

Introduction
Type 2 diabetes is a complex disease mainly characterized by impaired insulin action and insulin secretion [1, 2]. A major breakthrough in the understanding of mechanisms leading to type 2 diabetes has recently come from studies on the central nervous system (CNS) dependent regulation of glucose and fat metabolism. The concept that the CNS has an essential role in glucose homeostasis and insulin action is not new. In 1849, Claude Bernard reported that pricking the floor of the fourth ventricle in rabbits produced hyperglycemia [3].
The past decade has greatly increased knowledge about the ability of the CNS to regulate food intake, body weight and glucose homeostasis. It is now clear that the CNS, and in particular the hypothalamus, plays a pivotal role in regulating glucose homeostasis independently of its effects on body weight. Therefore, some of the molecular defects underlying type 2 diabetes may reside in the CNS, supporting the concept that type 2 diabetes is, at least in part, a hypothalamic disorder.
Central insulin actions
The effects of insulin on hepatic glucose fluxes may be divided into (i) direct effects on the liver mostly leading to rapid inhibition of glycogenolysis [4], and (ii) indirect effects mediated through extrahepatic actions such as the inhibition of lipolysis and reduction of glucagon levels [5-8]. More recently, it has been proposed that indirect effects of insulin on glucose production also include the activation of hypothalamic insulin signaling [9]. Insulin can act in the hypothalamus to modulate feeding behavior [10-12], neuropeptide Y expression [13, 14], hypoglycemia counter-regulation [15, 16] and autonomic outflow [17, 18]. These findings suggest that the brain is an insulin target with regard to energy homeostasis in mammals.
The activation of insulin receptors in the hypothalamus, in particular in the arcuate nucleus (ARC), plays an important role in the regulation of glucose homeostasis (Figure 1). Using pharmacological and molecular manipulations of the hypothalamic insulin action, Obici and co-workers demonstrated that the activation of insulin signaling in the ARC in the absence of elevated systemic insulin levels, is sufficient to decrease blood glucose levels via a substantial inhibition of endogenous glucose production [9]. Conversely, the blockade of insulin action in the ARC by insulin antibodies, or by decreasing insulin receptors by antisense oligonucleotides, or by inhibiting the insulin-dependent activation of phosphatidylinositol-3-kinase (PI3K), leads to a reduced ability of circulating insulin to suppress endogenous glucose production. Overall, these findings suggest that hypothalamic action of insulin is required for the full inhibitory effect of systemic insulin on endogenous glucose production and requires an intact insulin signaling cascade involving the activation of the insulin receptor, insulin receptor substrate (IRS), and PI3K [9, 19].
Figure 1.
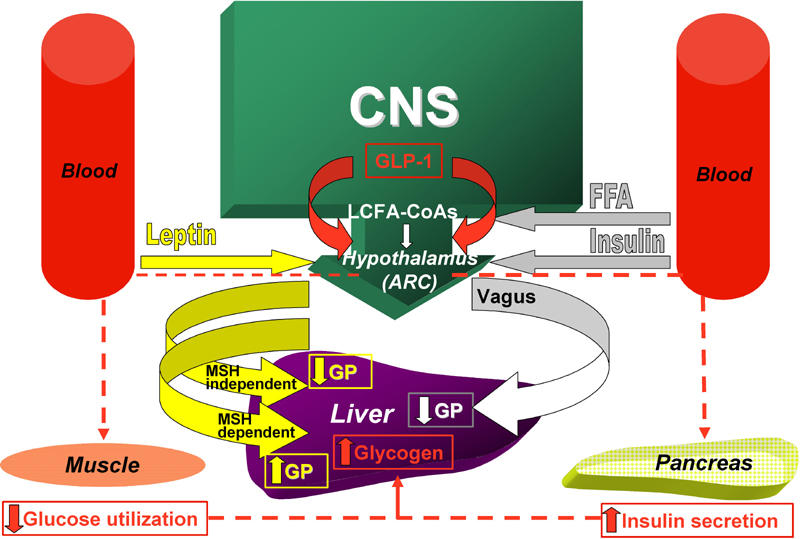
The activation of insulin signaling in hypothalamic arcuate nucleus (ARC) is sufficient to decrease blood glucose levels via the inhibition of hepatic glucose production (GP). This effect is mediated by the activation of efferent vagal fibers which innervate the liver. The effects of central leptin on glucose homeostasis involve both melanocortin-dependent and melanocortin-independent pathways. Acute activation of hypothalamic melanocortin receptors, results in an increased rate of hepatic gluconeogenesis. The effect of central leptin on the suppression of glucose production and glycogenolysis is mediated by the stimulation of melanocortin-independent pathways involving insulin-like pathways such as the PI3K signaling cascade. Glucagon-like peptide-1 (GLP-1) is produced in a discrete set of hindbrain neurons that project to a specific population of GLP-1 receptor-containing cells in the hypothalamus. During hyperglycemia following a meal, GLP-1 inhibits muscle glucose utilization and increases insulin secretion to favor hepatic glycogen storage. The intracellular pool of long-chain fatty acids (LCFA-CoAs) is under the control of several biochemical events including a free fatty acids (FFA) flux. Accumulation of LCFA-CoAs reduces hepatic glucose production.
