

Abstract
Functional polarization of T helper (Th) subsets of lymphocytes has been implicated in promoting or conferring risk to Type 1 diabetes mellitus (T1DM) development in human and diabetic animal models. It is assumed that an immoderate preponderance of type 1 immunity establishes the prerequisite for this development. Over the past years, various immune-intervention strategies have been tested to protect diabetic animals from developing overt diabetes. These protocols implicate a protective mechanism that is attributed to a change in the set of autoreactive Th cells from their Th1 to the Th2 phenotype. The studies were aimed at improving the effectiveness of Th2 cells to secrete the principal cytokines, IL-4 and IL-10, in order to mediate protection from diabetes in NOD mice. In contrast, some immune-modulation protocols utilizing non-specific reagents report that diabetes protection is apparently attributed to preferential survival of both Th1 and Th2 cells, rather than via a shift from their Th1 to Th2 phenotypes. Even though we know that excessive immune responses against self antigens are also controlled and terminated by regulatory T cells, this article focuses on the polarization of Th effector cells and discusses the controversial findings regarding the Th1/Th2 hypothesis to draw a conclusion on its relevance in T1DM from the existing knowledge.
Keywords: type 1 diabetes, Th cell, polarization
Introduction
The mammalian immune system fights against infectious agents by generating humoral (antibody) and cellular immune responses. The cellular arm of this defense system is equipped with effector and regulatory functions mediated by T lymphocytes of the CD4 and CD8 subsets and their secreted cytokines. Cytokines are substances composed of proteins or peptides that function as important inflammatory mediators, growth factors and chemical information transmitters among immune cells for coordinating immune responses. Information transmission through cytokines is achieved by binding to receptors carried on the surface of immune cells. The identification of antigen-specific murine CD4 T cell clones that secret non-overlapping patterns of cytokines has led to the sub-classification of CD4 cells into T-helper 1 (Th1) and Th2 categories, with Th1 cells predominantly producing Interleukin-2 (IL-2), interferon-γ (IFN-γ) and tumor necrosis factor-β (TNF-β). Th2 cells release the principal cytokines that include IL-4, IL-5 and IL-10 [1, 2]. The assumed differences in the cytokine profiles of each Th cell subset are heavily based on experiments in animal models and in vitro studies. Following these earlier in vitro discoveries, the functional aspects of Th1 and Th2 subsets of CD4 cells have then been shown to exist in vivo in both mice and humans [3, 4].
Antigen-specific Th1 and Th2 cells are derived from a naïve Th precursor (Thp) cell. Their differentiation is initiated via contact with an antigen-presenting cell (APC) that has taken up antigens, and present fragments of the respective antigen in association with membrane-bound major histocompatibility complex (MHC) class II molecules. Upon engagement of its T cell receptor (TCRαβ) with the antigenic peptide/MHC complex on the APC together with the required costimulation signal provided through ligation of its accessory molecules, such as CD28 with B7-1 and B7-2, Thp undergoes differentiation to become an uncommitted cell type designated as Th0 that produces IL-4 and IFN-γ [3, 5]. However, multiple cytokine-secreting activities of Th0 cells have been suggested to be attributed not to the clonal expansion of these cells but rather to a mixed population of differentiated Th cells that exhibit these capabilities [6]. For the Th0 cell, the cytokine environment around it then influences its development into either the Th1 or Th2 subset [3] (Figure 1). This process of Th cell differentiation into the committed types of Th cells is called polarization.
Figure 1. Th cell polarization.
