

Abstract
Activating mutations in the KCNJ11 gene encoding the Kir6.2 subunit of ATP-sensitive potassium channel have been described in patients with permanent neonatal diabetes mellitus (PNDM). The main pathophysiological feature of PNDM associated with Kir6.2 mutations is a profound defect in insulin secretion. However, the expression of Kir6.2 protein is not limited to β-cells; it also includes skeletal muscles, heart, brain, and peripheral nerves. Thus, the hypothesis that Kir6.2 mutations may influence insulin sensitivity in humans seems justified. Moreover, this notion is additionally supported by an animal model of Kir6.2 knock-out mice. Four adult carriers of a Kir6.2 mutation from the Polish population (mean age 31.5 years, range 20-50) were available for this study that aimed to evaluate their insulin sensitivity by the hyperinsulinemic euglycemic clamp technique. Three subjects carried the R201H mutation and one patient was a carrier of the K170N mutation. In addition, eight healthy volunteers with normal glucose tolerance were examined for comparison (mean age 31.0 years, range 20-41). The mean M value, i.e. the amount of metabolized glucose, for PNDM cases equaled 4.49 mg/(kg x min) (range 2.76-6.66) and was significantly lower than in the control group (9.64 mg/(kg x min), range 4.59-18.00). This observation suggests that impaired insulin sensitivity, in addition to profoundly decreased insulin secretion, contributes to the clinical picture of PNDM resulting from mutations in the Kir6.2 gene. An additional factor that might influence insulin sensitivity in our diabetes patients is glucose toxicity that may have appeared due to poor metabolic control prior to the examination (mean HbA1c = 8.95%). The intriguing question to be answered in the future is whether an improvement in insulin action could be seen following the transfer of Kir6.2 mutation carriers to sulphonylurea compounds.
Keywords: neonatal diabetes, mutation, Kir6.2 gene, insulin sensitivity
Introduction
Recently, substantial progress has been made in understanding the molecular background of permanent neonatal diabetes mellitus (PNDM). Almost half of the patients diagnosed with diabetes before 6 months of age have a mutation in the KCNJ11 gene encoding the Kir6.2 subunit of the ATP-sensitive potassium channel [1]. This protein is expressed in β-cells and also in some other tissues, such as skeletal muscles, heart, peripheral nerves, and brain. These mutations cause early onset diabetes by the reduction of potassium channel sensitivity to ATP. This results in a permanent activation of the channel and its opening causing cellular membrane hyperpolarization. In this state, insulin secretion by β-cells is almost completely suppressed. Thus, it is not surprising that the Kir6.2 mutation carriers are characterized by insulin deficiency, which is usually very profound, and that they are clinically insulin-dependent, hence treated with insulin from disease onset [2]. The birth weight of subjects with Kir6.2 mutations is low and they are frequently prone to ketoacidosis. While the clinical picture may imitate early onset type 1 diabetes, the autoimmunological markers are not present. In 25% of patients with Kir6.2 mutations, features such as muscle weakness, developmental delay and epilepsy may be present in addition to diabetes. These extra-pancreatic symptoms reflect the pattern of tissue expression of the Kir6.2 protein. Interestingly, the initial reports showed that PNDM associated with Kir6.2 responded very well to sulphonylurea agents [2, 3]. Sulfonylurea derivatives bind to the potassium channels causing their closure and improve responsiveness to glucose [2]. It has not yet been determined whether the symptoms related to other organs, such as epilepsy or psychomotorical delay, can be also improved by the action of these compounds [2].
While there is little doubt that the major mechanism of PNDM associated with the Kir6.2 gene is a profound defect in insulin secretion, no data is available on the influence of its mutations on insulin sensitivity. The hypothesis that Kir6.2 mutations may indeed influence insulin action is justified by the fact that this protein is expressed in skeletal muscles and the heart [2]. Thus, certain severe mutations that cause apparent muscle weakness, as well as those not associated with such clinical presentation, may influence glucose uptake and alter insulin sensitivity. This notion is further supported by the animal model, in which Kir6.2 knock-out mice show increased glucose uptake by skeletal muscles and adipose tissue [4]. One may speculate that the activating mutations in humans could potentially have the opposite effect. It should also be pointed out that some monogenic forms of diabetes resulting primarily from a severe β-cell defect, for example MODY5 that is a consequence of HNF1β mutations, are characterized by altered insulin sensitivity [5]. The aim of this study was to evaluate insulin sensitivity in adult Kir6.2 mutation carriers with PNDM prior to their transfer to sulphonylurea agents.
