B cells. They took T cells from the FT transplanted NOD.scid mice at 6 weeks of age, T cells from 6-week-old NOD.μMT-/- mice and T cells from normal NOD mice and transferred each of these types of cells to NOD.scid recipients. Insulitis and diabetes developed in all the recipient groups. Diabetes was found in more than 30% of the recipients receiving T cells from NOD.FT transplanted mice, more than 20% in the recipients receiving μMT-/- T cells and more than 60% in the recipients of NOD T cells [36]. Similar results were obtained when T cells from young 5-week old NOD mice were transferred to NOD.scid mice, and caused insulitis and diabetes [37]. In these circumstances, where the transferred cells undergo homeostatic proliferation and rapid expansion, unrelated to antigen-specific stimulation and due to the lymphopenic environment, diabetes can occur in the absence of B cells. Other factors, such as the absence of lymphocytes in the scid recipients, may also increase antigen presentation by dendritic cells and macrophages. Thus, although under normal circumstances B cells clearly play a prominent role, they are not absolutely necessary if there are other means by which T cells can expand.
The stimulation of CD8 T cell expansion may be a critical function of B cells. This was indicated using a model system, where the major goal was to study the role of CD8 T cells in autoimmune diabetes development [28]. NOD mice that express the costimulatory molecule B7.1 on the islets using the rat insulin promoter (RIP) develop accelerated diabetes as early as 3 weeks of age [38]. When these mice were crossed with NOD.μMT-/- mice, there was no effect on the development of this much accelerated diabetes. In these mice, there is a great expansion of CD8 T cells within the islets, compared with normal NOD mice and B cells are not required for the disease to occur. Again, this is an instance where other factors, for example the expression of costimulatory molecules, have allowed the expansion of potentially diabetogenic T cells [28].
Thus, it has been established, by removal of B cells, that this subset of cells plays a role in autoimmune diabetes in the NOD mouse, although, under some circumstances, they may not be absolutely required for the development of disease.
Loss of tolerance in autoreactive B cells
Many studies have confirmed the importance of a loss of T cell tolerance in autoimmune diabetes. As B cells also clearly play a role, it is possible that defective B cell tolerance is another factor affecting the disease.
A number of mechanisms of B cell tolerance exist to prevent autoreactive B cells from causing autoimmune disease. There are several possible outcomes of B cell encounter with self-antigen. These include clonal deletion, B cell receptor editing, clonal anergy and ignorance. The mode of tolerance of any given autoreactive B cell is likely to be determined by multiple factors, such as affinity of the receptor for the antigen and cross-linking of the receptor. Whether the antigenic determinant is membrane bound or soluble will therefore have a bearing, as membrane-bound antigens cross-link receptors more effectively. In addition, other genetic factors may also govern autoreactive B cell responses.
Clonal deletion
Self-reactive B cells can undergo apoptosis and death (i.e. clonal deletion). Using mice that expressed a B cell transgene recognizing H-2Kb, Nemazee and Burki showed that in mice expressing Kb, the development of B cells was blocked and immature B cells were eliminated in the bone marrow [39]. B cells may also be deleted when they encounter antigen in the periphery [40]. Silveira and colleagues have demonstrated that clonal deletion of B cells recognizing membrane-bound antigens takes place in NOD mice [41]. They demonstrated clonal deletion in BCR transgenic mice specific for Hen Egg Lysozyme (IgHEL) and membrane-bound HEL (mHEL) [42]. On both NOD and B6 genetic backgrounds, very few transgenic IgHEL cells were present in the periphery [41], indicating no defect in clonal deletion in NOD mice.
Deletion of autoreactive B cells is dependent on the ability of antigens to cross link the B cell receptor, and there is a difference in the ability of monomeric or oligomeric soluble antigens to tolerize B cells compared with multi-valent membrane-bound antigen [42]. Soluble antigens are less likely to be able to cross link the BCR. In mice that expressed B cells transgenic for HEL (IgHEL) together with soluble HEL (sHEL), clonal deletion was not seen. Instead, B cells from these mice are tolerized by anergy discussed below [43]. Similarly, when experiments were done in NOD mice expressing IgHEL and soluble HEL (sHEL), (compared with IgHEL+mHEL), B cells expressing the IgHEL were not deleted [41]. Thus there is a difference in the responses leading to this deletional tolerance dependent on whether the autoantigen is membrane bound or soluble. Although these mechanisms have been defined using transgenic mice, there may be important implications for physiological antigens, for example insulin is a soluble antigen, whereas other islet autoantigens are not.
B cell receptor editing
In some circumstances, B cells may alter the BCR by the process of receptor editing, thus preventing deletion [44]. In B cell development, the immunoglobulin heavy chain rearranges first. Then, the light chain rearranges so that production of surface IgM can occur. If there is no cross linking of the receptor, rearrangement of the light chain will terminate and the B cell will continue development. However, if self-antigen to which the immunoglobulin reacts strongly is encountered, the B cell development will stop and further light chain rearrangement will continue. This secondary light chain rearrangement can rescue the immature B cells by replacing the already rearranged light chain with another sequence. This mechanism was investigated using the Ig3-83 transgenic mice where the transgene recognizes MHC class I H-2Kk and MHC class I H-2Kb [44]. As with clonal deletion, this mechanism of tolerance is likely to occur following encounter with antigens to which the B cells bind strongly. This receptor-editing phenomenon was also investigated in these Ig3-83 transgenic mice comparing the B6 and NOD genetic backgrounds [41]. It was shown that receptor editing occurs to a similar extent in mice on both the B6 and the NOD genetic background and thus it was deduced that this mechanism of tolerance is not defective in NOD mice. Thus both clonal deletion and receptor editing appear to operate normally as B cell tolerance mechanisms in NOD mice.
B cell anergy
If B cells encounter weakly binding antigens such as in the case of small soluble proteins or continuous encounter with a soluble antigen, this may induce anergy and the B cells are rendered unresponsive. Goodnow and colleagues had demonstrated anergy as a mechanism of B cell tolerance using the BCR transgenic model specific for Hen Egg Lysozyme (IgHEL) and soluble HEL (sHEL) [43]. Anergic mature B cells cannot be activated by antigen, due to a block in signal transduction [45]. However, anergy can be reversed by treatment with LPS and self-reactive B cells (in this case, anti-HEL reactive B cells) may thus be reactivated [46].
Functional anergy was demonstrated in the transgenic mice expressing IgHEL and sHEL in mice backcrossed onto the NOD background [41]. However, this anergy could be reversed when an antigenic stimulus was accompanied by anti-CD40 stimulation. Autoreactive anti-double stranded DNA-specific anergic B cells can also be stimulated when T cell "help" factors are provided [47]. Autoreactive diabetogenic T cells in NOD mice have defects in tolerance induction [48-51]. It was suggested that these T cells provide CD40-mediated signals that could reverse the weak anergic state induced in B cells by engagement of a soluble cell autoantigen during their development [47]. These autoreactive B cells could then amplify diabetogenic CD4 T cell responses.
Clonal ignorance
B cell ignorance is an alternative mechanism of protection from autoreactivity of B cells with a low binding affinity for self-antigen. These B cells do not recognize their antigen because there is a low concentration or the antigen is sequestered and the B cell receptor is not activated. B cells able to produce autoantibodies to insulin have been postulated to be controlled by this mechanism [52], although later studies have suggested other mechanisms are more important (see below).