Insulin acts in the ARC hyperpolarizing rat hypothalamic glucose-responsive neurons by opening and activating ATP-sensitive potassium channels (KATP channels) [20] and this is per se sufficient to lower blood glucose via inhibition of hepatic glucose output and gluconeogenesis [19]. Delivery of a KATP channel blocker to the ARC, such as glybenclamide, abolishes the central effects of insulin on endogenous glucose production, and prevents, in part, the suppression of endogenous glucose production by circulating insulin [9, 19].
Pocai and co-workers showed that insulin acts on KATP channels in hypothalamic neurons to control hepatic glucose production by decreasing glucose-6-phosphatase and phosphoenolpyruvate kinase expression in the liver [19]. The sulfonylurea receptor subunit (SUR1) of the KATP channels is expressed in the mediobasal hypothalamus and is required for the assembly of the associated pore-forming subunits (Kir6x) [21]. Interestingly, in SUR1 null mice, the ability of insulin to suppress hepatic gluconeogenesis and endogenous glucose production is impaired [19].
Overall, these data indicate that the central effects of insulin on the suppression of endogenous glucose production are mediated by the activation of IRS, PI3K and KATP channels in the ARC. This input is relayed to the motor nucleus of the vagus nerve in the brainstem and leads to the activation of the efferent vagal fibers which innervate the liver. Indeed, hepatic branch vagotomy leads to a loss of about 50% of the insulin inhibitory effect on glucose production [22]. However, this effect is not blocked by selective vagal deafferentation, suggesting that only the efferent vagal fibers are required for the neural and insulin-mediated inhibition of glucose production [19].
A recent report suggests that brain insulin action is responsible for the increase of hepatic IL-6 in the liver and that IL-6-STAT3 signaling in the liver contributes to central insulin action, leading to the suppression of hepatic glucose production [23]. The phosphorylation of hepatic STAT3 induced by glucose loading was attenuated in mice lacking the brain insulin receptors (NIRKO mice), indicating that insulin action in the brain contributes to postprandial activation of hepatic STAT3. Injection of insulin into the cerebral ventricle of wild-type mice resulted in liver STAT3 phosphorylation in a time-dependent manner, demonstrating a direct link between insulin action in the brain and hepatic STAT3 signaling. The intracerebroventricular (ICV) infusion of insulin did not induce the phosphorylation of IR or Akt in the liver but increased hepatic IL-6. Previous reports indicated that IL-6 activates STAT3 and inhibits the expression of gluconeogenic genes in cultured hepatocytes [24, 25]. In a mouse model, transplantation of an IL-6 producing tumor resulted in the inhibition of the expression of glucose-6-phosphatase (G6pc), a key enzyme in the gluconeogenic pathway in the liver [26]. Inoue and co-workers investigated whether IL-6 was involved in the phosphorylation of hepatic STAT3 induced by insulin action in the brain [23]. Administration of antibodies that neutralize circulating IL-6 in mice, prevented liver STAT3 phosphorylation induced by a bolus injection of IL-6, euglycemic hyperinsulinemic clamp, or ICV insulin infusion. Moreover, STAT3 phosphorylation in the liver induced by the ICV insulin infusion was inhibited in mice lacking IL-6 (IL-6KO mice). These results indicate that IL-6 is essential for phosphorylation of hepatic STAT3 induced by brain-insulin action. Moreover, inhibition of hepatic glucose production assessed by an euglycemic hyperinsulinemic clamp was also attenuated in IL-6KO mice, which is consistent with the notion that IL-6 contributes to the suppression of hepatic glucose production.