Methods
The Nationwide Registry was established in 2005 for the task of dissecting the genetic background of PNDM in Poland. So far, 33 subjects have been included, all diagnosed before the end of 6 months of age (range 1-26 weeks). This constitutes one of the world’s largest national PNDM collections. To date, we have examined the KCNJ11 gene in 25 insulin-treated, diabetic patients (age at examination: 1-50 years) by automatic sequencing. So far, we have identified eleven patients with heterozygous missense diabetes causing mutations (M.T. Malecki, unpublished data). Three adult patients with R210H, two females and one male (Pol13, Pol14, and Pol19, respectively), and one female with K170N (Pol17), were available for examination of insulin sensitivity by body glucose uptake measured by the hyperinsulinemic euglycemic clamp technique as described elsewhere [6]. No evidence of additional extra-pancreatic features was detected on physical and neurological examination of the patients. For comparison, we performed hyperinsulinemic euglycemic clamp in eight adult non-obese, and non-elderly, volunteers with normal glucose tolerance as confirmed by the WHO test. All the study subjects were Caucasian residents of Poland. The clinical characteristics of both study groups is presented in Table 1. All participants gave their informed consent. The study protocol was approved by the Ethical Committee of the Jagiellonian University, Medical College. The project was conducted according to the rules of Helsinki Declaration as revised in 2000.
Table 1. Clinical characteristics of the study groups and the results of hyperinsulinemic euglycemic clamp.
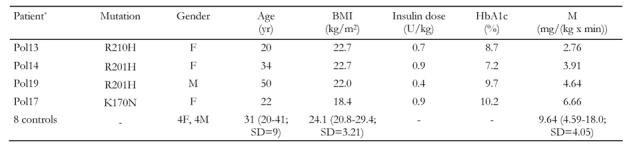
For controls the mean values, range and standard deviations for age, BMI, and M parameters are shown. BMI: body mass index. M: metabolized glucose. * Patient’s no. in the Polish PNDM registry.
All diabetic patients were on insulin from diagnosis until the study entry. Pol13 and Pol17 were treated with an intensive regimen of multiple daily insulin injections. Pol14 was on two daily injections of premix, biphasic human insulin. Pol19 was treated with one daily injection of mixed intermediate and long-acting porcine insulin. The clamp study was performed in the Department of Metabolic Diseases, Jagiellonian University, Krakow, Poland. All individuals were examined after at least eight hours of overnight fasting. The diabetic patients were hospitalized one day before the examination. They did not receive their morning dose of insulin prior to the clamp. Control individuals were admitted in the morning on the day of examination. The left antecubital veins were catheterized for infusions and the right forearms were placed in a heated cuff to obtain arterialized venous blood samples from the right antecubital veins. Blood samples were taken every 5 minutes. A primed, continuous insulin infusion, adjusted for the total body surface area, was started in order to achieve stable plasma insulin concentrations. Blood glucose concentrations of 5.0 mmol/l were maintained by a variable rate of 20% glucose infusion according to the Furler algorithm [7]. The equilibration period was at least 30 minutes. M values were calculated during the last 60 minutes of the clamp, when plasma glucose levels were maintained at 5.0 mmol/l. Comparisons between groups were made using the Student's t-test for independent variables. Homogeneity of variances was tested with Levene's test, and normality was tested with Shapiro-Wilk's W test. A value of p < 0.05 was considered statistically significant.
Results and Discussion
M values of the patients, as well as the control group, are shown in Table 1. Mean M in the PNDM subjects was 4.49 mg/(kg x min). This value in the controls reached 9.64 mg/(kg x min) and it was significantly higher than in the patient group (p = 0.04). Moreover, for all mutation carriers, the M value was below the mean for the controls (range 2.76 to 6.66 mg/(kg x min)). Interestingly, the K170N mutation carrier had better insulin sensitivity than all other diabetic subjects who carried the most common Kir6.2 mutation, which is an R201H substitution.
Our initial report constitutes the first description of insulin sensitivity in subjects with PNDM due to mutations in the Kir6.2 gene. We found that the insulin sensitivity was lower in the adult PNDM subjects than in healthy adult, non-obese, and non-elderly volunteers from the same Polish population. It should be pointed out that the M values measured in our control group correspond with the levels described in healthy, young subjects from other populations [8]. The K170N mutation carrier had a higher M value than any R201H mutation carrier examined in this study. While the nature of the mutations may influence, not only the degree of β-cell dysfunction, but also insulin resistance, there are other factors that could have contributed to this observation such as low BMI (18.4 kg/m2) and the young age of this subject. The molecular basis for decreased insulin sensitivity in carriers of activating KCNJ11 mutation is uncertain. In the animal-model study, lack of a functional Kir6.2 subunit resulted in an increased response to insulin in muscle and fatty cells [4]. This finding seems to be in line with the results of our report in humans and, taken together, both studies support the hypothesis that the Kir6.2 mutations may directly alter peripheral insulin sensitivity.
