

Abstract
We studied the role of morphine in anoxia/reoxygenation injury to hepatocytes. Overnight cultured rat hepatocytes were incubated in anoxic buffer at pH 6.2 for 4 h and reoxygenated at pH 7.4 for 2 h to simulate anoxia/reoxygenation. Some hepatocytes were preincubated with 50 μM morphine for 10 min prior to onset of anoxia/reoxygenation. To study the effect of morphine on nitric oxide (NO), hepatocytes were loaded with 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM). Changes in NO concentration were assessed with a multi-well fluorescence reader and confocal microscopy. Morphine substantially improved cell viability after reoxygenation and increased NO generation, which was blocked by ATP-sensitive potassium channel blockers. Confocal images revealed that the increase in NO occurred mainly at the cytosol. However, treatment with opioid receptor antagonists did not reverse cytoprotection by morphine. These results indicate that morphine prevents anoxia/reoxygenation injury to hepatocytes. Protective mechanisms are associated with the potassium channels and NO, but are independent of opioid receptor-mediated signaling.
1. Introduction
Opiates have been used clinically for more than a century [1] . Morphine, a classic synthetic opiate, is still the main stay of treating acute pain from surgery, angina, myocardial infarction, and trauma [2]. Recently, this classic opioid analgesic has been shown to protect the heart against anoxia/reoxygenation injury and the protection is mediated via stimulation of opioid receptors [3; 4]. Fryer et al. reported that morphine-induced cardioprotection resulted from the opening of the ATP-sensitive potassium (KATP) channels through the activation of G protein [5]. It has also been suggested that a direct stimulation of opioid receptors activates the mitochondrial KATP channels [3]. Although morphine appears to protect neurons against lethal reperfusion injury [6], it is currently unknown whether morphine confers protection against anoxia/reoxygenation injury to the liver.
The mechanisms underlying opiate-induced protection may be multifactorial. Nitric oxide (NO) may be one important protective mechanism in ischemic tissues. Recently, we showed that reperfusion with NO donors prevents onset of the mitochondrial permeability transition (MPT) and subsequent cell death in hepatocytes [7]. NO also improves tissue viability after anoxia/reoxygenation by modulating the activity of protein kinases [8] and mitochondrial Ca2+ loading [9]. Furthermore, NO synthase can be activated during the late phase of ischemic preconditioning in myocardium [10]. However, it remains unknown how opiates modulate hepatocellular NO.
Accordingly, the goals of the present study were to examine the role of morphine in anoxia/reoxygenation injury to cultured hepatocytes and to investigate the mechanisms of morphine-induced cytoprotection. Our results show that morphine improves hepatocellular viability after anoxia/reoxygenation. The mechanism underlying this protection involves the activation of the KATP channels and subsequent increase in intracellular NO concentration.
2. Materials and methods
2.1. Hepatocyte isolation and culture
The animal protocol was approved by the Institutional Animal Care and Use Committee. Hepatocytes were isolated from one-day fasted male Sprague-Dawley rats (average body weight 250 to 300 g) by collagenase perfusion method, as described previously [11]. Cell viability was greater than 90%, as determined by trypan blue exclusion. The cells were resuspended in Waymouth’s MB-752/1 medium containing 100 units/ml penicillin, 100 μg/ml streptomycin, 10% fetal calf serum, 100 nM insulin, and 100 nM dexamethasone. For cell viability assays, aliquots (1 ml) of 1.5 x 105 hepatocytes were plated onto 24-well microtiter plates (Falcon, Lincoln Park, NJ). For confocal microscopic studies, 3 ml of 1.5 x 105 cells were cultured on 40-mm round glass coverslips in 60-mm culture dishes. Multi-well plates and glass coverslips were coated with 0.1% Type 1 rat-tail collagen and air-dried before cell culture. Hepatocytes were overnight (19 – 22 h) cultured in humidified 5% CO2 / 95% air at 37ºC. The culture was washed once with a fresh Krebs-Ringer-N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES) buffer (KRH) to remove unattached cells prior to use. All experiments were carried out in KRH buffer containing (in mM) 115 NaCl, 5 KCl, 2 CaCl2, 1 KH2PO4, 1.2 MgSO4, and 25 Na-HEPES at pH 7.4 or pH 6.2.