The relative importance of hypothalamic insulin signaling for the overall control of liver glucose production has been a matter of debate lately, since a recent study demonstrated that a 4-fold rise in cranial insulin levels does not change acute hepatic glucose output in dogs [26]. Therefore, apparent differences between rodents and dogs concerning acute and chronic control of glucose homeostasis must be carefully addressed in future work.
Central leptin actions
Leptin, the product of the ob gene, is a circulating hormone produced by white adipose tissue that has potent effects on feeding behavior, thermogenesis and neuroendocrine responses [27-29]. The severe obesity caused by leptin absence in rodents and humans [30] makes it clear that leptin is a fundamental hormone regulating energy homeostasis. Abundant evidence [31, 32], including the brain-specific knockout of the leptin receptor [33], indicates that the CNS is the site for leptin anti-obesity actions. The leptin receptor mRNA is highly expressed in the hypothalamus, including the ARC, the ventromedial nucleus and the dorsomedial nucleus [34-37].
In addition to its effects on food intake and body weight, leptin has potent effects on glucose homeostasis. Conditions of adipose tissue deficiency, both in animal models and in humans with lipodystrophy, are associated with severe insulin resistance. Leptin administration has impressive effects in improving insulin action and glucose homeostasis [38, 39].
To investigate the functional significance of the leptin receptor in the ARC, Coppari and co-workers used a mouse model that allows site-selective restoration of the leptin receptor expression in leptin receptor null animals. Restoration of leptin receptors in the ARC had modest effects on body weight, but remarkably improved glucose homeostasis.
Numerous lines of evidence suggest that central leptin action, as well as insulin, regulates energy balance and glucose homeostasis via neural pathways that are only partially overlapping (Figure 1).
The binding of leptin to its receptor (LepR) in the ARC leads to the activation of the signaling cascade involving Janus kinase, signal transducer and activator of transcription 3 (Jak-STAT3) [40, 41]. Recent data suggest that leptin, like insulin, activates the IRS-PI3K pathway [42, 43]. Delivery of a functional LepR to the ARC of LepR-deficient rats improves peripheral insulin sensitivity via a mechanism requiring the activation of PI3K [44].
Another candidate pathway for regulating glucose homeostasis is the central melanocortin system [45-47]. Melanocortin signaling acutely affects insulin levels and glucose uptake, and humans with MC4R mutations are extremely insulin resistant [47].
Activation of the leptin receptor in the ARC leads to stimulation of neurons producing anorectic peptides (melanocortins) and the inhibition of neurons co-producing the orectic peptides NPY/AGRP. These data suggest that leptin effects on peripheral glucose metabolism could be mediated by these downstream neural pathways. It has been demonstrated that the effects of central leptin on glucose homeostasis involve both melanocortin-dependent and melanocortin-independent mechanisms [48]. Acute activation of hypothalamic melanocortin receptors, results in an increased rate of hepatic gluconeogenesis and the increased expression of the rate-limiting gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (Glc6Pase) [48]. ICV administration of leptin increases the rate of hepatic gluconeogenesis and the expression of PEPCK and Glc6Pase, without changing glucose production, due to the simultaneous and compensatory inhibition of hepatic glycogenolysis [48, 49]. By contrast, the ICV co-administration of leptin and a melanocortin antagonist, which prevents leptin-induced activation of melanocortin receptors, abolishes the leptin-induced increase in gluconeogenic fluxes, without affecting leptin's ability to suppress glycogenolysis, and results in reduced hepatic glucose production [48].