In the previous sections, B cell tolerance has been discussed in the context of model antigens. However, B cell tolerance has also been investigated in mice in relation to the natural endogenous soluble antigen, insulin. As discussed in the sections above, autoantibodies to insulin and proinsulin have been demonstrated in both humans and mice. Insulin is a small soluble molecule that circulates in low concentrations in response to glucose following ingestion. Rojas and colleagues have immunized a BALB/c mouse with insulin and induced an anti-insulin response, producing an IgG mAb125. Subsequently, the Ig heavy chain VH125 and Ig light chain Vκ125 genes were cloned. Transgenic mice expressing the genes encoding the insulin-reactive 125 antibody were generated [53]. On the B6 genetic background, the investigators showed that the transgenic B cells were not clonally deleted, although there was a reduction in surface expression of the transgenic Ig. B cells were also not ignorant of the endogenous insulin as they produced insulin-specific antibodies. However, when immunized with beef and pork proinsulin, they failed to increase the anti-insulin antibody levels indicating that the B cells were anergic. Furthermore, these mice showed no evidence of insulitis and anti-insulin antibody had no effect on glucose homeostasis. When the Ig transgenic mice were backcrossed to the NOD genetic background [54], the transgenic B cells were also able to develop and, as in the B6 background, had impaired responses to standard stimulators of B cells such as lipopolysaccharide, anti-CD40 and IL-4. However, although the transgenic B cells did not proliferate, maturation and upregulation of the costimulatory molecule CD86 occurred in the NOD mice [54]. Thus, they appeared to be anergic but were still able to mature, migrate to the follicles in lymphoid organs and to the pancreas and upregulate costimulatory molecules that are vital for full stimulation of T cells.
Thus, on the NOD genetic background predisposed to developing spontaneous autoimmune diabetes, clonal deletion and receptor editing appear to be normal but there is an alteration in the induction of anergy in two different transgenic models. This may contribute to B cell effects on the development of diabetes.
B cells as antigen-presenting cells
B cells are one of three types of specialized APC in the immune system, including dendritic cells and macrophages, each of which has a distinct role. The most potent APC for activation of naïve T cells are dendritic cells (DCs). Immature DCs encounter antigens in peripheral tissues or solid organs and transport the antigens to draining lymph nodes. DCs acquire high levels of costimulatory molecules and MHC class I and II molecules during this process, and thus they can strongly activate T cells. Macrophages that have major phagocytic capabilities are part of the first line of defense against invading organisms. They can also upregulate the expression of MHC and costimulatory molecules during the phagocytosis. The activated macrophages stimulate cells of the adaptive immune response, such as T cells. However, they do not usually deal effectively with soluble antigens.
B lymphocytes play a different role as APC. Although B cells do not present antigens as efficiently as DCs, they are adapted to bind soluble antigens specifically via cell surface immunoglobulin and thus can present soluble proteins much more effectively. The specificity of the immunoglobulin directs processing of the protein [55]. B cells constitutively express high levels of MHC molecules and antigen-specific B cells may therefore be very important in diversifying the immune response, presenting sequestered antigens to CD4 T cells [56]. B cells do not constitutively express costimulatory molecules and therefore are unlikely to initiate responses to soluble self-proteins. However, in some circumstances where B cells upregulate costimulatory molecules, they can also efficiently activate naïve T cells. Antigen-specific T cells, in turn, are able to activate antigen-specific B cells and these can then further stimulate T cells to break self-tolerance [57, 58].
B cells as APC in diabetes
B cell function and the production of antibodies, including autoantibodies, are closely linked to CD4 T cells (Figure 1). CD4 T cells provide an activation signal to the B cells through CD154 (CD40L), expressed on the surface of the CD4 T cell, which interacts with and activates the B cell expressing CD40 (Figure 1). In addition, the production of antibodies also implicates CD4 T cells in providing the cytokine and other costimulatory signals for isotype switching from IgM to IgG subclasses. Thus the presence of autoantibodies is likely to indicate CD4 recognition of antigen presented by B cells (Figure 1).
B cell deficient μMT-/- mice on the NOD genetic background have been used to determine the role of B cells as antigen-presenting cells in the development of diabetes. Reconstitution of NOD.μMT-/- mice with immunoglobulin from diabetic NOD mice does not precipitate diabetes. Serreze and colleagues purified immunoglobulin from the sera of diabetic NOD mice and injected this Ig twice weekly between 8 and 20 wk of age. Circulating Ig levels were shown to be greater than in control treated mice, although less than in normal NOD mice. Neither insulitis nor diabetes was observed [33].
Antigen presentation function of B cells has been investigated in a variety of experimental systems. In μMT-/- mice on the B10A genetic background, Constant and colleagues [59] had shown that priming to a variety of exogenous protein antigens was deficient in these mice (except for keyhole limpet hemocyanin (KLH)). However, priming to peptide antigens was intact. This general defect in priming to protein antigens may not be obvious if only a limited number of antigens is used. In terms of the B cell role as antigen-presenting cells in the NOD mouse, it was found that T cells in NOD.μMT-/- mice had an impaired ability to respond to islet autoantigens. Spontaneous responses to autoantigens such as GAD and heat shock protein 60 (HSP60) were reduced compared to the responses to purified protein derivative (PPD) and mitogens [60]. In addition, immunized responses to GAD were also reduced in NOD.μMT-/- mice compared to KLH [33]. However, responses to GAD were restored when NOD.μMT-/- mice were reconstituted with syngeneic NOD bone marrow together with peripheral mature B cells, and the incidence of insulitis and diabetes increased [33]. Whilst the results suggested that specific responses to autoantigens were impaired in these studies, it should be noted that, overall, primed responses to protein antigens are generally impaired in μMT-/- mice. In addition, B cells could also be providing other stimuli for responses to autoantigens, other than their role as APCs.
Noorchashm and colleagues generated chimeric NOD.BCIID mice that were deficient in I-Ag7 in the B cell compartment in a scheme shown in Figure 2 [29]. The investigators postulated that the defect and the reduction in insulitis and diabetes was related to the class II deficiency in the B cell compartment. However, it should be noted that there was also a change in the MHC class I region in the B cells in these mice and therefore, it cannot be concluded that this is solely on the basis of lack of I-Ag7 on B cells.
Figure 2. Generation of NOD mice that have a class II deficiency on the B cells (NOD.BCIID mice).