2.2. Anoxia/reoxygenation of cultured rat hepatocytes
Hepatocytes were incubated in KRH buffer at pH 6.2 in an anaerobic chamber (Coy Laboratory Products, Ann Arbor, MI) for 4 h to simulate the anoxia and acidosis of tissue ischemia. To simulate reoxygenation and recovery to normal intracellular pH after reperfusion, anaerobic KRH buffer at pH 6.2 was replaced with aerobic KRH buffer at pH 7.4 after 4 h of anoxia [7]. Hepatocytes were reoxygenated for up to 2 h at 37ºC. Atmosphere in the anaerobic chamber was kept anoxic with 90% N2 / 10% H2. Back diffusion of oxygen into the anaerobic chamber was prevented by converting oxygen into water vapor using hydrogen and a heated palladium catalyst. Oxygen tension in the chamber was routinely monitored by a gas analyzer (Model 10, Coy Laboratory Products) and was less than 0.001 Torr.
2.3. Cell viability assay
Cell viability was determined by propidium iodide fluorometry using a multi-well plate reader (Fluostar, BMG Lab Technologies, Offenburg, Germany), as described previously [7]. Briefly, hepatocytes cultured on 24-well plates were washed once with a fresh KRH buffer. KRH buffer containing 30 μM propidium iodide were added to each well and the cells were further incubated at 37ºC. Fluorescence from each well was measured at excitation and emission wavelengths of 544 nm and 590 nm, respectively. The background fluorescence (A) was assessed at 20 min after addition of propidium iodide and then at intervals thereafter. Experiments were terminated by permeabilizing plasma membranes with 375 μM digitonin to label nuclei with propidium iodide. A final fluorescence (B) was measured at 20 min after digitonin treatment. The percentage of viable cells (V) was calculated as V=100(B-X)/(B-A), where X is the fluorescence at any given time.
2.4. Measurement of NO with DAF-FM
NO in hepatocytes was fluorometrically assessed with 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM). This fluorophore selectively reacts with NO to form a highly fluorescent benzotriazole derivative [12]. To measure intracellular NO, hepatocytes cultured on multi-well plates or coverslips were incubated in KRH buffer (pH 7.4) with 10 μM DAF-FM diacetate, a cell-permeant form of DAF-FM, at 37ºC for 1 h. After loading, the cells were washed twice with fresh KRH and returned to the incubator at 37ºC for another 30 min to complete the deesterification of diacetate moiety. Endogenous esterases hydrolyze the diacetate ester, trapping free DAF-FM inside the cells. Intracellular NO concentrations were fluorometrically quantified using multi-well fluorescence scanner. Fluorescence from each well was measured at excitation and emission wavelengths of 485 nm and 505–512 nm, respectively. The background fluorescence was measured and subtracted from each fluorescence reading.
2.5. Imaging of NO with laser scanning confocal microscopy
Hepatocytes on coverslips were loaded with DAF-FM, as described above. To localize the mitochondria, some hepatocytes were loaded with 300 nM Mitotracker Red (MTR) at 37ºC for 1 h in KRH buffer. After loading, the cells were rinsed with fresh KRH and incubated at 37ºC for 30 min before use. Hepatocytes loaded with fluorescence dyes were mounted on the stage of a Zeiss LSM 410 laser scanning confocal microscope equipped with a 63 x N.A. 1.4 oil-immersion planapochromat lens. The green DAF-FM fluorescence was excited 488 nm line of an argon-krypton laser and imaged through a 515 nm long-pass filter. The red MTR fluorescence was excited at 568 nm. The respective green and red fluorescence was separated using a 560 nm reflecting mirror and passed through 514 to 540 band pass and 590 nm long-pass filters. Temperature was maintained at 35–37ºC with an air-curtain incubator. Hepatocytes on the microscope stage were treated with 50 μM morphine for 10 min. For some experiments, hepatocytes were incubated with various inhibitors for 20 min before morphine treatment.