The neural circuits mediating leptin action on hepatic glucose fluxes are still unknown, the melanocortin-independent effects of central leptin closely resemble the effects of central insulin on hepatic glucose fluxes [19]. Since both leptin and insulin activate PI3K in the hypothalamus, and this signaling pathway has been implicated in the regulation of both feeding behavior and glucose homeostasis [9, 50], it could be speculated that the effect of central leptin on the suppression of glucose production and glycogenolysis is mediated by the stimulation of melanocortin-indepen-dent pathways involving insulin-like pathways such as the PI3K signaling cascade.
Central GLP-1 actions
There is increasing evidence that glucagon-like peptide-1 (GLP-1) plays a role in the regulation of glucose homeostasis, and its potential use in the treatment of diabetes has been proposed. Blockade or reduction of GLP-1 determined abnormally high glucose levels following glucose administration [51, 52].
Besides the gut, the other major site of GLP-1 production is the brain. GLP-1 is produced in a discrete set of hindbrain neurons that project to a specific population of GLP-1 receptor-containing cells in the brain stem, hypothalamus and midbrain. The CNS and peripheral actions of GLP-1 are distinct and independent. Some preliminary studies demonstrated that ICV administration of GLP-1 activates primarily the central CRH-containing neurons of the hypothalamo-pituitary-adrenocortical axis and this activation may be responsible for the reduction of plasma glucose levels [53]. Knauf and colleagues demonstrated by using GLP-1 receptor antagonists in the cerebroventricular system of mice, that during hyperglycemia, brain GLP-1 inhibited muscle glucose utilization and increased insulin secretion to favor hepatic glycogen stores [54]. Overall, these data suggest that central GLP-1 signaling plays a role in the control of blood glucose levels.
Similar effects were observed in mice with GLP-1 receptor knockout, and were reduced by selective muscle denervation, suggesting that GLP-1 activity in the brain induces peripheral neural signals which regulate systemic glucose metabolism. Interestingly, when the CNS GLP-1 receptors were blocked, there was a significant attenuation of meal-induced insulin secretion [54]. The authors propose that during hyperglycemia following a meal, CNS GLP-1 inhibits muscle glucose utilization and increases insulin secretion to favor hepatic glycogen storage (Figure 1).
Neuronal biochemical sensors
Neuronal biochemical sensors play a special role in sensing the nutritional status of the body [55, 56]. Recent evidence supports the notion that specific neural circuits in the ARC respond to increased availability of circulating nutrients by activating efferent pathways that lead to the suppression of endogenous glucose production [57-59].
The central administration of macronutrients such as glucose or oleic acid decreases blood glucose and insulin levels [57, 58, 60]. Like central insulin action, the central effect of circulating macronutrients on glucose production is mediated by the activation of KATP channels in the ARC [57, 59].
Elevations of plasma levels of free fatty acids (FFA) or glucose, activate hypothalamic centers which in turn suppress endogenous glucose production (Figure 1). Several lines of evidence indicate that lipid metabolism in neurons plays a pivotal role in mediating the hypothalamic responses to fuel availability. Consistent with this, ICV administration of fatty acid synthase inhibitors reduces food intake, NPY expression and blood glucose levels [61].
The enzyme carnitine palmitoyltransferase-1 (CPT-I) regulates the entry of long-chain fatty acids (LCFAs) into mitochondria, the site where LCFAs undergo beta-oxidation. Hypothalamic inhibition of CPT-1, decreases food intake and suppresses endogenous glucose production [62] via a neural pathway which requires the activation of KATP channels localized in the ARC and efferent vagal fibers innervating the liver [22, 58]. This finding indicates that changes in the rate of lipid oxidation in selective hypothalamic neurons signal nutrient availability to the hypothalamus, which in turn modulates the exogenous and endogenous flux of nutrients into the circulation (Figure 1) [63].