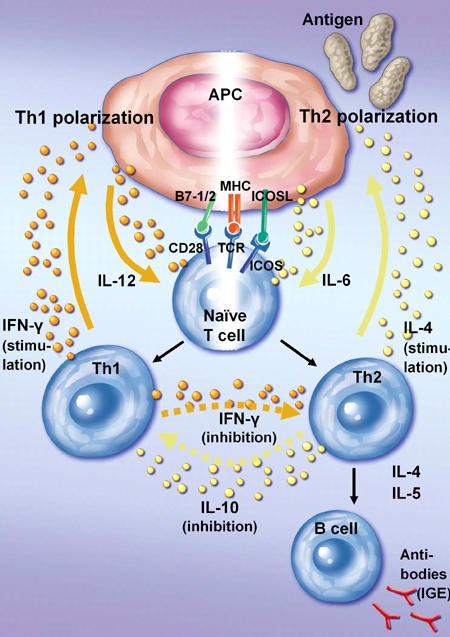
Polarization towards Th1 is achieved via antigen recognition of a naive T cell by its TCR together with a costimulation signal provided by B7-1 and B7-2. In contrast, Th2 activation heavily relies on costimulatory engagement of ICOS and ICOSL. APCs support Th1(Th2) cell polarization by releasing IL-12 (IL-6). Upon Th1 (Th2) cell activation these cells secrete IFN-γ (IL-4) to stimulate APCs to secrete more IL-12 (IL-6). IFN-γ secretion by Th1 cells also inhibits Th2 cell activation. Th2 cells in turn can secrete IL-10 to suppress the activation of Th1 cells. In addition, cytokine secretion by Th2 cells activates B cells to produce antibodies.
Preferential exposure to IFN-γ, and other type 1 cytokines, such as IL-12 and TNF-α produced by phagocytic cells, induces APCs to release more IL-12, which in turn initiates further Th1 polarization by an autocrine loop, whereas IL-4 produced by activated Th2 cells and mast cells directs polarized Th0 development into the Th2 type. Effector Th2 cells regulate the functions of other cell types, which include the help to antigen-specific B cells to produce antibodies (IgE) and to modulate the activities of mast cells and eosinophils. In contrast, by releasing IL-10, Th2 cells can also downregulate the production of IFN-γ by Th1 cells. In turn, the proliferation of Th2 cells may be inhibited by IFN-γ secreted by Th1 cells (Figure 1). Cytokines produced by Th1 and Th2 cells are thus antagonistic to each other.
After an effective response to foreign antigens the immune system normally returns to a state of rest, such that the number and functional status of the antigen-specific Th1 and Th2 cells are reset to approximately the preimmunization state. This process of immune balance maintenance is termed homeostasis [7]. In case of immune overreaction of T cells directed against self antigens, such as in T1DM, it is assumed that an imbalance in Th1/Th2 homeostasis contributes to the onset and progression of the disease [8, 9]. However, the Th1/Th2 hypotheses have been criticized, as increasing evidence has emerged that the interrelations of immune-responsible cells and the associated immunomodulatory molecules are far more complicated and interlocked than expressed by these simplistic stereotypes [10].
The relevance of the Th1/Th2 balance in T1DM
T1DM is an organ-specific autoimmune disease manifested by extensive T cell-mediated damage of pancreatic islet β-cells. Diseased pancreases of diabetic patients at the time of diagnosis are infiltrated by T lymphocytes of both CD4 and CD8 subsets [11, 12]. Within the roles of these cells in T1DM development, polarization of the Th cell-secreted cytokines has been suggested to contribute to disease development. In this regard, observation has been made from a few studies in human diabetics at disease onset that gross impairment of their Th2 function exists since their peripheral blood mononuclear cells (PBMC) produced decreased IL-4 levels in response to polyclonal activators, such as phytohemagglutinin and anti-CD3 antibodies [13]. The cytokine production capability of Th2 cells, which infiltrate the pancreas of diabetic patients, appears to be impaired as judged by the secretion of either IL-4 or IL-10. [14]. According to studies conducted in prediabetic patients involving high-risk first-degree relatives of diabetic children and recently diagnosed T1DM children, cellular responses against the islet cell antigen glutamic acid decarboxylase 65 (GAD65) are apparently deviated towards the Th1 phenotype [15, 16].
Studies conducted in different diabetic animal models have similarly demonstrated that autoreactive CD4 and CD8 participated in T1DM development [17-19]. Evidence that polarized Th function correlates with disease development has been reported in diabetes-prone NOD mice and BioBreeding (BB) rats [20-22]. A higher IFN-γ/IL-4 ratio is found to correlate with destructive insulitis, while a low ratio has been associated with non-destructive insulitis [23]. Recently, it has been demonstrated that the imbalance in Th1 and Th2 cytokine secretion actually contributes to the gender difference in T1DM development in NOD mice. T cells from disease-susceptible young female mice produce more IFN-γ (Th1) whilst those T cells from disease-resistant young male animals exhibit higher IL-4-secreting activity (Th2) [24].