2.6. Materials
Morphine sulfate was purchased from Abbott Laboratories (Chicago, IL). DAF-FM diacetate, Mitotracker Red, and 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO) were purchased from Molecular Probes (Eugene, OR). 7-Benzylidenenaltrexone (BNTX) was purchased from Tocris Cookson Inc. (Ellisville, MO). All other chemicals were of analytical grade and purchased from Sigma Chemical Co. (St. Louis, MO).
2.7. Statistical analysis
The data were expressed as mean ± SE and obtained from at least 3 separate cell preparations. Significant difference was determined by using the Student’s t-test at P < 0.05.
3. Results
3.1. Cytoprotection of morphine against anoxia/reoxygenation injury to hepatocytes
To simulate the anoxia and tissue acidosis of ischemia, cultured rat hepatocytes were incubated in the anaerobic KRH buffer at pH 6.2. After 4 h of anoxia, the cells were incubated in aerobic KRH buffer at pH 7.4 to simulate the reoxygenation and restoration of normal pH after reperfusion. Cell viability decreased after anoxia/reoxygenation and was less than 30% after 2 h of reoxygenation. In contrast, administration of morphine (50 μM) to hepatocytes for 10 min before anoxia substantially reduced reoxygenation-induced cell killing (P=0.0002) (Fig. 1A). Morphine at lower concentrations (10 – 20 μM) did not prevent cell death, while morphine at higher concentration (200 μM) was toxic (data not shown).
Fig. 1. Protection by morphine against anoxia/reoxygenation injury to hepatocytes.

Overnight cultured hepatocytes were incubated in anoxic KRH buffer at pH 6.2 for 4 h, and reoxygenated in aerobic KRH buffer at pH 7.4 at 37ºC for 2 h to simulate ischemia/reperfusion. (A) Some hepatocytes were treated with 50 μM morphine for 10 min prior to anoxia. Cell viability was fluorometrically assessed by measuring propidium iodide uptake. * P < 0.05 vs. Control. (B) Hepatocytes were preincubated for 20 min in KRH containing either 0.1 μM BNTX, a selective opioid δ1 receptor antagonist, or 10 μM naloxone, a nonselective opioid receptor antagonist. The cells were then incubated with morphine for another 10 min. After 4 h of anoxia, cell viability was measured over the time of reoxygenation. Values are mean ± SE from 8 separate cell isolations.
Stimulation of endogenous opioid receptors has been suggested as a major signaling pathway in pharmacological preconditioning [4]. To test whether cytoprotection by morphine is associated with the activation of opioid receptors, hepatocytes were preincubated with 0.1 μM 7-benzylidenenaltrexone (BNTX) [13], a selective opioid δ1 receptor antagonist, and 10 μM naloxone [4], a nonselective opioid receptor antagonist, for 20 min before the addition of morphine (Fig. 1B). Cell viability was fluorometrically determined after anoxia/reoxygenation. Addition of BNTX and naloxone did not affect morphine-mediated cytoprotection. After 2 h of reoxygenation, the viability in the presence of BNTX and naloxone was similar to that by morphine alone. These data suggest that the protective mechanisms of morphine are not related to the stimulation of opioid receptors.
3.2. Protective role of NO in anoxia/reoxygenation injury
NO is an important signaling molecule in cytoprotection against apoptotic cell death and ischemic stress to hepatocytes [7] and to myocytes [14]. Morphine and opioid receptor stimulation activate NO synthases and release NO [15]. To test whether cytoprotection by morphine is associated with NO, hepatocytes were pretreated with 100 μM 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO), a water soluble NO scavenger [16], at 37ºC for 20 min. Hepatocytes were then treated with 50 μM morphine. After 10 min, cells were subjected to anoxia/reoxygenation, as described above. Some hepatocytes were treated with carboxy-PTIO alone prior to anoxia/reoxygenation. Cell viability after reoxygenation was fluorometrically assayed by measuring propidium iodide uptake (Fig. 2A). Pretreatment with carboxy-PTIO reversed morphine-induced protection, although carboxy-PTIO alone did not affect cell viability. This suggests that NO may be involved in morphine-induced protection against reoxygenation injury to hepatocytes. To further determine the role of NO, changes in intracellular NO concentration was assessed before and after morphine treatment. Hepatocytes cultured on multi-well plates were loaded with 10 μM DAF-FM at 37°C for 1 h. The cells were then incubated in KRH buffer for 10 min in the presence or absence of 50 μM morphine. Some hepatocytes were incubated with 100 μM carboxy-PTIO for 20 min before morphine treatment. Morphine increased DAF-FM fluorescence more than 9-fold, compared to control (888.2 ± 70.9 with morphine vs. 93.4 ± 4.4 without morphine), which was reversed by carboxy-PTIO (Fig. 2B).