In cells, as well as neurons, cellular oxidation of long-chain fatty acyl-CoAs is regulated by the cellular levels of malonyl-CoA due to its role as a potent inhibitor of CPT-1 activity [64]. Neuronal levels of malonyl-CoA act as neural sensors of fuel availability and regulators of energy balance and glucose homeostasis.
The variation of the intracellular pool of LCFA-CoAs is controlled by several biochemical events including the following: (i) FFA and glucose flux, (ii) key metabolic enzymes such as acetyl-CoA carboxylase (ACC), an enzyme that converts acetyl-CoA, generated from glycolysis, to malonyl-CoA, and (iii) malonyl-CoA decarboxilase (MCD) (an enzyme that converts malonyl-CoA to acetyl-CoA). These events are able to lower malonyl-CoA levels, prevent the accumulation of LCFA-CoAs by derepressing CPT-1 activity and increasing LCFA-CoAs oxidation. MCD overexpression in the ARC leads to reduced accumulation of LCFA-CoAs, increased food intake, and increased endogenous glucose production [65]. These results support the notion that in the CNS, malonyl-CoA act as a neural sensor of fuel availability and a regulator of energy balance and glucose homeostasis.
Concluding remarks
The hypothalamus is emerging as a critical site for the integration of nutritional, endocrine, and neural cues signaling the body's metabolic and nutritional status. These signals should normally activate a negative feedback loop between the availability of nutrients and their intake and metabolism. It has been postulated that an onset of hypothalamic resistance to multiple signals, such as leptin, insulin, and fatty acids, could contribute to the susceptibility to weight gain and insulin resistance in predisposed individuals and animals.
The concept that the CNS has a primary role in the control of insulin sensitivity and glucose homeostasis suggests that diabetes might be viewed as a disorder with underlying defects in the CNS. The knowledge of the central pathways involved in glucose metabolism may help to fully understand the pathophysiology of this complex disease. In addition, the hypothalamic circuits that regulate insulin action might become the targets of future preventive and therapeutic strategies.
However, targeting the hypothalamus for diabetic prevention and therapy is challenging. The CNS blood-brain barrier is able to block the flux of many substances which are administered systemically. In addition, some molecules which are potentially beneficial for the central treatment of insulin resistance, may have adverse effects on peripheral target tissues.
We are probably at the beginning of a new era of investigation to improve our understanding of the complex neuronal network involved in the regulation of glucose homeostasis.
References
- 1.Taylor SI. Deconstructing type 2 diabetes. Cell. 1999;97:9–12. doi: 10.1016/s0092-8674(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 2.Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 3.Elmquist JK, Marcus JN. Rethinking the central causes of diabetes. Nat Med. 2003;9:645–647. doi: 10.1038/nm0603-645. [DOI] [PubMed] [Google Scholar]
- 4.Sindelar DK, Chu CA, Venson P, Donahue EP, Neal DW, Cherrington AD. Basal hepatic glucose production is regulated by the portal vein insulin concentration. Diabetes. 1998;47(4):523–529. doi: 10.2337/diabetes.47.4.523. [DOI] [PubMed] [Google Scholar]
- 5.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acids induced inhibition of glucose uptake. J Clin Invest. 1994;93:438–446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis GF, Vranic M, Harley P, Giacca A. Fatty acids mediate the acute extrahepatic effects of insulin on hepatic glucose production in humans. Diabetes. 1997;46:111–119. doi: 10.2337/diab.46.7.1111. [DOI] [PubMed] [Google Scholar]
- 7.Rebrin K, Steil GM, Getty L, Bergman RN. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes. 1995;44:1038–1045. doi: 10.2337/diab.44.9.1038. [DOI] [PubMed] [Google Scholar]
- 8.Rebrin K, Steil GM, Mittelman SD, Bergman RN. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest. 1996;98:741–749. doi: 10.1172/JCI118846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 10.Woods SC, Lotter EC, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature. 1979;282:503–505. doi: 10.1038/282503a0. [DOI] [PubMed] [Google Scholar]