Pancreatic Th1 vs. Th2 cytokine environment in T1DM development
Human pancreatic islet cells have been reported to exhibit differential responses upon exposure to Th1 and Th2 cytokines. In the presence of the principal Th1 cytokine, IFN-γ, human islet cells are found to release an increased level of nitric oxide [25], a reactive oxygen species (ROS) that can impair insulin secretion and cause β-cell dysfunction by antioxidant depletion of tissue cells during pancreatic inflammation [26]. ROS production in turn may also have a significant effect on macrophage vitality and function [27]. Murata and co-workers found that macrophages can directly induce polarization towards type 1 or 2 immunity by interacting as APCs with Th cells in response to their antioxidant level as judged by their inherent level of oxidized glutathione [28]. Macrophages with most of their glutathione in non-oxidized form tend to promote Th1 development, while those with more oxidized glutathione content preferentially set the Th cell to differentiate along the Th2 pathway [28].
To exploit the cytokine-created autocrine loop in Th2 cell polarization, several studies have been carried out in murine models to create a Th2 cytokine-enriched environment to halt T1DM development. Administration of exogenous IL-10 offers disease protection in NOD mice [29]. Interestingly, the source of IL-10 production has been reported to lead to different outcomes in disease development. Constitutive expression of the IL-10 transgene by pancreatic islet α- and β-cells as a means to create an IL-10-enriched environment in order to increase the differentiation of Th2 cells in the organ promoted accelerated inflammation and diabetes development [30, 31]. In another experimental set up, the IL-10 transgene was allowed to be expressed in Th2 cells under the control of the IL-4-specific transcription factor c-Maf, such that the cytokine is released only when the Th2 cells become activated. It turned out that IL-10-dependent induced inflammation of the pancreas was not found in the IL-10.NOD mice. On the other hand, IL-10 production alone by pancreatic Th2 cells has been noted to be ineffective in providing significant protection against T1DM onset in these animals [32].
The role of IL-4 in T1DM development has been the subject of extensive investigations. Similar to the exogenous administration of IL-10, IL-4 injection is reported to be capable of halting T1DM progression in NOD mice [33, 34]. Unlike the phenomenon observed in pancreatic IL-10-expressing transgenic mice, a constitutive release of IL-4 from pancreatic β-cells is able to prevent insulitis development in NOD-IL-4 transgenic mice [35]. A subsequent study demonstrated that this phenomenon was attributed to pancreas-infiltrated GAD65-specific Th2 cells that have acquired an enhanced capability to downregulate the diabetogenic activity of Th1 cells [36]. Interestingly, regulated IL-4 release by Th2 cells under the control of c-Maf has not been shown to be effective in protecting NOD mice from developing diabetes, despite being capable of halting disease progression in the Ins-HA/TCR-SFE and RIP-LCMV-NP diabetic mouse models [37].
Th1 to Th2 shift as an immune-intervention strategy for T1DM treatment?
Various immunomodulation protocols have shown effectivity in the treatment of T1DM in animal models. They involved immunosuppression by the administration of antilymphocyte serum, cyclosporine, and monoclonal antibodies to T cells [38], as well as the modulation of immune pathways by the application of non-specific immunostimulants [39, 40] and designed epitopes aimed at the tolerization of autoreactive T cells [41]. For the non-specific immunotherapeutic approaches, protective mechanisms can be attributed to the induction of Th2 cells that suppress autoreactive Th1 cells, possibly through a Th1 to Th2 shift mechanism. With respect to the T cell pool in the NOD mice, it is interesting to note that CD4 and CD8 cells of the animals have been reported to display resistance to activation-induced cell death (ACID) [42]. A more recent study showed that CD4 cells in NOD mice required a much higher threshold of stimulation through TCR signaling to undergo multiple cycles of cell division needed to differentiate into mature T cells before they die by ACID [43]. Depending on the immuno-stimulants that were used to induce a non-specific immunomodulation, protection against diabetes development in NOD mice could apparently be attributed to preferential ACID of autoreactive Th1 cells rather than through a Th1 to Th2 phenotype shift of the autoreactive T cells. In this light, existing islet Th2 cells may be able to mediate prevention of T1DM by downregulating surviving diabetogenic Th1 cells [44].
