

Abstract
The most useful information about the anatomy of human memory comes from cases where there has been extensive neuropsychological testing followed by detailed post-mortem neurohistological analysis. To our knowledge, only eight such cases have been reported (four with medial temporal lobe damage and four with diencephalic damage). Here we present neuropsychological and post-mortem neurohistological findings for one patient (NC) with bilateral damage to the medial temporal lobe and two patients (MG, PN) with diencephalic damage due to bilateral thalamic infarction and Korsakoff’s syndrome, respectively. All three patients exhibited a similar phenotype of amnesia with markedly impaired declarative memory (anterograde and retrograde) but normal performance on tests of nondeclarative memory (e.g., priming and adaptation-level effects) as well as on tests of other cognitive functions. Patient NC had damage to the hippocampus (dentate gyrus and the CA1 and CA3 fields) and layer III of the entorhinal cortex, but with relative sparing of the CA2 field and the subiculum. Patient MG had damage to the internal medullary lamina and mediodorsal thalamic nuclei. Patient PN had damage to the mammillary nuclei, mammillothalamic tracts, and the anterior thalamic nuclei. These findings illuminate several issues regarding the relation between diencephalic and medial temporal lobe amnesia, the status of recognition memory in amnesia, and the neuroanatomy of memory.
From the earliest descriptions of memory-impaired patients (Ribot 1881), studies of human amnesia have provided useful insight into the structure and organization of human memory (Scoville and Milner 1957; Talland 1965; Milner et al. 1998; Baddeley 2001; Schacter 2001). It is now understood that damage to either the medial temporal lobe (MTL) or diencephalon impairs declarative memory (conscious knowledge about facts and events) while sparing nondeclarative memory and other cognitive functions (Victor et al. 1989; Eichenbaum and Cohen 2001; Squire et al. 2004).
The most informative reports about the functional neuroanatomy of human memory come from cases where there has been an opportunity to carry out extensive neuropsychological testing as well as post-mortem neurohistological analysis. To our knowledge, only eight such cases have been reported. In four of these cases, there was damage within the MTL (Zola-Morgan et al. 1986; Rempel-Clower et al. 1996). These patients demonstrated that bilateral damage limited to the CA1 field of the hippocampus is sufficient to cause significant memory impairment. Furthermore, more extensive damage to the hippocampus or other MTL structures causes more severe memory impairment (also see Corkin et al. 1997; Stefanacci et al. 2000; Squire et al. 2004). Work with nonhuman primates reached similar conclusions (Zola-Morgan et al. 1994).
The four other cases were patients with Korsakoff’s syndrome, an example of diencephalic amnesia (Mair et al. 1979; Mayes et al. 1988). These patients were reported to have damage to the mammillary nuclei and a “band of gliosis” in the region of the paratenial nucleus. Subsequent discussion about the location and extent of damage in these four patients raised the suggestion that the “band of gliosis” actually denoted damage to the mediodorsal thalamic nucleus (MD) rather than the paratenial nucleus (Victor et al. 1989; Harding et al. 2000). Other neurohistological studies of Korsakoff’s syndrome emphasized the role of either the MD (Victor et al. 1989) or the anterior thalamic nuclei (Harding et al. 2000) in memory impairment, but these studies did not report neuropsychological findings for the patients.
Thus, in the case of diencephalic amnesia, the neurohistological evidence has not converged on a single view about what damage is necessary or sufficient to impair memory. Notably, the previously studied patients with diencephalic amnesia had Korsakoff’s syndrome, a condition caused by thiamine deficiency and usually in conjunction with alcoholism (Victor et al. 1989). Neurohistological studies of patients with diencephalic amnesia not related to Korsakoff’s syndrome would be useful. In addition, there have been no neurohistological studies that included both patients with diencephalic amnesia and patients with medial temporal lobe amnesia. Such studies, involving patients who had been assessed during life with the same neuropsychological tests, would illuminate the relationship between these two forms of amnesia.
In the present study, we report neuropsychological and post-mortem neurohistological findings from three patients. One patient (NC) had damage to the MTL. Two patients (MG and PN) had damage to the diencephalon due to bilateral thalamic infarction or Korsakoff’s syndrome, respectively. We discuss the findings from these patients and their implications for the functional neuroanatomy of human memory.
Results
Neuropsychological data from the patients are described first, followed by the post-mortem neuropathological findings. As mentioned above, all three patients were tested on multiple occasions. NC was tested for 12 yr, MG for 7 yr, and PN for 21 yr. No significant changes in memory or cognitive abilities were noted in any patient during the years they were studied.
Neuropsychological findings
Some of the data for patients NC, MG, and PN (see Tables 1 and 2) are presented together with comparable data for four patients (H) with histologically confirmed lesions of the hippocampus (RB from Zola-Morgan et al. 1986; GD, LM, and WH from Rempel-Clower et al. 1996), and five patients (KOR) with alcoholic Korsakoff’s syndrome (for BL and DM, see Delis et al. 1992; for RC, NF, and JW, see Reber and Squire 1994).
Table 1.
Characteristics of amnesic patients

(WAIS-R) Wechsler Adult Intelligence Scale-Revised; (WMS-R) Wechsler Memory Scale-Revised. The full-scale WAIS-R and each of the five indices of the WMR-R yield a mean score of 100 in the normal population with a standard deviation of 15. The WMS-R does not provide numerical scores for individuals who score below 50. Therefore, values below 50 were scored as 50 for computing means. Mean scores are also provided for four patients with histologically confirmed damage to the hippocampus (H: GD, WH, and LM from Rempel-Clower et al. 1996; RB from Zola-Morgan et al. 1986) and five patients with Korsakoff’s syndrome (KOR). RB was not tested on the WMS-R. Scores for MG on the WMS-R are the average of two tests. “Age” is the age at testing.
Table 2.