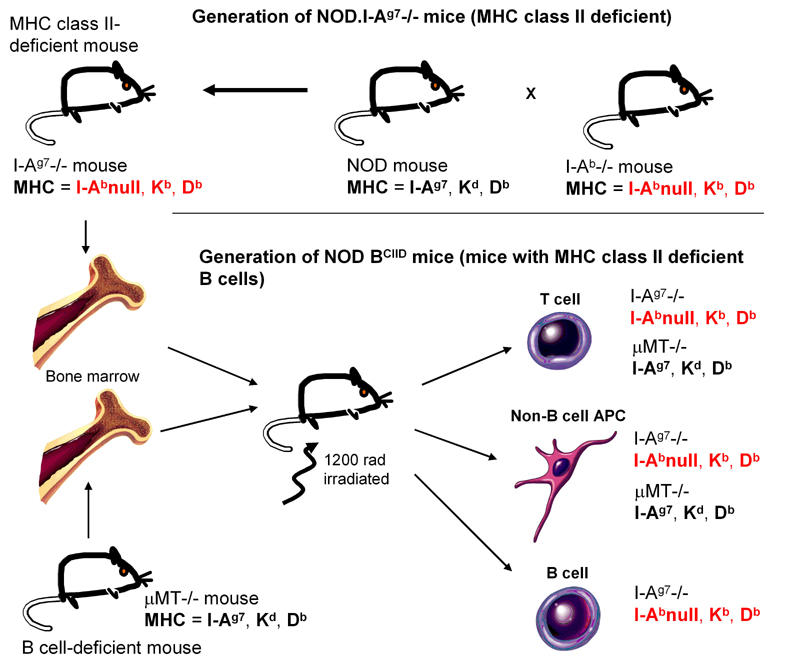
NOD mice were crossed with I-Ab-/- B6 mice on the H-2b genetic background (KbDb) to generate NOD mice expressing the class II knockout mutation. Since MHC class I is in strong linkage disequilibrium with MHC class II, the introduction of the MHC class II knockout mutation introduces not only I-Ab-/- but also alters the MHC class I molecules of the NOD haplotype (KdDb) to KbDb. NOD female mice then were irradiated and reconstituted with bone marrow from these NOD.I-Ab-/- mice and NOD.µMT-/- mice. This generated mice that had T cells and non-B cell antigen-presenting cells from both NOD I-Ab-/- mice and NOD.µMT-/- mice. Therefore, the resulting mice expressed the NOD I-Ag7 (derived from the NOD.µMT-/- mice) on the non-B cell antigen-presenting cells. However, in the B cell compartment, the mice would only express B cells from the NOD I-Ab-/- mice as NOD.µMT-/- mice do not have mature B cells. Thus the B cells would not express MHC class II and the MHC class I molecules are de-rived from the H-2b haplotype. All the other cells in the NOD BCIID mice express MHC molecules of both H-2g7 (KdDbI-Ag7) and I-Ab-/- (KbDb) haplotypes.
Thus far, B cells have been considered as facilitators of development of diabetes by direct antigen presentation. It is possible that they may also have a disease modulatory role. Natural killer T (NKT) cells express an invariant Vα14Jα281 T cell receptor and are important modulators of diabetes and other autoimmune diseases (reviewed in [61]). Bezbradica and colleagues have recently examined activation of NKT cells by dendritic cells and B cells. NOD NKT cells produce lower levels of cytokines compared with equivalent stimulation in B6 mice using α-galactosyl-ceramide (αGalCer). They have demonstrated that NOD DC activate NKT cells poorly compared with DC in B6 mice. However, in NOD mice, B cells were able to elicit IL-4 production from NKT cells [62]. Treatment with αGalCer has previously been shown to be able to protect NOD mice from diabetes [63, 64]. This stimulation of NKT cells with concomitant cytokine production may provide another explanation for the efficacy of this treatment.
LPS-activated B cells have protected NOD mice from diabetes when transferred into young, 4-week-old NOD mice [65]. These B cells produced anti-inflammatory cytokines such as TGF-β and upregulated Fas ligand which could trigger apoptosis of activated T cells. Spontaneous Th1 responses as well as antigen-presenting function were reduced. The investigators suggested that these findings may explain different incidences of diabetes in "natural environments", which are not LPS-free, compared with specific pathogen-free facilities [65]. They also indicate that B cells may play a pivotal role in later activation or reduction of pathogenic T cell responses.
B cells as antigen-specific APC
Thus far, it is clear that there is an important contribution of B cells to the development of diabetes. A question then arises as to whether islet antigen specific B cells are required for B cell facilitation of disease. Hulbert and colleagues [66] generated a B cell transgenic mouse that expressed IgMa transgenes encoding VH genes related to anti-insulin mab125 into the germline of NOD mice. Transgenic Ig light chain genes were not used in these mice; endogenous light chains could therefore be selected. They had previously shown that the VH125 contained two amino acid replacements necessary for insulin binding [52]. Although the heavy chain is fixed, the mice are able to select a diversity of light chains. This contrasts with the mice discussed above in the B cell tolerance section that expressed both transgenic Ig heavy and light chains [53]. The expression of this heavy chain transgene favored the selection of an insulin-reactive B cell repertoire in the NOD mouse, a phenomenon not found in the equivalent mouse on a B6 genetic background. The mice had increased insulitis and accelerated diabetes compared to the non-transgenic littermate controls. A control transgenic mouse expressing a fixed heavy chain that did not have increased insulin-binding capability developed a lower incidence of insulitis and diabetes.
Silveira and colleagues [67] generated B cell-deficient mice that expressed a fixed Ig transgene for HEL. These mice were protected from insulitis and diabetes in a similar manner to the non-transgenic B cell-deficient mice. Using GAD responses, they showed that NOD T cells were not able to generate responses to GAD in the presence of transgenic B lymphocytes but could do so with non-transgenic NOD lymphocytes [67]. They postulated that this was likely to occur because the transgenic B lymphocytes, with a fixed BCR specificity, were unable to non-specifically take up and present GAD sufficiently for T cell recognition.
Wong and colleagues also used a transgenic mouse expressing an IgMa heavy chain construct VH186.2, reactive to the hapten (4-hydroxy-3-nitrophenyl) acetyl (NP) [35]. Antibodies of limited diversity could be selected by use of endogenous light chains. To test whether secreted antibody was necessary for the B cell role in diabetes development, two forms of this transgene were constructed. In the first, the variable-diversity-joining (VDJ) region was ligated to a constant region from which the secreted exon and polyadenylation site had been excised, resulting in production of only membrane-bound but not secreted Ig (MIg). A control construct was identical except that the secreted exon was intact (M+SIg). It was found that in the MIg mice there was an increase in the amount of insulitis and a small but statistically significant increase in the incidence of diabetes, although the incidence was not restored to the level seen in normal NOD mice. It was noted, however, that the M+SIg mice, whilst they could produce anti-islet antibodies, the level was much lower than in normal NOD mice. This suggested that the reduced Ig repertoire resulting from the fixed heavy chain transgenes used here was not sufficient to produce the full spectrum of islet-specific antigen receptors to allow appropriate diversity [35]. This confirmed that antigen specificity of the B cells is important. The antigen-presenting role of B cells was separated from the ability to secrete antibody in the development of autoimmune diabetes. The data clearly showed that antibody secretion is not essential for some of the diabetes-promoting role of B cells.
B cell characteristics in NOD mice
The above observations have been made using transgenic models. Unmanipulated NOD mice have a higher frequency of activated B cells compared to non diabetes-prone strains of mice [68]. This was indicated by a higher frequency of CD69+B220+ B cells in mice as young as 14 days of age. The CD69+ B cells showed an increase in the level of MHC class II, and the costimulatory molecules B7.1 and B7.2 [68]. It was suggested, by comparison of H-2g7-expressing mouse strains with non-H-2g7 strains that the activated phenotype was correlated with the H-2 haplotype. This could be related to T cell activation of the B cells following TCR-MHC/Ag interactions [68]. B cells from NOD mice, as well as the H-2g7 insulitis-prone but diabetes-resistant NOR and NOD.B6Idd4B congenic mice, were also found to be more resistant to activation-induced cell death and hyper-responsive [69].