Fig. 2. Role of NO in morphine-induced cytoprotection.

(A) Hepatocytes were preincubated in KRH buffer (pH 7.4) containing 100 μM carboxy-PTIO, an NO chelator, for 20 min prior to the addition of morphine. Following 10 min with morphine, the cells were subjected to 4 h of anoxia and 2 h of reoxygenation. Some hepatocytes were incubated with carboxy-PTIO alone. Cell viability was measured with the time of reoxygenation. Values are mean ± SE from 8 separate cell isolations. * P < 0.05 vs. Control. (B) To measure intracellular NO, hepatocytes were incubated in KRH buffer (pH 7.4) containing 10 μM DAF-FM diacetate at 37ºC for 1 h and further incubated with morphine for 10 min. Some hepatocytes were pretreated with carboxy-PTIO for 20 min. NO was fluorometrically quantified using a multi-well fluorescence scanner. Fluorescence at 10 min after morphine treatment was measured and the background fluorescence was subtracted. Values are mean ± SE from 8 separate cell isolations. * P < 0.05 vs. no addition (Normoxia).
3.3. Confocal imaging of NO
To investigate NO localization, hepatocytes cultured on glass coverslips were labeled with DAF-FM. Some cells were co-loaded with DAF-FM and MTR to monitor NO and localization of the mitochondria, respectively. Representative baseline image of DAF-FM was shown in Fig. 3A (left panel). Most of the green fluorescence of DAF-FM was localized at the cytosol and nuclei whereas the mitochondria remained as dark voids. Subsequently, 100 μM S-nitroso-N-acetyl–penicillamine (SNAP), an NO donor, was added to saturate DAF-FM in all compartments and to reveal intracellular distribution of the fluorophore (right panel, Fig. 3A). Within 2 min, all compartments including the mitochondria became fluorescent. This experiment, therefore, ruled out the possibility of unequal loading of DAF-FM and further demonstrated background level of NO was substantially low in the mitochondria. Confocal images with DAF-FM and MTR revealed that morphine substantially increased cytosolic NO (Fig. 3B).
Fig. 3. Confocal microscopy with DAF-FM.
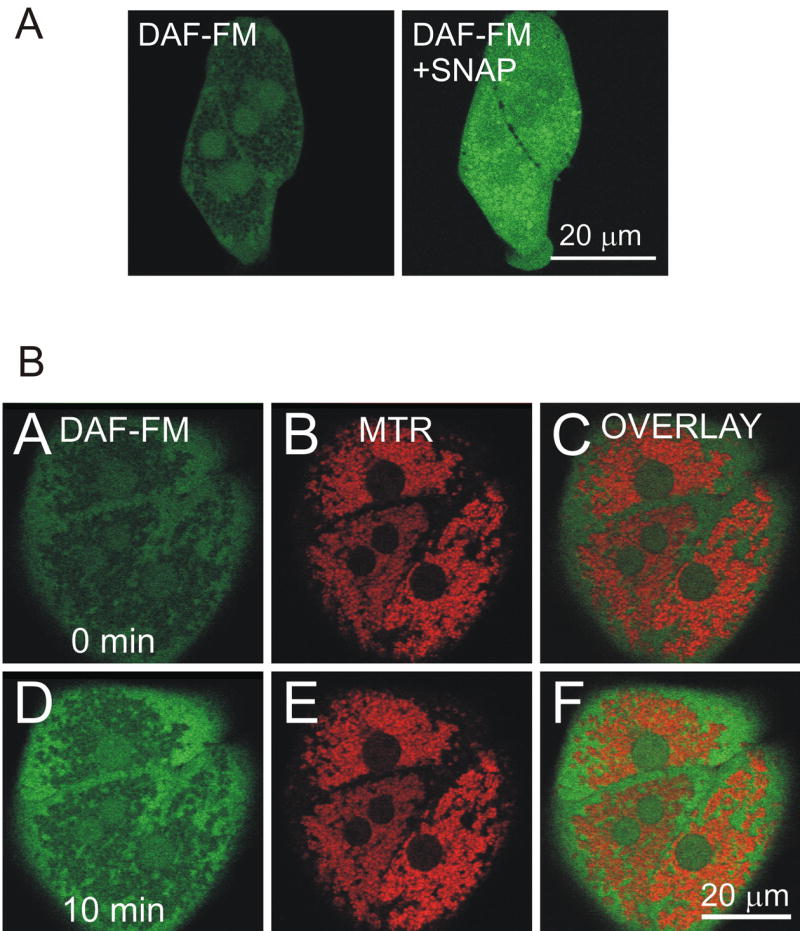
Hepatocytes were loaded with DAF-FM, as described in Materials and methods. The green fluorescence of DAF-FM was imaged by laser scanning confocal microscopy. (A) Representative image at the end of DAF-FM loading. Note that DAF-FM fluorescence was evident in the cytosol and nuclei, leaving the mitochondria as dark voids (left panel). Hepatocytes were further incubated with 100 μM SNAP, an NO donor, for 1–2 min to visualize dye distribution inside the cells (right panel). Note that all compartments became fluorescent with SNAP, indicating that DAF-FM was localized in all