Memory test performance

aThe diagram recall score is based on delayed (10–15 min) reproduction of the Rey-Osterrieth figure (Osterrieth 1944) (maximum score: 36).
bThe paired associates score is the number of words recalled on three successive trials (maximum score: 10/trial).
cThe word recall score is the percentage of words recalled out of 15 across five successive study-test trials (Rey 1964).
dThe word recognition score is the percentage of words identified correctly across five successive study-test trials (yes/no recognition of 15 new words and 15 old words). The mean percent hit rate was 48, 56, and 95 for NC, PN, and MG, respectively. The mean percent false alarm rate was 7, 13, and 29, respectively.
eThe score for words and faces is based on a 24-h recognition test of 50 words or 50 faces (modified from Warrington 1984) (maximum score: 50; chance: 25).
Mean scores ± standard errors are also provided for four patients with histologically confirmed hippocampal damage (H), five patients with Korsakoff’s syndrome (KOR), and eight controls (CON) from Squire and Shimamura (1986). RB was tested only on the diagram recall test and the paired associates test. Scores for PN on Words and Faces are the average of two tests.
Table 1 shows scores for the full-scale Wechsler Adult Intelligence Scale-Revised (WAIS-R) and the Wechsler Memory Scale-Revised (WMS-R) for patients NC, MG, and PN as well as the H and KOR groups just described. NC, MG, and PN had WAIS-R scores in the normal range but were impaired on the WMS-R, especially on the delay memory index.
Anterograde memory
Table 2 shows scores for six measures of anterograde (declarative) memory function. The scores of eight control subjects (mean age = 50.9 yr) are also included (Squire and Shimamura 1986). Patients NC, MG, and PN were each impaired at learning new material. Figure 1 shows copies and delayed reproductions of the Rey-Osterrieth figure (Osterrieth 1944) for the patients and a typical control. Although the patients copied the figure accurately, they were unable to reproduce it after a 10–15-min delay (Fig. 1; see Table 2 for scores).
Figure 1.

The Rey-Osterrieth figure. Participants copied the Rey-Osterrieth figure (Osterrieth 1944) and then reproduced the figure from memory 10–15 min later, without forewarning. Normal copies (top) and impaired reproductions (bottom) are shown for patients NC, MG, and PN, along with the copy and reproduction of a typical control (CON).
Figure 2 shows the performance of the three patients and controls (n = 19) on tests of recall and recognition memory for 20-word lists tested after delays of 15 sec, 10 min, 2 h, or 1 d (Haist et al. 1992). The patients were impaired at both recall (10% vs. 31%, P < 0.05) and recognition memory (66% vs. 91%, P < 0.05).
Figure 2.

Recall and recognition. The three amnesic patients (AMN) and controls (CON, n = 19) studied eight lists of 20 words each on eight separate days (Haist et al. 1992). Each participant took four free-recall tests and four two-alternative, forced-choice recognition tests after delays of 15 sec, 10 min, 2 h, or 1 d.
Retrograde memory
The three patients and controls were asked 92 questions about public events that occurred between 1950 and 1987 (Squire et al. 1989b; NC was designated K1 and PN was designated K5). Participants took a recall test (Fig. 3A, left) and also a four-alternative, recognition memory test for the same questions (Fig. 3A, right). All three patients were impaired on questions about the most recent time periods (especially on the recall test) but performed as well as controls on questions about more remote time periods. MG became amnesic in 1986 and thus exhibits retrograde amnesia covering 10–15 yr. NC and PN were diagnosed in 1979, but the time of onset of their amnesia is uncertain. Accordingly, some of their impairment on these tests must reflect anterograde amnesia, and it is difficult to know how much of the impairment reflects retrograde amnesia.
Figure 3.

Retrograde memory. (A) Public events. The patients (dark squares) and controls (white squares; n = 8 healthy controls for NC and MG; n = 9 alcoholic controls for PN) were given recall and four-alternative recognition memory tests for public events that occurred between 1950 and 1987. The standard errors range from 3.7% to 8.5% for recall and from 2.8% to 5.3% for recognition. Data are from Squire et al. 1989). (B) Autobiographical memory. The three amnesic patients (AMN) and controls (CON, n = 5) were given 10 cue words (e.g., nail, book) and asked to recall, in response to each cue word, an autobiographical episode from any past period (Crovitz and Schiffman 1974). Narratives were rated from 0 (no memory) to 3 (detailed episodic memory specific to time and place). Data for controls are from MacKinnon and Squire (1989).
Figure 3B shows performance of the patients and controls (n = 5) on a test of autobiographical memory (Crovitz and Schiffman 1974). Participants were read 10 concrete nouns one at a time and asked to recollect an autobiographical memory associated with each word. Each narrative was rated from 0 (no memory) to 3 (detailed episodic memory specific to time and place). Scores are the sum of the ratings of the 10 narratives (max = 30). NC, MG, and PN performed similarly to controls (scores of 28.3 and 29.4, respectively). The amnesic patients (unlike controls) drew a disproportionate number of their autobiographical memories from the very remote past (also see MacKinnon and Squire 1989).
Nondeclarative memory
Figure 4 shows performance on two tests of nondeclarative memory. For the test of priming, the three patients and controls (n = 15) saw eight words and eight nonwords one at a time (Haist et al. 1991). After 1 min, participants were asked to identify briefly presented words and nonwords (half new and half old). Patients and controls exhibited a similar advantage for identifying old items relative to new items (21.9% and 24%, respectively, for words; 19.0% and 19.9% for nonwords). Thus, the patients exhibited normal priming.
Figure 4.