Insulitis in NOD mice consists of a large number of both CD4 and CD8 T cells as well as B cells. In general, the B cells and the CD4 T cells are to be found clustered in the same areas, while CD8 T cells are more dispersed. Islet-infiltrating B cells express an increase in B7.1 and B7.2 suggesting a role in local costimulation [70]. This is in keeping with the observation that if B7 costimulation is provided locally on islet cells, the presence of B cells in the development of diabetes is no longer required [28]. Islet-infiltrating B cells also produce TNF-α [70] and this could contribute to inflammation in the islets and recruitment of other inflammatory cells.
Noorchashm and colleagues examined CD4 T cell activation using anti-CD3 stimulation in NOD mice. Using CFSE labeling, they found that NOD CD4 T cells responded less well to anti-CD3 stimulation compared with B6 CD4 T cells, a phenomenon related to the non-MHC genes in the NOD genetic background and not to the expression of the MHC haplotype [71]. Both B cell and non-B cell antigen-presenting cells were tested for their ability to effectively activate CD4 T cells. Their experiments indicated, using anti-CD3 as a stimulus, that B cell costimulation was required for effective CD4 T cell activation. Costimulation by B cells in the pancreatic lymph nodes is important [34] in addition to the activation of T cells by B cells within the islets [70].
Increased activation, and particularly the provision of costimulatory signals by B cells to T cells may be particularly important in the interaction between B cells and T cells in promoting autoimmunity.
The B cell repertoire contributes to the development of autoimmune diabetes
The repertoire of B cells in NOD mice is likely to contribute to the propensity to develop autoimmunity. Leijon and colleagues had found that the germline IgVH locus was identical to B6 mice. However, the pattern of VH gene utilization differed considerably between these two mouse strains. In NOD mice, the neonatal type of VH genes was expressed in adult mice contrasting with a different repertoire in B6 mice [72, 73].
Natural autoantibodies of the IgM, IgG and IgA classes are found in all normal individuals and react with a variety of serum proteins, cell surface structures and intracellular structures. They are encoded by germline genes and are characteristically reactive to multiple antigens and do not undergo affinity maturation in normal individuals [74]. Natural autoantibodies are involved in a variety of functions that include immune regulation, homeostasis and repertoire selection, resistance to infections, transport and functional modulation of biologically active molecules [74]. Thomas and colleagues have found that insulin-binding autoantibodies in NOD mice have the characteristics of a natural autoantibody repertoire [75]. They had characterized the structure and function of insulin autoantibodies of B cell hybridomas generated from the spleen in the absence of mitogen or antigen stimulation. They found that insulin-binding mAb in NOD mice are of low avidity and they are able to interact with many other antigens including DNA and immunoglobulin. The V region genes were also mainly germline (unmutated) and identical to B genes expressed by antibodies in other systemic autoimmune disease such as in SLE [75].
Natural antibodies are mainly produced by the B-1 subset of B cells that express the surface marker CD5 and they are enriched in the peritoneal and pleural cavities. The characteristics of islet-infiltrating B cells in NOD mice have been compared with B cells from other lymphoid tissues. Divergent observations of the nature of these B cells have been made. When B cells from islet infiltrates, spleen, pancreatic lymph nodes and peritoneal cells were examined for the presence of CD5, Hussain and Delovitch did not find any increase in the percentage of CD5 B-1 cells in the islet infiltrate compared to the spleen and pancreatic lymph nodes [70]. However, Kendall and colleagues did observe an increase in CD5+ B cells in the islet infiltrate [76]. These CD5+ B cells can be specifically eliminated by the use of intraperitoneal hypotonic lysis. When NOD mice were treated weekly with hypotonic lysis for 4 weeks, there was a delay in the onset of diabetes in the treated mice [76]. This treatment had the effect of reducing B cells in the pancreatic infiltrate without affecting the T cells. Furthermore, the investigators found that the infiltrating B cells were redistributed, remaining around the periphery of the islets and appeared to be less invasive [76]. They suggested that the delay in onset of diabetes following hypotonic lysis treatment occurred because of a reduction of antigen-presenting function by the B cells at the actual site of the autoimmune attack.
Active selection of autoantibody repertoire in single chain Ig transgenic mice is altered in NOD mice. When the heavy chain transgene of the insulin-recognizing 125 antibody, described above, was expressed without the light chain in B6 mice, no insulin-binding B cells were detected in the transgenic mice. In contrast, when these mice were backcrossed onto the NOD background, selection of appropriate light chains to allow insulin binding occurred and the NOD 125Tg mice expressed B cells producing autoantibodies that were able to bind insulin [66]. Interestingly, a number of different κ-light chains were selected that were clonally independent and not due to oligoclonal expansion [77]. The most frequently expressed Vκ1 and Vκ9 genes selected to pair with the 125Tg were shared with the autoimmune model, lupus-prone New Zealand Black/BINJ mice. It was suggested that the polymorphisms seen in these selected Vκ-alleles may code for alterations in structure that could skew the antigenic repertoire recognized by B cells in NOD mice [77].
Thus, there are differences in the B cell repertoire in NOD mice, expressing a more fetal pattern of VH usage, a natural antibody repertoire that can bind insulin but also other autoantigens, a possible increase in CD5 B-1 cells that infiltrate the islets and a propensity to select for B cell receptors that may skew the BCR repertoire towards autoantigenic specificities.
B cells produce autoantibodies
The use of autoantibodies as important markers for the future development of diabetes has been discussed earlier. They indicate autoreactive B cell activity. The known diabetes-related autoantigens are T-dependent antigens - production of the autoantibodies by B cells is dependent on activation by CD4 T cells. Although, the autoantibodies in diabetes are not thought to be directly pathogenic, there is evidence that they play an important role in the etiology of diabetes. Greeley and colleagues performed an elegant experiment whereby they bred NOD.μMT-/- females with NOD males to generate offspring that were NOD.μMT+/-, and therefore B cell-sufficient. However, the mothers, homozygous for the B cell deficiency, did not produce antibodies and therefore could not transmit autoantibodies to their offspring [78]. The offspring from these parents developed a lower incidence of diabetes (< 25% at 40 weeks) compared with offspring (> 50% at 40 weeks) that were also NOD.μMT+/-, but from NOD females mated with NOD.μMT-/- males [78]. In the latter case, the mothers were B cell sufficient and could produce antibodies and transmit them to the offspring. This suggested that transplacental antibodies or antibodies transferred during lactation play a role in the disease development. Further, to demonstrate that the antibodies specifically from the NOD female transmitted to the offspring were important, NOD female mice were superovulated, mated with a male NOD mouse and the embryos were transferred to surrogate DBA/2 females or control NOD females. A lower incidence of diabetes in both male and female offspring was found in the mice born of the DBA/2 surrogate mothers [78]. These experiments provide compelling evidence that maternally transmitted antibodies from NOD mice can influence the development of disease, and insulin autoantibodies are clearly measurable as transmitted from the mother to the offspring [78]. The study does not, however, indicate how the antibodies may be affecting the disease process. There may also be other factors involved in this process of facilitation of diabetes in the early phases of the process. This was shown by Kagohashi and colleagues [79] who also performed embryo transfer of NOD embryos into surrogate non-diabetes prone strains ICR (designated NOD/ICR) and DBA/2J (designated NOD/DBA) [79] and examined insulitis. Interestingly, they found that NOD/ICR and NOD/DBA offspring developed insulitis significantly earlier than the offspring transplanted into NOD mothers (designated NOD/NOD). However, despite the early insulitis, similar to the study by Greeley and colleagues [78], diabetes was significantly suppressed in NOD/ICR and NOD/DBA offspring in comparison with NOD/NOD offspring. Insulin autoantibodies (IAAs) were undetectable in ICR and DBA/2J surrogate mothers and in NOD/ICR and NOD/DBA offspring at the onset of insulitis, suggesting that maternal factors other than transmitted IAAs induced the earlier onset of insulitis [79].