compartments. (B) Hepatocytes on glass coverslips were co-loaded with 10 μM DAF-FM and 300 nM Mitotracker Red (MTR). Simultaneous images of green DAF-FM (left panels), red MTR (middle panels), and overlay (right panels) fluorescence were collected by confocal microscopy. Images were collected before (A, B, and C) and after 50 μM morphine addition (D, E, and F). Note that morphine substantially increased cytosolic NO.
3.4. Relationship between NO and the KATP Channels
Opioids activate the opening of the ATP-sensitive potassium (KATP) channels and protect myocardium against ischemia/reperfusion injury [5]. To investigate the relationship between morphine, NO, and the KATP channels, hepatocytes were incubated with KATP channel blockers, 5-hydroxydecanoate (5-HD) and glibenclamide [17], for 20 min before morphine treatment. Cell viability was examined after reoxygenation. Pretreatment of hepatocytes with 200 μM 5-HD abolished cytoprotection of morphine (Fig. 4A). Similar effects were also observed upon pretreatment with 50 μM glibenclamide. Pretreatment of hepatocytes with 5-HD or glibenclamide alone did not affect cell viability after reoxygenation (Fig. 4B).
Fig. 4. Effects of the KATP channel blockers on morphine-induced cytoprotection and NO increase.

(A) Hepatocytes were preincubated for 20 min in KRH buffer containing 200 μM 5-HD or 50 μM glibenclamide, the KATP channel blockers. The cells were then treated with morphine for 10 min and subjected to simulated anoxia/reoxygenation, as described in Fig. 1. The KATP channel blockers abolished the protection by morphine. Values are mean ± SE. * P < 0.05 vs. morphine. (B) Some hepatocytes were treated with 5-HD or glibenclamide (GC) alone and exposed to simulated anoxia/reoxygenation, showing that the KATP channel blockers alone did not affect cell viability. (C) NO was fluorometrically measured by DAF-FM. *P<0.05 vs. Control.
To correlate the KATP channels with NO, hepatocytes were labeled with DAF-FM in the presence of 5-HD and glibenclamide. Changes in fluorescence were monitored after addition of morphine (Fig. 4C). DAF-FM Fluorometry showed that 5-HD and glibenclamide blocked morphine-induced increase in NO, suggesting that opening of KATP channels may be an event upstream to morphine-mediated increase in NO. Confocal images also confirmed that the cytosolic increase in NO after morphine treatment was prevented by these KATP channel blockers (Supplementary Fig).
4. Discussion
In this study, we demonstrate for the first time that morphine improves hepatocyte survival after anoxia/reoxygenation through KATP channels/NO-dependent pathway. This novel cytoprotection is independent of opioid receptor stimulation.