Nondeclarative memory. (A) Priming. The three amnesic patients (AMN) and controls (CON, n = 15) saw either 16 words (for an average of 50 msec) or 16 nonwords (for 117 msec), half of which had been presented earlier. The facilitation score is the percentage of previously viewed items that could be correctly identified (i.e., named) minus the percentage of novel items that could be correctly identified. Across four separate study-test sequences, patients and controls exhibited a similar advantage for previously viewed items. Data are from Haist et al. (1991). (B) Adaptation-level effects. On three different days, the three patients and controls (n = 11) lifted 10 identical-appearing objects that as a group were light, neutral, or heavy. After a delay, participants lifted the 10 neutral objects and rated each object’s weight from 1 (extremely light) to 9 (extremely heavy). When participants lifted light objects first, they subsequently perceived the neutral set as heavy (light bias). When participants lifted the heavy objects first, they subsequently perceived the neutral set as light (heavy bias). Patients and controls exhibited these effects to the same degree. Data are from Benzing and Squire (1989).
For the test of adaptation-level effects, the three patients and controls (n = 11) lifted 10 identical-appearing objects four times with one hand (Benzing and Squire 1989). After 20–25 min, participants lifted another 10 identical-appearing objects with their other hand once and rated the weight of each object from 1 to 9. Altogether, participants took three different tests in which the first set of objects was lighter than, identical to, or heavier than the second set of objects. Both patients and controls were biased in the same way by their experience with the first set of objects. Thus, the patients exhibited normal adaptation-level effects.
NC, MG, and PN participated in several other studies that demonstrated intact nondeclarative memory. Intact abilities included artificial grammar learning (MG, PN) (Knowlton et al. 1992), cognitive skill learning (NC, MG, and PN) (Squire and Frambach 1990), facilitated reading speed (MG, PN) (Musen et al. 1990), priming of object naming (NC, MG, PN) (Cave and Squire 1992), and probabilistic classification learning (MG, PN) (Reber et al. 1996).
Performance on other cognitive tests
The memory impairment in NC, MG, and PN appeared largely in the absence of other cognitive deficits. All three scored within normal range on the WAIS-R (Table 1). All three also scored similarly to controls on the Dementia Rating Scale (Mattis 1976), when the memory subscale was excluded (NC, 110; MG, 119; PN, 112; mean of 11 controls = 115.3; maximum = 119; controls from Janowsky et al. 1989).
The patients also took the Boston Naming Test (Kaplan et al. 1983), the Wisconsin Card Sorting Test (WCST) (Heaton 1981), and the Verbal Fluency Test (Benton and Hamsher 1976). On the Boston Naming Test, which asks participants to name line drawings of 60 objects, the patients performed similarly to controls (NC, 51; MG, 53; PN, 53; mean of six controls = 55.8 ± 1.4, range = 49–58; controls from Squire et al. 1990).
On the WCST, which is sensitive to frontal lobe dysfunction, NC, MG, and PN sorted 4, 6, and 3 categories, respectively (maximum = 6). The Verbal Fluency Test is also sensitive to frontal lobe dysfunction and asks participants to provide as many words as possible in 1 min beginning with the letter F (and then A and S). The patients performed similarly to controls (NC, 31; MG, 32; PN, 35; mean of six controls = 37.5 ± 3.7, range 30–55; controls from Squire et al. 1990).
In summary, all three patients exhibited marked impairment on tests of declarative memory but performed normally on tests of nondeclarative memory and on other tests of cognitive function.
Neuropathological findings
Gross appearance of the brains
All three brains were examined prior to histological analysis. The brain of patient NC appeared normal with no infarcts, atrophy, or sulcal widening. The mammillary nuclei appeared to be normally sized or perhaps slightly reduced in volume. Inspection of blocks of her brain indicated some dilation of the ventricles. Extreme atrophy of the hippocampal formation was also noted bilaterally. MG’s brain exhibited marked atrophy and evidence of recent infarcts in the right hemisphere along most of its rostral-caudal extent. The mammillary nuclei were present and of normal size. The brain of patient PN exhibited signs of sulcal widening, especially in the frontal lobes. A small tumor, thought to be a neurofibroma or meningioma, was noted superficial to the right temporal lobe. There was also evidence of a recent, left-sided, occipital infarct. Finally, her mammillary nuclei appeared shrunken.
Patient NC
Medial temporal lobe
The primary finding in NC’s brain was extensive damage to the entire rostral-caudal extent of the hippocampal formation bilaterally (Fig. 5A–D). The CA1 and CA3 fields appear almost completely acellular. The dentate gyrus is extensively damaged as well, and there appears to be a complete loss of granule cells and hilar cells. The CA2 field and the subiculum appear relatively spared. The perirhinal (Fig. 5E) and parahippocampal cortices appear normal. The entorhinal cortex shows some loss of layer III cells (Fig. 5F).
Figure 5.

Coronal sections through the left and right hippocampal region of NC rostrally (A,B) and caudally (C,D). The subiculum (S) and presubiculum (PreS) appear normal (A–D). Arrowheads indicate nearly complete cell loss in the CA1 fields bilaterally (A–D). Similarly, arrows indicate nearly complete cell loss in the CA3 fields bilaterally (B,C,D). The dentate gyrus (DG) also shows nearly complete cell loss bilaterally (B,C,D). In contrast, CA2 is relatively spared (B,C,D). Furthermore, the entire hippocampal formation appears reduced in volume, failing to fill the ventricle as expected (C,D). Perirhinal cortex (E) appears normal, but entorhinal cortex (ERC) appears to have some cell loss in layer III (F). Layer IV is absent from entorhinal cortex and is not indicated here. Scale bar, 1 mm (A–D) or 0.5 mm (E,F). (S) Subiculum; (PreS) presubiculum; (DG) dentate gyrus; (ERC) entorhinal cortex.
Diencephalon
NC’s diencephalon appears normal. The mammillary nuclei are present bilaterally and show no signs of pathology. The mammillothalamic tract and the thalamus bilaterally also appear normal.
Patient MG
Medial temporal lobe
MG’s medial temporal lobes appear normal, including the hippocampal region, and the entorhinal, perirhinal, and parahippocampal cortices.
Diencephalon
Within the last 2 wk of her life, MG suffered a right-sided stroke. The consequences of this stroke were evident throughout the right hemisphere. There was extensive damage in the region of the right thalamus and internal capsule, and we could not distinguish between damage to the right thalamus from the original infarct that caused amnesia and damage from the more recent infarct. Accordingly, analysis of the thalamus was restricted to the left hemisphere.