There are a number of mechanisms by which autoantibodies could facilitate the development of disease. It is possible that islet-specific antibodies could indirectly promote disease by enhancing antigen presentation by DCs. Alternatively, they may cause tissue damage that releases islet cell antigens, although, as indicated earlier, there is no direct evidence for this. Finally, they could also stimulate antibody-dependent cell-mediated cytotoxicity, although again, there is currently no evidence for this notion in pathogenesis of diabetes.
B cells as antigen-presenting cells and the production of autoantibodies together influence the pathogenesis of diabetes
In addition to the ability of B cells to specifically process and present antigens by virtue of the expression of antigen-specific Ig on the surface, secreted antibody with the same specificity can also enhance the antigen presentation function. Reijonen and colleagues have found that antibodies to GAD65 can modulate presentation of GAD65 to T cells [80]. They used a T cell hybridoma T33.1, which recognizes the GAD65 amino acids 274-286 presented by HLA-DRB1*0401. Using sera from patients known to be GAD65 antibody positive, they tested antigen-presenting cells exposed to recombinant human GAD65 alone or complexed with GAD65Ab for the ability to stimulate the GAD-peptide specific T cell hybridoma. Sera from GAD65 Ab-negative patients were used as controls. Stimulation of the T cell hybridoma was considerably enhanced with sera from patients with high GAD65 autoantibody levels [80]. Sera from GAD65 Ab-negative subjects had no effect. Furthermore, the experiments indicated that uptake of antibody-complexed GAD65 was mediated through the Fc receptor (FcR) because blocking FcR inhibited the enhanced stimulation [80].
The specificity of the B cell receptor may also influence the presentation of soluble autoantigens. It has been proposed that both enhancement and inhibition of the presentation of T cell epitopes that are spatially related to antibody epitopes could occur [81]. Jaume and colleagues have studied a panel of B cell lines producing antibodies that are related to immunodominant GAD65 T cell epitopes recognized by T cell hybridomas restricted to HLA DRB1*0401 [82]. Each of the B cell lines bound to a GAD65 conformational epitope distinct from each other and they all expressed HLA DRB1*0401. The HLA DRB1*0401-expressing Priess cell line was used as a control line that can take up GAD65 and present it non-specifically. Using this system, they showed that presentation of T cell epitopes that were closely related to the conformational antibody epitope was blocked or inhibited. The individual hybridomas were able to present T cell epitopes that were distant from their antibody epitopes with enhanced efficiency, compared to the Priess cell line, indicating the increased efficiency of presentation of a protein recognized by the BCR [82]. The investigators suggested that binding of a GAD65 autoantibody to its epitope stabilizes and sequesters the epitope, thus masking any T cell epitopes in the antibody-binding region. This would have the effect of suppressing presentation of T cell epitopes in the region of GAD bound by antibody and favor presentation of epitopes distant from the antibody-binding area. Banga and colleagues have made similar observations [83].
Other aspects of B cell function contributing to their role in the development of diabetes
It should be noted that, in the quest to determine the role of this important group of cells, it is likely that multiple functions of B cells are important. Although removal of specific types of B cell function will give us indication of the crucial roles, other factors may be involved.
B cells could play roles other than as APC, for example, by promoting normal lymphoid architecture and follicular dendritic cell (FDC) formation [84]. B and T cells that proliferate in the early phases of the immune response migrate to a primary lymphoid follicle in the spleen and form a germinal center (GC). GCs are composed mainly of highly proliferating B cells and antigen-specific T cells that help the B cells. In the primary follicle, resting B cells cluster around the FDC which acts to attract both naïve and activated B cells into the follicle. In examining antigen specific B cells in diabetes, Silveira and colleagues [67] investigated the formation of GCs in mice where the B cells expressed a transgene that recognizes HEL. B cells from these mice could not recognize diabetes autoantigens but were able to form normal GCs. However, the mice did not develop diabetes. This implies that GC formation, while important, is not a major function of B cells in the pathogenesis of diabetes in this model system. However, it does not rule out the possible role of islet autoantibody-specific germinal centers in the pathogenesis of the disease.
B cells as a possible therapeutic target?
As discussed in this review, there is much evidence for the importance of B cells in autoimmune diabetes in the NOD mouse model. However, there is very little information about human B cells in the development of T1DM although the autoantibodies have been much studied. If B cells are important in the pathogenesis of human T1DM, then perhaps, in a directed fashion, they could be a possible therapeutic target.
CD20 is a transmembrane protein expressed on the surface of almost all B cells. Rituximab (anti-human CD20) is a human-mouse chimeric monoclonal antibody that targets human CD20 and causes rapid and specific B cell depletion [85]. It was the first monoclonal antibody treatment approved for clinical use by the Food and Drug Administration for the treatment of cancer. In the last decade, Rituximab has been given to many B cell lymphoma patients [86]. Rituximab has also been explored for treating autoimmune diseases, such as rheumatoid arthritis, SLE, autoimmune hemolytic anemia and immune thrombocytopenic purpura, and showed clear efficacy [87-92]. Unlike T1DM, all these autoimmune disorders are primarily B cell-medi-ated diseases, although T cells also play an important role in the pathogenesis. Interestingly, though antibodies may play a role in these diseases, clinical responses to the treatment were often unlinked to changes in antibody titers. Thus, the mechanism of Rituximab in autoimmune disease remains unclear. At the recent 65th ADA Scientific Meeting (2005), it was announced that a clinical trial will be launched soon to use Rituximab to treat patients with T1DM. T1DM is considered primarily a T cell-mediated autoimmune disease and the role of B cells in diabetes development has not been elucidated. Whether Rituximab would be useful in treating T1DM in vivo is basically not known, even in animal studies. Rituximab targets human CD20 and does not cross react with mouse CD20. There have been no equivalent anti-mouse CD20 antibodies until very recently [93]. Therefore, there are no data from efficacy testing of anti-CD20 treatment in animal models of autoimmune diabetes, for example to find out the timing of B cell targeting. In addition, there is also no indication of when B cells might be appropriately targeted for the best clinical outcome. For example, it may be necessary to deplete B cells at an early stage, and treatment at clinical onset may be too late to alter the course of the disease. Clearly, given the important roles that B cells have in immune responses in general, any therapeutic targeting would need to be very specific.
Summary and concluding remarks
It is clear from data derived from the NOD mouse model of autoimmune diabetes that B cells play an important role in the pathogenesis of the disease. It is likely that multiple facets of B cell action and their interactions with T cells are important and no single factor seems to be dominant. There are defects in tolerance that are likely to relate to background genes including failure to delete autoreactive B cells particularly those which respond to soluble antigens. Other failures of tolerance may include defective B cell proliferation separated from signals that promote B cell maturation and costimulatory function. B cells function as antigen-presenting cells, and their ability to present antigens relating to the specificity of the B cell receptor is likely to be an important promoter of disease. They accumulate in the islets as part of the inflammatory infiltrate and their ability to present antigens in situ, as well as providing costimulatory function, may be important. The BCR repertoire may also predispose towards the development of diabetes.