Conclusion
The Th1/Th2 hypothesis continues to be an attractive model to explain risk and show resistance to autoimmune diabetes development in both human and diabetic animal models. It appears to be important to establish the Th2 cytokine environment that is needed in the pancreas to deviate the diabetogenic function mediated by autoreactive Th1 cells to their protective Th2 phenotype. In this context, it is interesting that different cellular sources of the principal cytokines secreted by pancreatic islet and Th2 cells could generate different outcomes in protecting NOD mice from developing their spontaneous diabetes. Recent understanding in cell biology of CD4 cell subsets, and studies conducted in the NOD mice, have suggested that autoreactive Th1 and Th2 cells exhibit differential requirements for their activation, cell cycle progression to attain maturity, and subsequently die by ACID or switch to become memory cells. These observations have generated new interpretations for protection that is seen with immune-intervention protocols in diabetic animal models using non-specific immunostimulants. This knowledge may open up to the development of other novel immune-modulatory regimens for the future treatment of T1DM.
References
- 1.Kim J, Woods A, Becker-Dunn E, Bottomly K. Distinct functional phenotypes of cloned 1a-restricted helper T cells. J Exp Med. 1985;162:188–201. doi: 10.1084/jem.162.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mossman TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. 1. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 3.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 4.Romagnani S. Lymphokine production by human T cells in disease states. Ann Rev Immunol. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura T, Kamogawa Y, Bottomly K, Flavell RA. Polarization of IL-4- and IFN-gamma-producing CD4+ T cells following activation of naive CD4+ T cells. J Immunol. 1997;158:1085–1094. [PubMed] [Google Scholar]
- 6.Bucy RP, Karr L, Huang G, Li J, Carter D, Honjo K, Lemons JA, Murphy KM, Weaver CT. Single cell analysis of cytokine gene coexpression during CD4+ T cell phenotype development. Proc Natl Acad Sci U S A. 1995;92:7565–7569. doi: 10.1073/pnas.92.16.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbas AK, Murphy KM and Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 8.Lafaille JJ. The role of helper T cell subsets in autoimmune diseases. Cytokine Growth Factor Rev. 1998;9(2):139–151. doi: 10.1016/s1359-6101(98)00009-4. [DOI] [PubMed] [Google Scholar]
- 9.Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM: therapeutic intervention by immunostimulation? Diabetes. 1994;43:613–621. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- 10.Bot A, Smith KA, von Herrath M. Molecular and cellular control of T1/T2 immunity at the interface between antimicrobial defense and immune pathology. DNA Cell Biol. 2004;23(6):341–350. doi: 10.1089/104454904323145227. [DOI] [PubMed] [Google Scholar]
- 11.Hanninen A, Jalkanen S, Salmi M, Toikkanen S, Nikolakaros G, Simell O. Macrophages, T cell receptor usage, and endothelial cell activation in the pancreas at the onset of insulin-dependent diabetes mellitus. J Clin Invest. 1992;90:1901–1910. doi: 10.1172/JCI116067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imagawa A, Hanafusa T, Tamura S, Moriwaki M, Itoh N, Yamamoto K, Iwahashi H, Yamagata K, Waguri M, Nanmo T et al. Pancreatic biopsy as a procedure for detecting the in situ autoimmune phenomenon in type 1 diabetes. Diabetes. 2001;50:1269–1273. doi: 10.2337/diabetes.50.6.1269. [DOI] [PubMed] [Google Scholar]
- 13.Berman MA, Sandborg CI, Wang Z, Imfeld KL, Zaldivar F Jr, Dadufalza V, Buckingham BA. Decreased IL-4 production in new onset type 1 insulin-dependent diabetes mellitus. J Immunol. 1996;157(10):4690–4696. [PubMed] [Google Scholar]