MG’s mammillary nuclei and left mammillothalamic tract appear normal. Figure 6 shows the damage in the left thalamus. Figure 6A shows the most rostral extent of the lesion. In this section, the main damage is in the ventral lateral nucleus (VL, mostly likely the motor portion of VLa), and there is evidence of gliosis medially in the region of the paracentral nucleus (Pc). Figure 6B shows a section 2.5 mm caudally. At this level, the infarct has damaged the Pc, the central medial nucleus (CeM), and the central lateral nucleus (CL), which are all considered part of the internal medullary lamina (anterior intralaminar group) (Jones 1985). The CL is also considered part of the mediodorsal nucleus (MD, the densocellular division). Figure 6, C and D shows the lesion caudally 2.5 mm and 5.0 mm, respectively. The focus of the lesion in these sections is the CL, with some damage to the CM, MD, and VL as well. Finally, note that the overall shape of the thalamus is abnormal, suggesting that the tissue has shifted to fill space left by the lesion (Fig. 6D). The other thalamic nuclei, including the anterior nuclei, appear relatively normal.
Figure 6.

Approximately coronal sections through the left thalamus of MG. At each level, black arrows indicate the lesion. (A) Rostrally, the lesion begins in the ventral lateral nuclear complex (likely the motor portion of VLa). At this level, the anterior ventral nucleus (AV) and medial dorsal nucleus (MD) appear normal. The arrowhead and dashed oval indicate marked gliosis in the internal medullary lamina (in the region of Pc, the paracentral nucleus). (B) The lesion is still present 2.5 mm caudally, in the ventral lateral nuclear complex and also includes the internal medullary lamina inferomedially, notably Pc and the central medial nucleus (CeM). (C,D) Lastly, at 5.0 mm and 7.5 mm caudally, the lesion damages mainly the central lateral nucleus (CL). Also note that the thalamus does not protrude into the adjacent ventricle as expected (the dashed line and asterisk in D indicate the approximate, expected curvature of the superior thalamus). Overall, the rostral-caudal extent of the thalamic lesion is ∼1–1.5 cm. Scale bar, 1 mm. (AD) Anterior dorsal nucleus; (AV) anterior ventral nucleus; (CeM) central medial nucleus; (CL) central lateral nucleus; (CM) center médian nucleus; (LD) lateral dorsal nucleus; (LP) lateral posterior nucleus; (MD) medial dorsal nucleus; (Pc) paracentral nucleus; (VA) ventral anterior nucleus; (VL) ventral lateral nucleus; (VPL) ventral posterior lateral nucleus.
Patient PN
Medial temporal lobe
PN exhibits no evidence of pathology in the hippocampus proper, dentate gyrus, or subiculum. The entorhinal, perirhinal, and parahippocampal cortices are normal along their entire rostral-caudal extent.
Diencephalon
PN has extensive damage in the diencephalon. Figure 7 shows damage to the mammillary nuclei bilaterally (Fig. 7A,B), as well as the mammillothalamic tract and the anterior ventral nucleus on the right (Fig. 7C,D). There is similar damage in the left mammillothalamic tract and left thalamus. In the mammillary nuclei, gliosis is so pronounced that the few remaining cells are obscured (Fig. 7A,B). Figure 7, C and D shows that glia have also infiltrated the mammillothalamic tract along its course from the mammillary nuclei to the anterior thalamic nuclei (for comparison, see Fig. 7E for the normal mammillothalamic tract of NC). Within the thalamus itself, the anterior dorsal nucleus is damaged so extensively that it is difficult to identify. Also, the anterior ventral nucleus is reduced in volume and neuronal number. Furthermore, it is extremely gliotic, worse medially than laterally. This pattern of damage is suggestive of primary mammillary nuclei pathology with secondary, transneuronal damage to cells in the anterior nuclei receiving projections via the mammillothalamic tract. The other thalamic nuclei, including the MD and CL, appear normal.
Figure 7.

Coronal sections through the diencephalon of PN. The mammillary nuclei (A, right; B, left) show extensive gliosis as well as neuronal loss. In the thalamus (right side shown in C,D), the mammillothalamic tract shows marked gliosis along its course toward the anterior nuclei (arrowheads in C, D; E shows the normal right mammillothalamic tract for patient NC, for comparison). The anterior ventral nucleus (AV) also shows neuronal loss and extensive gliosis. The damage is more evident ventromedially than dorsolaterally. The anterior dorsal nucleus (AD) (which should be evident in D but cannot be detected) is extremely damaged, retaining only a few shrunken cells overwhelmed by glia. Scale bar, 0.5 mm (A,B) or 1 mm (C–E). (AD) Anterior dorsal nucleus; (AV) anterior ventral nucleus; (CeM) central medial nucleus; (MD) medial dorsal nucleus; (MN) mammillary nucleus; (VA) ventral anterior nucleus; (VL) ventral lateral nucleus.
Comparison of patients
Figure 8 shows the dentate gyrus and CA3 field (left), the CA2 field (middle), and the CA1 field (right) for each of the three patients. Only NC has damage to the dentate gyrus and the CA3 and CA1 fields. The CA2 field appears relatively normal in all three patients. Figure 9 shows one mammillary nucleus (Fig. 9A) and the anterior ventral nucleus (Fig. 9B) for each of the three patients. Only PN has damage to both the mammillary nuclei and the anterior thalamic nuclei (including the anterior dorsal and the anterior ventral nuclei).
Figure 8.

Hippocampal region. Coronal sections through the hippocampal region of each patient. The hippocampal region appears normal in patients MG and PN. In contrast, the hippocampal region of patient NC is markedly abnormal. The dentate gyrus (DG), the CA3 field, and the CA1 field show nearly complete cell loss in NC, although the CA2 field appears relatively normal. Arrows indicate the DG and the CA fields, but some panels include CA fields in addition to the one that is highlighted. Scale bar, 0.5 mm. (S) Subiculum; (DG) dentate gyrus.