As yet, it is not clear whether B cells play a similar role in human type 1 diabetes. Although there is a case report of autoimmune diabetes developing in a patient who was genetically deficient in B cells, the fact that diabetes could develop in the absence of B cells in this rare case does not necessarily imply that they do not normally play a role. With increasing experience using several models to study B cells in diabetes, it has become clear that B cells have diverse roles in the immunopathogenesis of autoimmune diabetes.
More studies will be required to define the role of B cells in the human disease. If they are found to be as important in humans as they clearly are in the mouse models, then antigen-specific B cells may well be a potential therapeutic target for therapy and prevention of type 1 diabetes.
Acknowledgments
FSW is a Wellcome Trust Senior Fellow in Clinical Science. LW is funded by grants from the Seaver Institute, American Diabetes Association and the Juvenile Diabetes Research Foundation.
References
- 1.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 2.Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29:267–274. doi: 10.1007/BF00452061. [DOI] [PubMed] [Google Scholar]
- 3.Foulis AK, McGill M, Farquharson MA. Insulitis in type 1 (insulin-dependent) diabetes mellitus in man--macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol. 1991;165:97–103. doi: 10.1002/path.1711650203. [DOI] [PubMed] [Google Scholar]
- 4.Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanenberg H, Kolb-Bachofen V, Kantwerk-Funke G, Kolb H. Macrophage infiltration precedes and is a prerequisite for lymphocytic insulitis in pancreatic islets of pre-diabetic BB rats. Diabetologia. 1989;32:126–134. doi: 10.1007/BF00505185. [DOI] [PubMed] [Google Scholar]
- 6.Verge CF, Stenger D, Bonifacio E, Colman PG, Pilcher C, Bingley PJ, Eisenbarth GS. Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: Combinatorial Islet Autoantibody Workshop. Diabetes. 1998;47:1857–1866. doi: 10.2337/diabetes.47.12.1857. [DOI] [PubMed] [Google Scholar]
- 7.Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 1983;222:1337–1339. doi: 10.1126/science.6362005. [DOI] [PubMed] [Google Scholar]
- 8.Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, DeCamilli P, Camilli PD. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–156. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 9.Baekkeskov S, Nielsen JH, Marner B, Bilde T, Ludvigsson J, Lernmark A. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature. 1982;298:167–169. doi: 10.1038/298167a0. [DOI] [PubMed] [Google Scholar]
- 10.Christie MR, Genovese S, Cassidy D, Bosi E, Brown TJ, Lai M, Bonifacio E, Bottazzo GF. Antibodies to islet 37k antigen, but not to glutamate decarboxylase, discriminate rapid progression to IDDM in endocrine autoimmunity. Diabetes. 1994;43:1254–1259. doi: 10.2337/diab.43.10.1254. [DOI] [PubMed] [Google Scholar]
- 11.Rabin DU, Pleasic SM, Shapiro JA, Yoo-Warren H, Oles J, Hicks JM, Goldstein DE, Rae PM. Islet cell antigen 512 is a diabetes-specific islet autoantigen related to protein tyrosine phosphatases. J Immunol. 1994;152:3183–3188. [PubMed] [Google Scholar]
- 12.Payton MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000-M(r) tryptic fragments of islet antigens in insulin-dependent diabetes to the protein tyrosine phosphatase-like molecule IA-2 (ICA512) J Clin Invest. 1995;96:1506–1511. doi: 10.1172/JCI118188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solimena M, Dirkx R Jr, Hermel JM, Pleasic-Williams S, Shapiro JA, Caron L, Rabin DU. ICA 512, an autoantigen of type I diabetes, is an intrinsic membrane protein of neurosecretory granules. Embo J. 1996;15:2102–2114. [PMC free article] [PubMed] [Google Scholar]
- 14.Atkinson MA, Maclaren NK. Islet cell autoantigens in insulin-dependent diabetes. J Clin Invest. 1993;92:1608–1616. doi: 10.1172/JCI116745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonifacio E, Lampasona V, Genovese S, Ferrari M, Bosi E. Identification of protein tyrosine phosphatase-like IA2 (islet cell antigen 512) as the insulin-dependent diabetes-related 37/40K autoantigen and a target of islet-cell antibodies. J Immunol. 1995;155:5419–5426. [PubMed] [Google Scholar]
- 16.Gardner SG, Gale EA, Williams AJ, Gillespie KM, Lawrence KE, Bottazzo GF, Bingley PJ. Progression to diabetes in relatives with islet autoantibodies. Is it inevitable? Diabetes Care. 1999;22:2049–2054. doi: 10.2337/diacare.22.12.2049. [DOI] [PubMed] [Google Scholar]
- 17.Franke B, Galloway TS, Wilkin TJ. Developments in the prediction of type 1 diabetes mellitus, with special reference to insulin autoantibodies. Diabetes Metab Res Rev. 2005;21:395–415. doi: 10.1002/dmrr.554. [DOI] [PubMed] [Google Scholar]
- 18.Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, Eisenbarth GS. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A. 2000;97:1701–1706. doi: 10.1073/pnas.040556697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonifacio E, Atkinson M, Eisenbarth G, Serreze D, Kay TW, Lee-Chan E, Singh B. International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes. 2001;50:2451–2458. doi: 10.2337/diabetes.50.11.2451. [DOI] [PubMed] [Google Scholar]
- 20.Bernard NF, Ertug F, Margolese H. High incidence of thyroiditis and anti-thyroid autoantibodies in NOD mice. Diabetes. 1992;41:40–46. doi: 10.2337/diab.41.1.40. [DOI] [PubMed] [Google Scholar]
- 21.Baxter AG, Mandel TE. Hemolytic anemia in non-obese diabetic mice. Eur J Immunol. 1991;21:2051–2055. doi: 10.1002/eji.1830210912. [DOI] [PubMed] [Google Scholar]
- 22.Carrillo J, Puertas MC, Alba A, Ampudia RM, Pastor X, Planas R, Riutort N, Alonso N, Pujol-Borrell R, Santamaria P et al. Islet-infiltrating B-cells in nonobese diabetic mice predominantly target nervous system elements. Diabetes. 2005;54:69–77. doi: 10.2337/diabetes.54.1.69. [DOI] [PubMed] [Google Scholar]
- 23.Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, Afifiyan F, Elford A, Jackowski G et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med. 2003;9:198–205. doi: 10.1038/nm818. [DOI] [PubMed] [Google Scholar]
- 24.Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, Roep BO. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345:1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 25.Kitamura D, Rajewsky K. Targeted disruption of mu chain membrane exon causes loss of heavy-chain allelic exclusion. Nature. 1992;356:154–156. doi: 10.1038/356154a0. [DOI] [PubMed] [Google Scholar]
- 26.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new "speed congenic" stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akashi T, Nagafuchi S, Anzai K, Kondo S, Kitamura D, Wakana S, Ono J, Kikuchi M, Niho Y, Watanabe T. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int Immunol. 1997;9:1159–1164. doi: 10.1093/intimm/9.8.1159. [DOI] [PubMed] [Google Scholar]