- 14.Farilla L, Dotta F, Di Mario U, Rapoport B, McLachlan SM. Presence of interleukin 4 or interleukin 10, but not both cytokines, in pancreatic tissue of two patients with recently diagnosed diabetes mellitus type I. Autoimmunity. 2000;32(3):161–166. doi: 10.3109/08916930008994088. [DOI] [PubMed] [Google Scholar]
- 15.Karlsson MG, Lawesson SS, Ludvigsson J. Th1-like dominance in high-risk first-degree relatives of type I diabetic patients. Diabetologia. 2000;43(6):742–749. doi: 10.1007/s001250051372. [DOI] [PubMed] [Google Scholar]
- 16.Karlsson Faresjo MG, Ernerudh J, Ludvigsson J. Cytokine profile in children during the first 3 months after the diagnosis of type 1 diabetes. Scand J Immunol. 2004;59(5):517–526. doi: 10.1111/j.0300-9475.2004.01420.x. [DOI] [PubMed] [Google Scholar]
- 17.Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Ly2+ T cells. J Exp Med. 1987;166:823–832. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller BJ, Appel MC, O'Neil JJ, Wicker LS. Both the Lyt-2 and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- 19.Chrsitianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T cells from diabetic versus prediabetic NOD.NON-thy-1a donors. Diabetes. 1993;43:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 20.Hirai H, Kaino K, Ito T, Kida K. Analysis of cytokine mRNA expression in pancreatic islets of nonobese diabetic mice. J Pediatr Endocrinol Metab. 2000;13(1):91–98. doi: 10.1515/jpem.2000.13.1.91. [DOI] [PubMed] [Google Scholar]
- 21.Rabinovitch A, Suarez-Pinzon WL, Sorensen O, Bleackley RC, Power RF. IFN-gamma gene expression in pancreatic islet-infiltrating mononuclear cells correlates with autoimmune diabetes in nonobese diabetic mice. J Immunol. 1995;154:4874–4882. [PubMed] [Google Scholar]
- 22.Rabinovitch A, Suarez-Pinzon WL, El-Sheikh A, Sorensen O, Power RF. Cytokine gene expression in pancreatic islet-infiltrating leukocytes of BB rats: expression of Th1 cytokines correlates with beta-cell destructive insulitis and IDDM. Diabetes. 1996;45:749–754. doi: 10.2337/diab.45.6.749. [DOI] [PubMed] [Google Scholar]
- 23.Kolb H. Benign vs destructive insulitis. Diabetes Metab Rev. 1997;13:139–146. doi: 10.1002/(sici)1099-0895(199709)13:3<139::aid-dmr190>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Bao M, Yang Y, Jun HS, Yoon JW. Molecular mechanisms for gender differences in susceptibility to T cell-mediated autoimmune diabetes in non-obese diabetic mice. J Immunol. 2002;168:5369–5375. doi: 10.4049/jimmunol.168.10.5369. [DOI] [PubMed] [Google Scholar]
- 25.Marselli L, Dotta F, Piro S, Santangelo C, Masini M, Lupi R, Realacci M, del Guerra S, Mosca F, Boggi U, Purrello F, Navalesi R, Marchetti P. Th2 cytokines have a partial, direct protective effect on the function and survival of isolated human islets exposed to combined proinflammatory and Th1 cytokines. J Clin Endocrinol Metab. 2001;86(10):4974–4978. doi: 10.1210/jcem.86.10.7938. [DOI] [PubMed] [Google Scholar]
- 26.Laybutt DR, Kaneto H, Hasenkamp W, Grey S, Jonas JC, Sgroi DC, Groff A, Ferran C, Bonner-Weir S, Sharma A, Weir GC. Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to beta-cell survival during chronic hyperglycemia. Diabetes. 2002;51:413–423. doi: 10.2337/diabetes.51.2.413. [DOI] [PubMed] [Google Scholar]
- 27.Murata Y, Amao M, Hamuro J. Sequential conversion of the redox status of macrophages dictates the pathological progression of autoimmune diabetes. Eur J Immunol. 2003;33(4):1001–1011. doi: 10.1002/eji.200323575. [DOI] [PubMed] [Google Scholar]
- 28.Murata Y, Shimamura T, Hamuro J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int Immunol. 2002;14(2):201–212. doi: 10.1093/intimm/14.2.201. [DOI] [PubMed] [Google Scholar]