Figure 9.

Diencephalon. (A) Coronal sections through the mammillary nucleus (MN) of each patient at low (A, top) and high (A, bottom) magnification. The MN of NC and MG appears normal. In contrast, the MN of PN is gliotic with neuronal loss. (B) Coronal sections through the anterior ventral nucleus (AV) of each patient at low (B, top) and high (B, bottom) magnification. The AV of NC appears normal. The AV of MG appears misshapen but otherwise appears normal (see Results). In contrast, the AV of PN is gliotic with neuronal loss. Scale bar, 0.5 mm (A, top and B, top) or 0.1 mm (A, bottom and B, bottom). (MN) Mammillary nucleus; (AV) anterior ventral nucleus.
Discussion
We have presented neuropsychological and neuropathological findings from three memory-impaired patients (NC, MG, and PN). Patient NC had extensive bilateral damage to the hippocampus (including the dentate gyrus and the CA1 and CA3 fields) and some loss of neurons in layer III of entorhinal cortex. There was no damage to the mammillary nuclei, mammillothalamic tracts, or thalamus. Patient MG had a bilateral stroke that damaged several thalamic nuclei, including nuclei in the anterior intralaminar group of the internal medullary lamina and the mediodorsal nuclei (MD). There was no damage to the mammillary nuclei, mammillothalamic tracts, or medial temporal lobes. Patient PN had alcoholic Korsakoff’s syndrome with damage bilaterally to the mammillary nuclei, mammillothalamic tracts, and anterior nuclei of the thalamus (including the anterior dorsal and the anterior ventral nuclei). All three patients exhibited a similar phenotype of amnesia with marked impairment on tests of declarative memory (anterograde and retrograde memory) but normal performance on tests of nondeclarative memory (e.g., priming and adaptation-level effects) as well as on tests of other cognitive functions (e.g., the WAIS-R and the Dementia Rating Scale).
It is notable that neither PN nor MG exhibited a “band of gliosis” (or any other damage) adjacent to the third ventricle, which was reported in two previous neuropathological studies involving four patients with well-characterized, diencephalic amnesia due to Korsakoff’s syndrome (Mair et al. 1979; Mayes et al. 1988). The precise location and extent of the band of gliosis in the previous studies is somewhat unclear. Mair et al. (1979) described it as located between the subependymal zone and the MD, in the region of (and extending ventrally from) the paratenial nucleus. These investigators also refer to this region as the midline region of the thalamus. Mayes et al. (1988) reported that damage in their patients was very similar to damage in the cases described by Mair et al. (1979).
Note that the band of gliosis in those studies was not limited to the paratenial nucleus itself. Rather, it was located in the anterior and medial thalamus in proximity to the paratenial nucleus (and thus, in the region of the central medial nucleus [CeM], medioventral nucleus, anterior nuclei, and MD). Others have noted that the paratenial nucleus is not prominent in humans (Bentivoglio et al. 1993; Harding et al. 2000), and it has been suggested that the band of gliosis described previously (Mair et al. 1979; Mayes et al. 1988) actually denoted damage to the anterior portion of the MD (Victor et al. 1989; Harding et al. 2000).
The mediodorsal thalamic nucleus has been prominent in discussions of diencephalic amnesia. Indeed, in the largest histological study of Wernicke-Korsakoff’s syndrome, damage to the MD uniquely identified patients with memory impairment (Victor et al. 1989). The mammillary nuclei were also damaged in the patients that were studied, but a few of these patients did not have MD damage and appeared not to have memory impairment. Butters (1984) pointed out that these same observations are consistent with the conclusion that damage in Korsakoff’s syndrome must include both the mammillary nuclei and MD to cause memory impairment, because all of the patients with memory impairment had damage to both structures.
In other cases of diencephalic amnesia, different areas of the thalamus have been emphasized. For example, damage to the internal medullary lamina and the mammillothalamic tract has been associated with memory impairment in cases of thalamic trauma or infarction (von Cramon et al. 1985; Mori et al. 1986; Squire et al. 1989a). Furthermore, a recent study of Korsakoff’s syndrome emphasized the importance of gliosis and neuronal loss in the anterior thalamic nuclei (Harding et al. 2000). In the present study, MG had damage to the internal medullary lamina as well as damage to the MD. PN had damage to the mammillothalamic tracts as well as the anterior thalamic nuclei (and mammillary nuclei). Thus, our findings suggest that damage involving several different diencephalic nuclei and tracts can cause memory impairment, specifically damage to the MD and the internal medullary lamina (along with associated nuclei), as well as damage to the mammillary nuclei, the mammillothalamic tract, and the anterior thalamic nuclei. Accordingly, our findings help reconcile the various studies of diencephalic amnesia that have tended to emphasize particular structures and tracts. We suggest that memory impairment is caused either by damage to the MD and the adjacent internal medullary lamina, or by damage to the mammillary nuclei, the mammillothalamic tract, and the anterior thalamic nuclei.
These diencephalic nuclei and tracts are anatomically related to the MTL. The ventroamygdalofugal pathway, which passes through the internal medullary lamina, connects perirhinal cortex and the amygdala to the MD via the inferior thalamic peduncle (Mishkin 1982; Russchen et al. 1987; Graff-Radford et al. 1990). Furthermore, the hippocampal formation originates projections via the fornix to the mammillary nuclei, which, in turn, project to the anterior thalamic nuclei via the mammillothalamic tract. These connections may explain why diencephalic lesions and MTL lesions can cause a similar memory impairment, as discussed previously (Aggleton and Brown 1999). Findings from functional magnetic resonance imaging (fMRI) also point to an extended anatomical system supporting new learning that includes the medial temporal lobe, the diencephalon and ventrolateral prefrontal cortex (Caulo et al. 2005).