There are a few shortcomings of our study which should be discussed. First, the number of subjects included in our assessment was low. It should be pointed out, however, that PNDM associated with Kir6.2 mutations is a very rare phenomenon. In fact, we included in this study all adult patients with this form of diabetes identified so far in the Polish population (M. T. Malecki, unpublished data). Perhaps one should also mention the possible influence of glucose toxicity on the peripheral insulin action in our patients, since, as shown in Table 1, none of them had well-controlled diabetes at the time of examination (HbA1c ranging from 7.2 to 10.2%). The insulin sensitivity in type 1 diabetes subjects with poor metabolic control, as assessed by hyperinsulinemic euglycemic clamp, was previously reported to be similar to that in our PNDM group [9]. The phenomenon of the presence of insulin resistance and its subsequent normalization following intensification of insulin therapy in type 1 diabetes is well described [10-12]. Poorly controlled diabetes could therefore have contributed to the moderate insulin resistance in the examined PNDM subjects. Thus, we can not ascribe unequivocally the decreased insulin sensitivity solely to the direct influence of an altered potassium channel function in our PNDM patients. To confirm the insulin resistance in PNDM associated with Kir6.2 and to explain its pathogenesis, further investigations are needed. There are intriguing questions to be answered in the future, for example, whether the improvement in insulin action occurs following the transfer of Kir6.2 mutation carriers to sulphonylurea compounds. Furthermore, it would be interesting to assess insulin sensitivity in subjects with severe Kir6.2 mutations that involve muscles and other organs.
Acknowledgments
This study was supported by the Polish Ministry of Education and Science (Grant No. 2 P0E 136 29) and funds from the Jagiellonian University, Medical College (Grant 501/NKL/59/L).
References
- 1.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 2.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54:2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 3.Klupa T, Edghill EL, Nazim J, Sieradzki J, Ellard S, Hattersley AT, Malecki MT. The identification of a R201H mutation in KCNJ11, which encodes Kir6.2, and successful transfer to sustained-release sulphonylurea therapy in a subject with neonatal diabetes: evidence for heterogeneity of beta cell function among carriers of the R201H mutation. Diabetologia. 2005;48:1029–1031. doi: 10.1007/s00125-005-1731-5. [DOI] [PubMed] [Google Scholar]
- 4.Miki T, Minami K, Zhang L, Morita M, Gonoi T, Shiuchi T, Minokoshi Y, Renaud JM, Seino S. ATP-sensitive potassium channels participate in glucose uptake in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2002;283:E1178–E1184. doi: 10.1152/ajpendo.00313.2002. [DOI] [PubMed] [Google Scholar]
- 5.Brackenridge A, Pearson ER, Shojaee-Moradie F, Hattersley AT, Russell-Jones D, Umpleby AM. Contrasting insulin sensitivity of endogenous glucose production rate in subjects with hepatocyte nuclear factor-1beta and -1alpha mutations. Diabetes. 2006;55:405–411. doi: 10.2337/diabetes.55.02.06.db05-1019. [DOI] [PubMed] [Google Scholar]
- 6.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 7.Furler SM, Zelenka GS, Kraegen EW. Development and testing of a simple algorithm for a glucose clamp. Med Biol Eng Comput. 1986;24:365–370. doi: 10.1007/BF02442689. [DOI] [PubMed] [Google Scholar]
- 8.Monzillo LU, Hamdy O. Evaluation of insulin sensitivity in clinical practice and in research settings. Nutr Rev. 2003;61:397–412. doi: 10.1301/nr.2003.dec.397-412. [DOI] [PubMed] [Google Scholar]
- 9.DelPrato S, Nosadini R, Tiengo A, Tessari P, Avogaro A, Trevisan R, Valerio A, Muggeo M, Cobelli C, Toffolo G. Insulin-mediated glucose disposal in type I diabetes: evidence for insulin resistance. J Clin Endocrinol Metab. 1983;57:904–910. doi: 10.1210/jcem-57-5-904. [DOI] [PubMed] [Google Scholar]
- 10.Nijs HG, Radder JK, Frolich M, Krans HM. Insulin action is normalized in newly diagnosed type I diabetic patients after three months of insulin treatment. Metabolism. 1988;37:473–478. doi: 10.1016/0026-0495(88)90049-2. [DOI] [PubMed] [Google Scholar]
- 11.Linn T, Ortac K, Laube H, Federlin K. Intensive therapy in adult insulin-dependent diabetes mellitus is associated with improved insulin sensitivity and reserve: a randomized, controlled, prospective study over 5 years in newly diagnosed patients. Metabolism. 1996;45:1508–1513. doi: 10.1016/s0026-0495(96)90180-8. [DOI] [PubMed] [Google Scholar]
- 12.Lager I, Lonnroth P, von Schenck H, Smith U. Reversal of insulin resistance in type I diabetes after treatment with continuous subcutaneous insulin infusion. Br Med J (Clin Res Ed) 1983;287:1661–1664. doi: 10.1136/bmj.287.6406.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]