- 29.Pennine KL, Roque-Gaffney E. Monahan M. Recombinant human IL-10 prevents the onset of diabetes in the NOD mouse. J Immunol Immunopathol. 1995;71:169–175. doi: 10.1006/clin.1994.1068. [DOI] [PubMed] [Google Scholar]
- 30.Wogensen L, Haung X, Sarvetnick N. Leucocyte extravasation into the pancreatic tissue in transgenic mice expressing interleukin 10 in the islets of Langerhans. J Exp Med. 1993;178:175–185. doi: 10.1084/jem.178.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortani M, Yoshimoto K, Tashiro F, Hashimoto C, Miyazaki JI, Ii S, Kudo E, Iwahana H, Hayashi Y, Sano T, Itakura M. Transgenic expression of IL-10 in pancreatic islet A cells accelerates autoimmune insulitis and diabetes in non-obese diabetic mice. Nature Immunol. 1994;6:1927–1936. doi: 10.1093/intimm/6.12.1927. [DOI] [PubMed] [Google Scholar]
- 32.Pauza ME, Neal H, Hagenbaugh A, Hilde C, David L. T-cell production of an inducible interleukin-10 transgene provides limited protection from autoimmune diabetes. Diabetes. 1999;48:1948–1953. doi: 10.2337/diabetes.48.10.1948. [DOI] [PubMed] [Google Scholar]
- 33.Cameron MJ, Arreaza GA, Zucker P, Chensue SW, Strieter RM, Chakrabarti S, Delotvitch TL. IL-4 prevents insulitits and insulin-dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper-2 cell function. J Immunol. 1997;159:4686–4692. [PubMed] [Google Scholar]
- 34.Rapoport MJ, Jaramillo A, Zipris D, Lazarus AH, Serreze DV, Leiter EH, Cyopick P, Danska JS, Delovitch TL. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med. 1993;178(1):87–99. doi: 10.1084/jem.178.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mueller R, Krahl T, Sarvetnick N. Pancreatic expression of interleukin-4 abrogate insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 1996;184:1093–1099. doi: 10.1084/jem.184.3.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallichan WS, Blasa B, Davies JD, Sarvetnick N. Pancreatic IL-4 expression results in islet-reactive Th2 cells that inhibit diabetogenic lymphocytes in the nonobese diabetic mouse. J Immunol. 1999;163:1696–1703. [PubMed] [Google Scholar]
- 37.Pauza ME, Nguyen A, Wolfe T, Ho IC, Glimcher LH, von Herrath M, Lo D. Variable effects of transgenic c-Maf on autoimmune disbetes. Diabetes. 2001;50:39–46. doi: 10.2337/diabetes.50.1.39. [DOI] [PubMed] [Google Scholar]
- 38.Rossini AA, Greiner DL, Friedman HP, Mordes JP. Immunopathogenesis of diabetes mellitus. Diabetes Rev. 1993;1:43–75. [Google Scholar]
- 39.Shehadeh NN, LaRosa F and Lafferty KJ. Altered cytokine activity in adjuvant inhibition of autoimmune diabetes. J Autoimmun. 1993;6(3):291–300. doi: 10.1006/jaut.1993.1025. [DOI] [PubMed] [Google Scholar]
- 40.Singh B, Rabinovitch A. Influence of microbial agents on the development of autoimmune diabetes. Autoimmunity. 1993;15:209–213. doi: 10.3109/08916939309019929. [DOI] [PubMed] [Google Scholar]
- 41.Harrison LC, Hafler DA. Antigen-specific therapy for autoimmune disease. Curr Opin Immunol. 2000;12(6):704–711. doi: 10.1016/s0952-7915(00)00166-7. [DOI] [PubMed] [Google Scholar]
- 42.Leijon K, Hammastrom B, Holmberg D. Non-obese diabetic (NOD) mice display enhanced immune responses and prolonged survival of lymphoid cells. Int Immunol. 1994;6(2):339–345. doi: 10.1093/intimm/6.2.339. [DOI] [PubMed] [Google Scholar]
- 43.Noorchashm H, Moore DJ, Noto LE, Noorchashm N, Reed AJ, Reed AL, Song HK, Mozaffari R, Jevnikar AM, Barker CF, Naji A. Impaired CD4 T cell activation due to reliance upon B cell-mediated costimulation in nonobese diabetic (NOD) mice. J Immunol. 2000;165:4685–4696. doi: 10.4049/jimmunol.165.8.4685. [DOI] [PubMed] [Google Scholar]
- 44.Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez-Pinzon WL, Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol. 2001;166:1352–1359. doi: 10.4049/jimmunol.166.2.1352. [DOI] [PubMed] [Google Scholar]