After 4 h of simulated ischemia at pH 6.2, necrotic cell killing occurred after reoxygenation at pH 7.4. Administration with morphine prior to anoxia markedly improved cell survival (Fig. 1A). The protection was, however, not abrogated by two specific opioid receptor antagonists, naloxone [18] or BNTX [3], suggesting that the protection by morphine is independent of opioid receptors (Fig. 1B). Cytoprotection was attributed to NO because: 1) pretreatment of hepatocytes with carboxy-PTIO, a soluble NO chelator [16], reversed cytoprotection by morphine (Fig. 2A), 2) morphine increased NO more than 9-fold, as judged by fluorometry with DAF-FM, an NO-sensitive fluorophore [12], (Fig. 2B) and 3) confocal imaging with DAF-FM revealed a substantial increase in cytosolic NO after morphine treatment (Fig. 3). Pretreatment with 5-HD and glibenclamide, the KATP channel blockers, abolished both morphine-mediated cytoprotection (Fig. 4A) and NO elevation (Fig. 4C), suggesting a critical role of the KATP channels in NO-dependent protection by morphine. Furthermore, confocal microscopy of DAF-FM confirmed that KATP channel blockers inhibited morphine-induced NO increase (Supplementary Fig). These findings support the conclusion that morphine activates the KATP channels, which in turn increases hepatocellular NO and prevents necrotic cell death after reoxygenation.
Morphine is the first synthetic opioid agonist that has been shown to reduce ischemia/reperfusion injury to myocardium and neuron [6; 17]. The mechanisms underlying morphine-mediated cytoprotection are multifactorial, including opioid receptor signaling [13], activation of ERK and p38 MAP kinases [19], enhanced NO formation [20] and opening of the KATP channels [17]. The present study shows that a brief exposure to morphine suppresses lethal anoxia/reoxygenation injury to hepatocytes by triggering the KATP channel opening and subsequent NO release. Unlike cardiomyocytes, hepatocellular protection by morphine was not associated with opioid receptors. Recent report showed that the liver is lack of opioid receptors [21], which is consistent with our results. The absence of opioid receptors in the liver may also explain why the dose of morphine for hepatocellular protection (50 μM) was higher than that required for protecting cardiomyocytes (1 μM) in which delta- and kappa-opioid receptors play a major role [22].
In the present study, we show that inhibition of the KATP channels abolishes cytoprotection by morphine. Although it is not known how morphine stimulates opening of the KATP channels in the liver, beneficial roles of the KATP channels have been documented in ischemic livers [23]. Hepatic KATP channels may also regulate adenosine transport and cell proliferation [24; 25]. In the heart, NO has been suggested to open directly KATP channels, leading to protection against ischemia/reperfusion injury [26]. However, our study suggests that opening of the KATP channels is an event upstream to NO formation in hepatocytes because blockade of the KATP channels not only reversed cytoprotection, but also inhibited morphine-induced increase in NO. Further studies will be needed to determine the mechanisms underlying morphine-mediated opening of the KATP channels.
Morphine increased cytosolic NO, as measured by DAF-FM fluorometry and confocal microscopy. NO is a diffusible signaling gas that both promotes and prevents cell injury, depending on the cell type and the concentration [27]. In hepatocytes, NO prevents apoptosis by tumor necrosis factor α and Fas ligand [28; 29]. NO signaling is one important mechanism implicated in ischemic preconditioning in the heart and liver [30; 31]. NO-induced cytoprotection includes cGMP stimulation, s-nitrosylation of proteins and caspases, reaction with reactive oxygen species and effects on mitochondrial respiration [28; 29; 32; 33]. Recently, we showed that reoxygenation with supraphysiological concentrations of NO prevents onset of the MPT and consequent hepatocellular death by a guanylyl cyclase/cGMP/protein kinase G-mediated signaling cascade [7]. Since the MPT is a causative event initiating both necrotic and apoptotic cell death in ischemic hepatocytes [11; 34], the protection by morphine is likely associated with NO-mediated inhibition of mitochondrial inner membrane permeabilization. The exact mechanism warrant further investigation.
In conclusion, we show that morphine generates NO via activation of the KATP channels, a mechanism independent of opioid receptors. Through this novel signal transduction pathway, morphine protects hepatocytes against lethal anoxia/reoxygenation injury.