- 11.Woods SC, Seeley RJ, Porte DJ, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 12.Sipols AJ, Baskin DG, Schwartz MW. Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes. 1995;44:147–151. doi: 10.2337/diab.44.2.147. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz MW, Sipols AJ, Marks JL, Sanacora G, White JD, Scheurink A, Kahn SE, Baskin DG, Woods SC, Figlewicz DP. Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology. 1992;130:3608–3616. doi: 10.1210/endo.130.6.1597158. [DOI] [PubMed] [Google Scholar]
- 14.Sahu A, Dube MG, Phelps CP, Sninsky CA, Kalra PS, Kalra SP. Insulin and insulin-like growth factor II suppress neuropeptide Y release from the nerve terminals in the paraventricular nucleus: A putative hypothalamic site for energy homeostasis. Endocrinology. 1995;136:5718–5724. doi: 10.1210/endo.136.12.7588328. [DOI] [PubMed] [Google Scholar]
- 15.Davies SN, Colburn C, Dobbins R, Nadeau S, Neal D, Williams P, Cherrington AD. Evidence that the brain of the conscious dog is insulin sensitive. J Clin Invest. 1995;95:593–602. doi: 10.1172/JCI117703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams P, Cherrington AD. Brain of the conscious dog is sensitive to physiological changes in circulating insulin. Am J Physiol. 1997;272:E567–E575. doi: 10.1152/ajpendo.1997.272.4.E567. [DOI] [PubMed] [Google Scholar]
- 17.Liang CS. Insulin infusion in conscious dogs: effects on systemic and coronary hemodynamics, regional blood flows, and plasma catecholamines. J Clin Invest. 1982;69:1321–1336. doi: 10.1172/JCI110572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowe JW, Young JB, Minaker KL. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal men. Diabetes. 1981;30:219–225. doi: 10.2337/diab.30.3.219. [DOI] [PubMed] [Google Scholar]
- 19.Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature. 2005;434:1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 20.Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- 21.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, Teshigawara K, Matsuki Y, Watanabe E, Hiramatsu R, Notohara K, Katayose K, Okamura H, Kahn CR, Noda T, Takeda K, Akira S, Inui A, Kasuga M. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006;3(4):267–275. doi: 10.1016/j.cmet.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Christ B, Yazici E, Nath A. Phosphatidylinositol 3-kinase and protein kinase C contribute to the inhibition by interleukin 6 of phosphoenolpyruvate carboxykinase gene expression in cultured rat hepatocytes. Hepatology. 2000;31:461–468. doi: 10.1002/hep.510310228. [DOI] [PubMed] [Google Scholar]
- 25.Inoue H, Ogawa W, Ozaki M, Haga S, Matsumoto M, Furukawa K, Hashimoto N, Kido Y, Mori T, Sakaue H. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med. 2004;10:168–174. doi: 10.1038/nm980. [DOI] [PubMed] [Google Scholar]
- 26.Metzger S, Goldschmidt N, Barash V, Peretz T, Drize O, Shilyansky J, Shiloni E, and Chajek-Shaul T. Interleukin-6 secretion in mice is associated with reduced glucose-6-phosphatase and liver glycogen levels. Am J Physiol. 1997;273:E262–E267. doi: 10.1152/ajpendo.1997.273.2.E262. [DOI] [PubMed] [Google Scholar]
- 27.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104(4):531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 28.Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr Rev. 2002;60:S1–14. doi: 10.1301/002966402320634878. [DOI] [PubMed] [Google Scholar]
- 29.Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116(2):337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 30.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O'Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341(12):879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 31.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269(5223):546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 32.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269(5223):543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 33.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108(8):1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387(2-3):113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- 35.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138(10):4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 36.Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A. 1997;94(13):7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395(4):535–547. [PubMed] [Google Scholar]