In the present study, one of the patients (NC) had MTL damage. NC had bilateral damage to the CA1 and CA3 fields of the hippocampus, the dentate gyrus, and layer III of entorhinal cortex. This pattern of brain damage appears not to have been described previously in memory-impaired patients and may be related to NC’s history of seizures. Layer III of the entorhinal cortex is epileptogenic, and a prolonged history of seizure can result in layer III cell loss (Du et al. 1995; Schwarcz et al. 2000). Other evidence suggests that the CA2 field is resistant to damage by seizures (Amaral and Insausti 1990).
Early reports suggested that MTL damage and diencephalic damage might produce qualitatively different kinds of memory impairment (Huppert and Piercy 1979; Parkin 1984). The current view is that damage to either region produces the same core memory impairment (e.g., Victor et al. 1989; McKee and Squire 1992; Aggleton and Brown 1999; Van der Werf et al. 2000). Our findings confirm this view by demonstrating the same core amnesic syndrome in patients with MTL or diencephalic lesions, all of whom took the same memory tests.
There has been some debate about the status of recognition memory in amnesia. On the one hand, there is considerable evidence that recognition memory, like other declarative memory abilities (e.g., recall memory), is impaired by either MTL or diencephalic damage (Haist et al. 1992; Reed and Squire 1997; Kopelman and Stanhope 1998; Manns et al. 2003; Zoppelt et al. 2003; Wais et al. 2006) (also a single case, RG [Kishiyama et al. 2005). On the other hand, some studies have reported that recognition memory is normal (or near normal) following damage to these regions (Aggleton and Shaw 1996; Turriziani et al. 2004) (also the single cases ROB [Hanley et al. 2001], YR [Holdstock et al. 2002], MR [Bastin et al. 2004], and KN [Aggleton et al. 2005).
All the studies mentioned above depended on neuroimaging to determine the location and extent of brain damage. Notably, every study that has thoroughly documented brain damage in amnesic patients through post-mortem histological analysis has found impaired recognition memory. Thus, patient RB, who had damage limited to the CA1 field of the hippocampus bilaterally, was impaired on tests of two-alternative, forced-choice, and yes/no recognition memory (Zola-Morgan et al. 1986). In addition, patients GD (damage limited to the CA1 field of the hippocampus), LM (CA1–3, dentate gyrus, entorhinal cortex), and WH (CA1–3, dentate gyrus, entorhinal cortex, subiculum) were impaired on several different tests of recognition memory (Rempel-Clower et al. 1996; Reed and Squire 1997). Lastly, patients EA, BC, JH, and JW (Korsakoff’s syndrome with damage to the mammillary nuclei and a band of gliosis in the midline region of the thalamus) were also impaired on tests of recognition memory (Mair et al. 1979; Mayes et al. 1988). Similarly, all three patients in the present study were impaired on tests of recognition memory (Table 2; Fig. 2). Accordingly, studies of amnesic patients with histologically identified brain lesions are unanimous in finding that recognition memory is impaired following damage to the MTL or diencephalon.
Retrograde memory has also been well studied in patients with MTL or diencephalic lesions. Despite initial reports that retrograde memory impairment was extensive and ungraded (i.e., similar for recent and remote time periods) (Sanders and Warrington 1971; Mair et al. 1979), most studies have found that retrograde memory impairment is temporally graded (i.e., memory for the recent past is impaired more than memory for the remote past), whether after damage limited to the hippocampal region, after more extensive MTL damage, or after diencephalic damage (for review, see Squire and Alvarez 1995). The findings from our three patients confirm that damage to the MTL or diencephalon produces temporally graded retrograde amnesia (Fig. 3).
Nondeclarative memory is known to be intact in patients with damage to the MTL (Squire 1992). The status of nondeclarative memory in patients with well-documented damage to the diencephalon has been less thoroughly studied (as noted by Phaf et al. 2000; Swinnen et al. 2005). Indeed, neither of the two earlier neuropathological studies of patients with Korsakoff’s syndrome commented on the status of nondeclarative memory (Mair et al. 1979; Mayes et al. 1988). The present results confirm that nondeclarative memory is intact following diencephalic lesions. Thus, NC, MG, and PN all exhibited normal priming for words and nonwords (Fig. 4A) as well as normal adaptation-level effects (Fig. 4B), and they also performed normally on other tests of nondeclarative memory (see Results).
In summary, we have presented neuropsychological and neurohistological findings from one patient with damage to the MTL and two patients with diencephalic damage. Damage to the MTL or diencephalon produces a well-circumscribed amnesic syndrome characterized by a common impairment in anterograde and retrograde declarative memory (including recognition memory) and intact nondeclarative memory. The pattern of brain injury in patient NC (damage to the dentate gyrus, CA1 and CA3 fields, and layer III of entorhinal cortex with relative sparing of the CA2 field and the subiculum) has not been reported in previous neurohistological studies of memory impairment and likely is related to her history of seizures. The pattern of brain injury in patient PN (damage to the mammillary nuclei, mammillothalamic tracts, and anterior nuclei of the thalamus) differs from the pattern of brain injury reported in the two previous neurohistological studies of patients with well-characterized amnesia due to Korsakoff’s syndrome (Mair et al. 1979; Mayes et al. 1988) but is consistent with reports that emphasize the importance of the mammillothalamic tract and the anterior thalamic nuclei (e.g., von Cramon et al. 1985; Markowitsch 1988; Harding et al. 2000). MG had damage to the mediodorsal nucleus and the internal medullary lamina, consistent with reports that emphasize the importance of these structures (von Cramon et al. 1985; Victor et al. 1989). It has only rarely been possible to study the anatomy of memory by carrying out extensive neuropsychological testing during life followed by detailed post-mortem neurohistological analysis. The present report adds three cases to this useful literature.
Materials and Methods
Three amnesic patients (NC, MG, and PN) were studied in the laboratory for 7–21 yr prior to death. Their case histories are presented below.