Supplementary Material
References
- 1.Way EL. Review and overview of four decades of opiate research. Adv Biochem Psychopharmacol. 1979;20:3–27. [PubMed] [Google Scholar]
- 2.MacPherson RD. The pharmacological basis of contemporary pain management. Pharmacol Ther. 2000;88:163–185. doi: 10.1016/s0163-7258(00)00090-5. [DOI] [PubMed] [Google Scholar]
- 3.McPherson BC, Yao Z. Morphine Mimics Preconditioning via Free Radical Signals and Mitochondrial KATP Channels in Myocytes. Circulation. 2001;103:290–295. doi: 10.1161/01.cir.103.2.290. [DOI] [PubMed] [Google Scholar]
- 4.Schultz JE, Rose E, Yao Z, Gross GJ. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am J Physiol. 1995;268:H2157–H2161. doi: 10.1152/ajpheart.1995.268.5.H2157. [DOI] [PubMed] [Google Scholar]
- 5.Fryer RM, Hsu AK, Gross GJ. Mitochondrial KATP channel opening is important during index ischemia and following myocardial reperfusion in ischemic preconditioned rat hearts. J Mol Cell Cardiol. 2001;33:831–834. doi: 10.1006/jmcc.2001.1350. [DOI] [PubMed] [Google Scholar]
- 6.Lim YJ, Zheng S, Zuo Z. Morphine preconditions Purkinje cells against cell death under in vitro simulated ischemia-reperfusion conditions. Anesthesiology. 2004;100:562–568. doi: 10.1097/00000542-200403000-00015. [DOI] [PubMed] [Google Scholar]
- 7.Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ. Nitric oxide protects rat hepatocytes against reperfusion injury mediated by the mitochondrial permeability transition. Hepatology. 2004;39:1533–1543. doi: 10.1002/hep.20197. [DOI] [PubMed] [Google Scholar]
- 8.Ahmmed GU, Xu Y, Hong DP, Zhang Z, Eiserich J, Chiamvimonvat N. Nitric oxide modulates cardiac Na+ channel via protein kinase A and protein kinase. G Circ Res. 2001;89:1005–1013. doi: 10.1161/hh2301.100801. [DOI] [PubMed] [Google Scholar]
- 9.Rakhit RD, Mojet MH, Marber MS, Duchen MR. Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Circulation. 2001;103:2617–2623. doi: 10.1161/01.cir.103.21.2617. [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci U S A. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 12.Itoh Y, Ma FH, Hoshi H, Oka M, Noda K, Ukai Y, Kojima H, Nagano T, Toda N. Determination and bioimaging method for nitric oxide in biological specimens by diaminofluorescein fluorometry. Anal Biochem. 2000;287:203–209. doi: 10.1006/abio.2000.4859. [DOI] [PubMed] [Google Scholar]
- 13.Schultz JE, Hsu AK, Gross GJ. Ischemic preconditioning in the intact rat heart is mediated by delta1- but not mu- or kappa-opioid receptors. Circulation. 1998;97:1282–1289. doi: 10.1161/01.cir.97.13.1282. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Guo Y, Zhang SX, Wu WJ, Wang J, Bao W, Bolli R. Ischemic preconditioning upregulates inducible nitric oxide synthase in cardiac myocyte. J Mol Cell Cardiol. 2002;34:5–15. doi: 10.1006/jmcc.2001.1482. [DOI] [PubMed] [Google Scholar]
- 15.Fimiani C, Mattocks D, Cavani F, Salzet M, Deutsch DG, Pryor S, Bilfinger TV, Stefano GB. Morphine and anandamide stimulate intracellular calcium transients in human arterial endothelial cells: coupling to nitric oxide release. Cell Signal. 1999;11:189–193. doi: 10.1016/s0898-6568(98)00060-6. [DOI] [PubMed] [Google Scholar]
- 16.Amano F, Noda T. Improved detection of nitric oxide radical (NO. ) production in an activated macrophage culture with a radical scavenger, carboxy PTIO and Griess reagent. FEBS Lett. 1995;368:425–428. doi: 10.1016/0014-5793(95)00700-j. [DOI] [PubMed] [Google Scholar]
- 17.Schultz JE, Hsu AK, Gross GJ. Morphine mimics the cardioprotective effect of ischemic preconditioning via a glibenclamide-sensitive mechanism in the rat heart. Circ Res. 1996;78:1100–1104. doi: 10.1161/01.res.78.6.1100. [DOI] [PubMed] [Google Scholar]