- 38.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 39.Ebihara K, Ogawa Y, Masuzaki H, Shintani M, Miyanaga F, Aizawa-Abe M, Hayashi T, Hosoda K, Inoue G, Yoshimasa Y, Gavrilova O, Reitman ML, Nakao K. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes. 2001;50(6):1440–1448. doi: 10.2337/diabetes.50.6.1440. [DOI] [PubMed] [Google Scholar]
- 40.Bates SH, Myers MG. The role of leptin-STAT3 signaling in neuroendocrine function: an integrative perspective. J Mol Med. 2004;82:12–20. doi: 10.1007/s00109-003-0494-z. [DOI] [PubMed] [Google Scholar]
- 41.Bjorbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 42.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW. Intracellular signaling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 43.Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- 44.Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Marks DL, Cone RD. Central melanocortins and the regulation of weight during acute and chronic disease. Recent Prog Horm Res. 2001;56:359–375. doi: 10.1210/rp.56.1.359. [DOI] [PubMed] [Google Scholar]
- 46.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108(7):1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med Mar. 2003;348(12):1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 48.Gutierrez-Juarez R, Obici S, Rossetti L. Melanocortin-independent effects of leptin on hepatic glucose fluxes. J Biol Chem. 2004;279:49704–49715. doi: 10.1074/jbc.M408665200. [DOI] [PubMed] [Google Scholar]
- 49.Liu L, Karkanias GB, Morales JC, Hawkins M, Barzilai N, Wang J, Rossetti L. Intracerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J Biol Chem. 1998;273:31160–31167. doi: 10.1074/jbc.273.47.31160. [DOI] [PubMed] [Google Scholar]
- 50.Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- 51.Wang Z, Wang RM, Owji AA, Smith DM, Ghatei MA, Bloom SR. Glucagon-like peptide-1 is a physiological incretin in rat. J Clin Invest. 1995;95:417–421. doi: 10.1172/JCI117671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edwards CM, Todd JF, Mahmoudi M, Wang Z, Wang RM, Ghatei MA, Bloom SR. Glucagon-like peptide 1 has a physiological role in the control of postprandial glucose in humans: studies with the antagonist exendin 9-39. Diabetes. 1999;48:86–93. doi: 10.2337/diabetes.48.1.86. [DOI] [PubMed] [Google Scholar]
- 53.Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology. 1997;138:4445–4455. doi: 10.1210/endo.138.10.5270. [DOI] [PubMed] [Google Scholar]
- 54.Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, Benhamed F, Gremeaux T, Drucker DJ, Kahn CR, Girard J, Tanti JF, Delzenne NM, Postic C, Burcelin R. Brain glucagon-like peptide–1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest. 2005;115:3554–3563. doi: 10.1172/JCI25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci. 2005;8:579–584. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- 56.Obici S, Rossetti L. Mini review: nutrient sensing and the regulation of insulin action and energy balance. Endocrinology. 2003;144:5172–5178. doi: 10.1210/en.2003-0999. [DOI] [PubMed] [Google Scholar]
- 57.Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes. 2002;51:271–275. doi: 10.2337/diabetes.51.2.271. [DOI] [PubMed] [Google Scholar]
- 58.Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, Schwartz GJ, Rossetti L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- 59.Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–947. doi: 10.1126/science.1112085. [DOI] [PubMed] [Google Scholar]
- 60.Lam TK, van de WG, Giacca A. Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab. 2003;284:E281–E290. doi: 10.1152/ajpendo.00332.2002. [DOI] [PubMed] [Google Scholar]
- 61.Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 62.Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med. 2003;9:756–761. doi: 10.1038/nm873. [DOI] [PubMed] [Google Scholar]
- 63.Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, Rossetti L. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest. 2006;116(4):1081–1091. doi: 10.1172/JCI26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGarry JD, Takabayashi Y, Foster DW. The role of malonyl-CoA in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. J Biol Chem. 1978;253:8294–8300. [PubMed] [Google Scholar]
- 65.He W, Lam TK, Obici S, Rossetti L. Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci. 2006;9:227–233. doi: 10.1038/nn1626. [DOI] [PubMed] [Google Scholar]