- 28.Wong FS, Visintin I, Wen L, Granata J, Flavell R, Janeway CA. The role of lymphocyte subsets in accelerated diabetes in nonobese diabetic-rat insulin promoter-B7-1 (NOD-RIP-B7-1) mice. J Exp Med. 1998;187:1985–1993. doi: 10.1084/jem.187.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noorchashm H, Lieu YK, Noorchashm N, Rostami SY, Greeley SA, Schlachterman A, Song HK, Noto LE, Jevnikar AM, Barker CF et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. 1999;163:743–750. [PubMed] [Google Scholar]
- 30.Yang M, Charlton B, Gautam AM. Development of insulitis and diabetes in B cell-deficient NOD mice. J Autoimmun. 1997;10:257–260. doi: 10.1006/jaut.1997.0128. [DOI] [PubMed] [Google Scholar]
- 31.Forsgren S, Andersson A, Hillorn V, Soderstrom A, Holmberg D. Immunoglobulin-mediated prevention of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Scand J Immunol. 1991;34:445–451. doi: 10.1111/j.1365-3083.1991.tb01567.x. [DOI] [PubMed] [Google Scholar]
- 32.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46:941–946. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 33.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–3918. [PubMed] [Google Scholar]
- 34.Greeley SA, Moore DJ, Noorchashm H, Noto LE, Rostami SY, Schlachterman A, Song HK, Koeberlein B, Barker CF, Naji A. Impaired activation of islet-reactive CD4 T cells in pancreatic lymph nodes of B cell-deficient nonobese diabetic mice. J Immunol. 2001;167:4351–4357. doi: 10.4049/jimmunol.167.8.4351. [DOI] [PubMed] [Google Scholar]
- 35.Wong FS, Wen L, Tang M, Ramanathan M, Visintin I, Daugherty J, Hannum LG, Janeway CA Jr, Shlomchik MJ. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53:2581–2587. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
- 36.Chiu PP, Serreze DV, Danska JS. Development and function of diabetogenic T-cells in B-cell-deficient nonobese diabetic mice. Diabetes. 2001;50:763–770. doi: 10.2337/diabetes.50.4.763. [DOI] [PubMed] [Google Scholar]
- 37.Charlton B, Zhang MD, Slattery RM. B lymphocytes not required for progression from insulitis to diabetes in non-obese diabetic mice. Immunol Cell Biol. 2001;79:597–601. doi: 10.1046/j.1440-1711.2001.01045.x. [DOI] [PubMed] [Google Scholar]
- 38.Wong S, Guerder S, Visintin I, Reich EP, Swenson KE, Flavell RA, Janeway CA Jr. Expression of the co-stimulator molecule B7-1 in pancreatic beta-cells accelerates diabetes in the NOD mouse. Diabetes. 1995;44:326–329. doi: 10.2337/diab.44.3.326. [DOI] [PubMed] [Google Scholar]
- 39.Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 40.Russell DM, Dembic Z, Morahan G, Miller JF, Burki K, Nemazee D. Peripheral deletion of self-reactive B cells. Nature. 1991;354:308–311. doi: 10.1038/354308a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silveira PA, Dombrowsky J, Johnson E, Chapman HD, Nemazee D, Serreze DV. B cell selection defects underlie the development of diabetogenic APCs in nonobese diabetic mice. J Immunol. 2004;172:5086–5094. doi: 10.4049/jimmunol.172.8.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 43.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 44.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, Howard M, Goodnow CC. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425–438. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodnow CC, Brink R, Adams E. Breakdown of self-tolerance in anergic B lymphocytes. Nature. 1991;352:532–536. doi: 10.1038/352532a0. [DOI] [PubMed] [Google Scholar]
- 47.Noorchashm H, Bui A, Li HL, Eaton A, Mandik-Nayak L, Sokol C, Potts KM, Pure E, Erikson J. Characterization of anergic anti-DNA B cells: B cell anergy is a T cell-independent and potentially reversible process. Int Immunol. 1999;11:765–776. doi: 10.1093/intimm/11.5.765. [DOI] [PubMed] [Google Scholar]
- 48.Kishimoto H, Sprent J. A defect in central tolerance in NOD mice. Nat Immunol. 2001;2:1025–1031. doi: 10.1038/ni726. [DOI] [PubMed] [Google Scholar]
- 49.Kreuwel HT, Biggs JA, Pilip IM, Pamer EG, Lo D, Sherman LA. Defective CD8+ T cell peripheral tolerance in nonobese diabetic mice. J Immunol. 2001;167:1112–1117. doi: 10.4049/jimmunol.167.2.1112. [DOI] [PubMed] [Google Scholar]
- 50.Ridgway WM, Ito H, Fasso M, Yu C, Fathman CG. Analysis of the role of variation of major histocompatibility complex class II expression on nonobese diabetic (NOD) peripheral T cell response. J Exp Med. 1998;188:2267–2275. doi: 10.1084/jem.188.12.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity. 2005;22:385–396. doi: 10.1016/j.immuni.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 52.Thomas JW, Hulbert C. Somatically mutated B cell pool provides precursors for insulin antibodies. J Immunol. 1996;157:763–771. [PubMed] [Google Scholar]
- 53.Rojas M, Hulbert C, Thomas JW. Anergy and not clonal ignorance determines the fate of B cells that recognize a physiological autoantigen. J Immunol. 2001;166:3194–3200. doi: 10.4049/jimmunol.166.5.3194. [DOI] [PubMed] [Google Scholar]
- 54.Acevedo-Suarez CA, Hulbert C, Woodward EJ, Thomas JW. Uncoupling of anergy from developmental arrest in anti-insulin B cells supports the development of autoimmune diabetes. J Immunol. 2005;174:827–833. doi: 10.4049/jimmunol.174.2.827. [DOI] [PubMed] [Google Scholar]
- 55.Watts C, West MA, Reid PA, Davidson HW. Processing of immunoglobulin-associated antigen in B lymphocytes. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1:345–352. doi: 10.1101/sqb.1989.054.01.042. [DOI] [PubMed] [Google Scholar]
- 56.Mamula MJ, Janeway CA Jr. Do B cells drive the diversification of immune responses? Immunol Today. 1993;14:151–152. doi: 10.1016/0167-5699(93)90274-O. [DOI] [PubMed] [Google Scholar]
- 57.Lin RH, Mamula MJ, Hardin JA, Janeway CA Jr. Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mamula MJ, Lin RH, Janeway CA Jr, Hardin JA. Breaking T cell tolerance with foreign and self co-immunogens. A study of autoimmune B and T cell epitopes of cytochrome c. J Immunol. 1992;149:789–795. [PubMed] [Google Scholar]
- 59.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 60.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol. 1998;161:1163–1168. [PubMed] [Google Scholar]
- 61.Hammond KJ, Kronenberg M. Natural killer T cells: natural or unnatural regulators of autoimmunity? Curr Opin Immunol. 2003;15:683–689. doi: 10.1016/j.coi.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 62.Bezbradica JS, Stanic AK, Matsuki N, Bour-Jordan H, Bluestone JA, Thomas JW, Unutmaz D, Van Kaer L, Joyce S. Distinct roles of dendritic cells and B cells in Va14Ja18 natural T cell activation in vivo. J Immunol. 2005;174:4696–4705. doi: 10.4049/jimmunol.174.8.4696. [DOI] [PubMed] [Google Scholar]
- 63.Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, Miura T, Haba T, Scherer DC, Wei J et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med. 2001;7:1052–1056. doi: 10.1038/nm0901-1052. [DOI] [PubMed] [Google Scholar]