- 18.Chien GL, Van Winkle DM. Naloxone blockade of myocardial ischemic preconditioning is stereoselective. J Mol Cell Cardiol. 1996;28:1895–1900. doi: 10.1006/jmcc.1996.0182. [DOI] [PubMed] [Google Scholar]
- 19.Fryer RM, Hsu AK, Gross GJ. ERK and p38 MAP kinase activation are components of opioid-induced delayed cardioprotection. Basic Res Cardiol. 2001;96:136–142. doi: 10.1007/s003950170063. [DOI] [PubMed] [Google Scholar]
- 20.Jiang X, Shi E, Nakajima Y, Sato S. COX-2 mediates morphine-induced delayed cardioprotection via an iNOS-dependent mechanism. Life Sci. 2006;78:2543–2549. doi: 10.1016/j.lfs.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 21.Jaume M, Jacquet S, Cavailles P, Mace G, Stephan L, Blanpied C, Demur C, Brousset P, Dietrich G. Opioid receptor blockade reduces Fas-induced hepatitis in mice. Hepatology. 2004;40:1136–1143. doi: 10.1002/hep.20428. [DOI] [PubMed] [Google Scholar]
- 22.Peart JN, Gross GJ. Exogenous activation of delta- and kappa-opioid receptors affords cardioprotection in isolated murine heart. Basic Res Cardiol. 2004;99:29–37. doi: 10.1007/s00395-003-0430-y. [DOI] [PubMed] [Google Scholar]
- 23.Hai S, Takemura S, Minamiyama Y, Yamasaki K, Yamamoto S, Kodai S, Tanaka S, Hirohashi K, Suehiro S. Mitochondrial KATP channel opener prevents ischemia-reperfusion injury in rat liver. Transplant Proc. 2005;37:428–431. doi: 10.1016/j.transproceed.2004.12.112. [DOI] [PubMed] [Google Scholar]
- 24.Duflot S, Riera B, Fernandez-Veledo S, Casado V, Norman RI, Casado FJ, Lluis C, Franco R, Pastor-Anglada M. ATP-sensitive K+ channels regulate the concentrative adenosine transporter CNT2 following activation by A1 adenosine receptors. Mol Cell Biol. 2004;24:2710–2719. doi: 10.1128/MCB.24.7.2710-2719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malhi H, Irani AN, Rajvanshi P, Suadicani SO, Spray DC, McDonald TV, Gupta S. KATP channels regulate mitogenically induced proliferation in primary rat hepatocytes and human liver cell lines. Implications for liver growth control and potential therapeutic targeting. J Biol Chem. 2000;275:26050–26057. doi: 10.1074/jbc.M001576200. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki N, Sato T, Ohler A, O’Rourke B, Marban E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- 27.Schlossmann J, Feil R, Hofmann F. Signaling through NO and cGMP-dependent protein kinases. Ann Med. 2003;35:21–27. doi: 10.1080/07853890310004093. [DOI] [PubMed] [Google Scholar]
- 28.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Bombeck CA, Yang S, Kim YM, Billiar TR. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J Biol Chem. 1999;274:17325–17333. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- 30.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 31.Carini R, Grazia DC, Splendore R, Domenicotti C, Nitti MP, Pronzato MA, Albano E. Signal pathway responsible for hepatocyte preconditioning by nitric oxide Free. Radic Biol Med. 2003;34:1047–1055. doi: 10.1016/s0891-5849(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 32.Gumpricht E, Dahl R, Yerushalmi B, Devereaux MW, Sokol RJ. Nitric oxide ameliorates hydrophobic bile acid-induced apoptosis in isolated rat hepatocytes by non-mitochondrial pathways. J Biol Chem. 2002;277:25823–25830. doi: 10.1074/jbc.M112305200. [DOI] [PubMed] [Google Scholar]
- 33.Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, Anderson PG. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem. 2000;275:20474–20479. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 34.Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol. 1997;273:C1783–C1792. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.