Patient NC
Case history
Patient NC was a left-handed, Caucasian female born in 1943. She received 12 yr of education and subsequently worked as a nurse’s aide for 10 yr. NC reported at least a 14-yr history of alcoholism, although at autopsy her liver showed no signs of cirrhosis. She was not known to have drunk alcohol from 1982 to 1994, the period during which she participated in studies in our laboratory. NC had a history of smoking cigarettes (a pack per day for at least 25 yr). She was prescribed metoprolol, propranolol, and nitrostat for severe hypertension and angina, but these conditions remained uncontrolled.
NC’s psychiatric history is remarkable for childhood hospitalizations for “crazy” behavior. She received a diagnosis of schizo-affective disorder with paranoid-type schizophrenia symptoms and depressive mood disorder. She saw a psychiatrist regularly and controlled the disorder with, among other treatments, trazadone and doxepin.
Her neurological history is remarkable for a seizure disorder that she controlled with Dilantin and a history (self-reported) of frequent head trauma. NC’s neurological history is also remarkable for mild distal polyneuropathy likely related to alcohol abuse, weak right extensor planar responses, “wild” right arm drift, and left facial weakness. She received a diagnosis of sleep apnea shortly before her death.
In 1979, at the age of 36, NC was diagnosed with a severe memory disorder, initially identified as Korsakoff’s syndrome. At the time she was also noted to have poor performance on digit span and on a calculation task. These difficulties were attributed to attention problems, possibly due to her medications. Her memory impairment remained stable during the 12 yr that we tested her. We now suggest that the initial diagnosis of Korsakoff’s syndrome was incorrect, as our histological analysis (see Results) revealed bilateral damage to the hippocampal formation in the absence of diencephalic damage.
In 1992, NC experienced an episode of congestive heart failure accompanied by a small, subendocardial infarction. She was subsequently diagnosed with hypertensive cardiomyopathy. In January 1994, she was hospitalized for pneumonia and later discharged. One month later, she was found deceased in her board-and-care facility. She was 51 yr old. The cause of death was reported as coronary artery disease, including near-complete occlusion of the left anterior descending and right coronary arteries. An autopsy revealed hepatic and pulmonary congestion, as well as arteriolar nephrosclerosis.
Acquisition and preparation of tissue
NC’s death was unattended, so the exact interval between death and removal of the brain is unknown (4–10 h). The brain was placed in cold 10% formalin in 0.1 M phoshate buffer for 3 mo. Photographs of the whole brain were taken, and then the brain was cut into ∼1-cm-thick coronal blocks. Each block was photographed and inspected. Blocks were then returned to cold 10% formalin in 0.1 M phosphate buffer for 2 mo. The blocks were then placed in a cryoproctectant solution of 10% glycerin and 10% formalin in 0.1 M phosphate buffer. After 2 d, the blocks were moved into a cyroprotectant solution of 20% glycerin and 10% formalin in 0.1 M phosphate buffer for 2 mo. Then, whole coronal blocks were frozen between glass slides by placing them in −70°C isopentene for 45 min and stored at −70°C.
Patient MG
Case history
MG was a right-handed, Caucasian female born in 1932. MG had a history of cigarette smoking (∼1.5 packs per day for as long as 30 yr) with no history of alcohol use. She had carotid artery occlusive disease, severe hypertension, and angina controlled with clonidine and nifedipine. Her neurological history is further significant for transient ischemia attacks (TIAs) that resulted in fainting spells.
In 1985, MG underwent bilateral carotid and femoral endarterectomy. In 1986, she was admitted to the hospital where an MRI revealed a symmetric, bithalamic stroke (Fig. 10). At that time, she was noted to have significant anterograde memory loss. She participated in our studies from 1987 to 1994.
Figure 10.
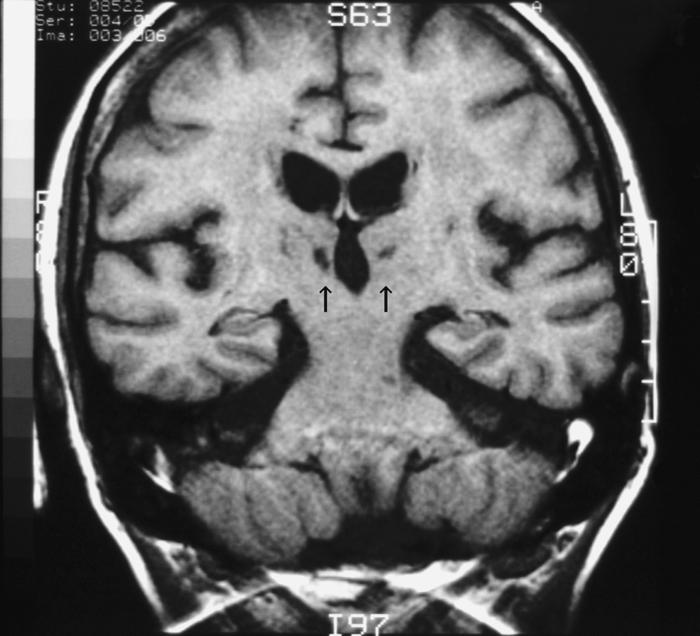
MRI of patient MG. In 1986, magnetic resonance images of the brain of patient MG demonstrated a bilateral infarction in the medial thalamus, likely in the vascular territory of the thalamic-subthalamic arteries (also known as the thalamoperforating arteries or paramedian thalamic arteries). Arrows indicate the lesion in the thalamus.
In June 1994, angiography revealed at least 50% carotid blockage bilaterally, worse on the right. Head CT revealed evidence of small vessel white matter disease and a new, nonhemorrhagic infarct dating to the recent 6 mo. Importantly, she was noted to have no musculoskeletal abnormalities at this time.