- 64.Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, Kronenberg M, Koezuka Y, Delovitch TL, Gombert JM et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057–1062. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- 65.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 66.Hulbert C, Riseili B, Rojas M, Thomas JW. B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol. 2001;167:5535–5538. doi: 10.4049/jimmunol.167.10.5535. [DOI] [PubMed] [Google Scholar]
- 67.Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur J Immunol. 2002;32:3657–3666. doi: 10.1002/1521-4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 68.Chiu PP, Jevnikar AM, Danska JS. Genetic control of T and B lymphocyte activation in nonobese diabetic mice. J Immunol. 2001;167:7169–7179. doi: 10.4049/jimmunol.167.12.7169. [DOI] [PubMed] [Google Scholar]
- 69.Hussain S, Salojin KV, Delovitch TL. Hyperresponsiveness, resistance to B-cell receptor-dependent activation-induced cell death, and accumulation of hyperactivated B-cells in islets is associated with the onset of insulitis but not type 1 diabetes. Diabetes. 2004;53:2003–2011. doi: 10.2337/diabetes.53.8.2003. [DOI] [PubMed] [Google Scholar]
- 70.Hussain S, Delovitch TL. Dysregulated B7-1 and B7-2 expression on nonobese diabetic mouse B cells is associated with increased T cell costimulation and the development of insulitis. J Immunol. 2005;174:680–687. doi: 10.4049/jimmunol.174.2.680. [DOI] [PubMed] [Google Scholar]
- 71.Noorchashm H, Moore DJ, Noto LE, Noorchashm N, Reed AJ, Reed AL, Song HK, Mozaffari R, Jevnikar AM, Barker CF et al. Impaired CD4 T cell activation due to reliance upon B cell-mediated costimulation in nonobese diabetic (NOD) mice. J Immunol. 2000;165:4685–4696. doi: 10.4049/jimmunol.165.8.4685. [DOI] [PubMed] [Google Scholar]
- 72.Leijon K, Freitas A, Holmberg D. Analysis of VH gene utilisation in the non-obese diabetic mouse. Autoimmunity. 1993;15:11–18. doi: 10.3109/08916939309004834. [DOI] [PubMed] [Google Scholar]
- 73.Andersson A, Ekstrand-Hammarstrom B, Eriksson B, Overmo C, Holmberg D. Neonatal treatment with monoclonal natural antibodies restores a normal pattern of VH gene utilization in the non-obese diabetic mouse. Int Immunol. 1994;6:623–630. doi: 10.1093/intimm/6.4.623. [DOI] [PubMed] [Google Scholar]
- 74.Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–818. doi: 10.1016/0952-7915(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 75.Thomas JW, Kendall PL, Mitchell HG. The natural autoantibody repertoire of nonobese diabetic mice is highly active. J Immunol. 2002;169:6617–6624. doi: 10.4049/jimmunol.169.11.6617. [DOI] [PubMed] [Google Scholar]
- 76.Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Peritoneal B cells govern the outcome of diabetes in non-obese diabetic mice. Eur J Immunol. 2004;34:2387–2395. doi: 10.1002/eji.200324744. [DOI] [PubMed] [Google Scholar]
- 77.Woodward EJ, Thomas JW. Multiple germline kappa light chains generate anti-insulin B cells in nonobese diabetic mice. J Immunol. 2005;175:1073–1079. doi: 10.4049/jimmunol.175.2.1073. [DOI] [PubMed] [Google Scholar]
- 78.Greeley SA, Katsumata M, Yu L, Eisenbarth GS, Moore DJ, Goodarzi H, Barker CF, Naji A, Noorchashm H. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med. 2002;8:399–402. doi: 10.1038/nm0402-399. [DOI] [PubMed] [Google Scholar]
- 79.Kagohashi Y, Udagawa J, Abiru N, Kobayashi M, Moriyama K, Otani H. Maternal factors in a model of type 1 diabetes differentially affect the development of insulitis and overt diabetes in offspring. Diabetes. 2005;54:2026–2031. doi: 10.2337/diabetes.54.7.2026. [DOI] [PubMed] [Google Scholar]
- 80.Reijonen H, Daniels TL, Lernmark A, Nepom GT. GAD65-specific autoantibodies enhance the presentation of an immunodominant T-cell epitope from GAD65. Diabetes. 2000;49:1621–1626. doi: 10.2337/diabetes.49.10.1621. [DOI] [PubMed] [Google Scholar]
- 81.Simitsek PD, Campbell DG, Lanzavecchia A, Fairweather N, Watts C. Modulation of antigen processing by bound antibodies can boost or suppress class II major histocompatibility complex presentation of different T cell determinants. J Exp Med. 1995;181:1957–1963. doi: 10.1084/jem.181.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jaume JC, Parry SL, Madec AM, Sonderstrup G, Baekkeskov S. Suppressive effect of glutamic acid decarboxylase 65-specific autoimmune B lymphocytes on processing of T cell determinants located within the antibody epitope. J Immunol. 2002;169:665–672. doi: 10.4049/jimmunol.169.2.665. [DOI] [PubMed] [Google Scholar]
- 83.Banga JP, Moore JK, Duhindan N, Madec AM, van Endert PM, Orgiazzi J, Endl J. Modulation of antigen presentation by autoreactive B cell clones specific for GAD65 from a type I diabetic patient. Clin Exp Immunol. 2004;135:74–84. doi: 10.1111/j.1365-2249.2004.02343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fu YX, Huang G, Wang Y, Chaplin DD. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin alpha-dependent fashion. J Exp Med. 1998;187:1009–1018. doi: 10.1084/jem.187.7.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leget GA, Czuczman MS. Use of rituximab, the new FDA-approved antibody. Curr Opin Oncol. 1998;10:548–551. doi: 10.1097/00001622-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 86.Johnson P, Glennie M. The mechanisms of action of rituximab in the elimination of tumor cells. Semin Oncol. 2003;30:3–8. doi: 10.1053/sonc.2003.50025. [DOI] [PubMed] [Google Scholar]
- 87.Edwards JC, Leandro MJ, Cambridge G. B lymphocyte depletion in rheumatoid arthritis: targeting of CD20. Curr Dir Autoimmun. 2005;8:175–192. doi: 10.1159/000082103. [DOI] [PubMed] [Google Scholar]
- 88.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 89.Heelan BT, Tormey V, Amlot P, Payne E, Mehta A, Webster AD. Effect of anti-CD20 (rituximab) on resistant thrombocytopenia in autoimmune lymphoproliferative syndrome. Br J Haematol. 2002;118:1078–1081. doi: 10.1046/j.1365-2141.2002.03753.x. [DOI] [PubMed] [Google Scholar]
- 90.Hensel M, Ho AD. Successful treatment of a patient with hairy cell leukemia and pentostatin-induced autoimmune thrombocytopenia with rituximab. Am J Hematol. 2003;73:37–40. doi: 10.1002/ajh.10309. [DOI] [PubMed] [Google Scholar]
- 91.Kneitz C, Wilhelm M, Tony HP. Effective B cell depletion with rituximab in the treatment of autoimmune diseases. Immunobiology. 2002;206:519–527. doi: 10.1078/0171-2985-00200. [DOI] [PubMed] [Google Scholar]
- 92.Perrotta S, Locatelli F, La Manna A, Cennamo L, De Stefano P, Nobili B. Anti-CD20 monoclonal antibody (Rituximab) for life-threatening autoimmune haemolytic anaemia in a patient with systemic lupus erythematosus. Br J Haematol. 2002;116:465–467. [PubMed] [Google Scholar]
- 93.Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]