On December 12, 1996, MG was admitted to the hospital with acute and chronic renal failure, urinary tract infection, and dehydration. After treatment she was noted to be ambulatory with a walker, but showing a “contracted left upper extremity.” MG was released from the hospital but returned 2 wk later in acute respiratory distress. She was intubated and ventilated. Examination revealed impending gangrene of the left lower leg due to obstruction of the iliac artery. At this time she also had left hemiparesis and a deformed left hand. These findings suggest that she had had a new, right-sided stroke, likely dating to the hospitalization of December 12. Honoring MG’s “do not resuscitate” order, her family requested extubation and death supervened. She was 64 yr old.
Acquisition and preparation of tissue
Approximately 4 h after death, the brain was removed and immediately placed in cold 10% formalin in 0.1 M phoshate buffer for 54 mo. In June 2001, photographs of the whole brain were taken, and then the brain was cut into ∼1-cm-thick coronal blocks. Blocks were then returned to cold 10% formalin in 0.1 M phosphate buffer for 1 mo. Each block was then photographed, and the blocks were placed in a cryoproctectant solution of 10% glycerin and 10% formalin in 0.1 M phosphate buffer. After 4 d, the blocks were moved into a cyroprotectant solution of 20% glycerin and 10% formalin in 0.1 M phosphate buffer for 2 d. Then, whole coronal blocks were frozen between glass slides by placing them in −70°C isopentene for 45 min and stored at −70°C.
Patient PN
Case history
PN was a right-handed, Caucasian female born in 1927. She was a lithographer and licensed vocational nurse with at least 11 yr of education. She had a history of iron deficiency anemia. PN reported that a car accident in about 1960 resulted in left-sided ambulatory difficulty secondary to a hip fracture. She denied brain injury from this accident. PN had multiple incidents of head trauma not associated with loss of consciousness, although one incident resulted in a left parietal skull fracture. Her psychiatric history is remarkable for two self-reported suicide attempts.
PN’s medical history is also remarkable for heavy alcohol abuse of unknown duration. She continued to binge drink while living in a board-and-care facility during the 1990s. In 1979, PN was diagnosed with significant memory impairment due to mild-to-moderate Korsakoff’s syndrome. A CT scan in 1985 demonstrated decreased tissue density in the thalamus and the caudate nucleus, and cortical atrophy as measured by increased fluid in the frontal sulcal and peri-Sylvian areas (Shimamura et al. 1988). An MRI in 1989 (Squire et al. 1990) revealed an enlarged lateral ventricle and a marked reduction in the volume of the mammillary nuclei (<5% of mean control volume). The temporal lobe, hippocampal formation, and parahippocampal gyrus were of normal size. PN participated in our studies from 1979 to 2000.
In January 2000, PN was admitted to the hospital after blood was noted in her stool. Endoscopy revealed damage to the lower portion of the stomach resulting from bile gastritis. The lower portion of her stomach was removed, and the remaining portion was connected to the duodenum. PN died in April 2000 of a vascular disorder of the intestine. She was 72 yr old.
Acquisition and preparation of tissue
Approximately 1 h after death, the carotid arteries were cannulated and the brain was perfused with 4% paraformaldehyde solution for ∼1 h. The brain was then removed, and a specimen of a right-sided temporal lobe tumor (thought to be a neurofibroma or meningioma) was taken for evaluation. The brain was then suspended in 4% paraformaldehyde in 0.01 M phosphate buffer for 1 d. Then, photographs of the whole brain were taken, and the brain was placed in fresh 4% paraformaldehyde in 0.01 M phosphate buffer. On the following day, the brain was cut into ∼1-cm-thick blocks and returned to 4% parafomaldehyde for 48 h. Then the blocks were placed into a cryoproctectant solution of 10% glycerin and 2% DMSO in 0.01 M phosphate buffer for 48 h. The blocks were then photographed and placed in 20% glycerin and 2% DMSO in 0.01 M phosphate buffer for 48 h. They were then frozen between glass slides by placing them in −70°C isopentane for 35 min and stored at −70°C.
Processing of brain tissue
After the brains were frozen, all three were processed in the same way. Whole coronal blocks were sectioned into left and right hemispheres along the midline commissures. The thalamus and medial temporal lobes were separated and processed separately. Continuous 50-μm sections were then cut through each frozen block using one of two freezing microtomes (a MICROM HM440e or a Reichert sliding microtome). Every tenth section was mounted on a gelatin-coated slide and stained with 0.25% thionin. The unmounted sections were stored in a cryoprotectant solution of 30% ethylene glycol and 25% glycerol in 0.01 M phosphate buffer.
Evaluation of brain tissue
Analysis involved qualitative assessment of the medial temporal lobe and diencephalon bilaterally.
Acknowledgments
This study was supported by the Medical Research of the Department of Veterans Affairs, National Institute of Mental Health (NIMH) Grant MH24600, the Metropolitan Life Foundation, and NIMH Training Grant 5-T32-MH20002 (J.J.G.). We thank David Amaral, Jeffrey Bennett, Ryan Bromm, Jennifer Frascino, Veronica Galván, E.G. Jones, Cecelia Manzanares, Lisa Stefanacci, Alisha West, Stuart Zola and Joyce Zouzounis for assistance.
Footnotes
Article published online before print. Article and publication date are at http://www.learnmem.org/cgi/doi/10.1101/lm.357406
References
- Aggleton J.P., Brown M.W. Episodic memory, amnesia, and the hippocampal–anterior thalamic axis. Behav. Brain Sci. 1999;22:425–444. [PubMed] [Google Scholar]
- Aggleton J.P., Shaw C. Amnesia and recognition memory: A re-analysis of psychometric data. Neuropsychologia. 1996;34:51–62. doi: 10.1016/0028-3932(95)00150-6. [DOI] [PubMed] [Google Scholar]
- Aggleton J.P., Vann S.D., Denby C., Dix S., Mayes A.R., Roberts N., Yonelinas A.P. Sparing of the familiarity component of recognition memory in a patient with hippocampal pathology. Neuropsychologia. 2005;43:1810–1823. doi: 10.1016/j.neuropsychologia.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Amaral D.G., Insausti R. Hippocampal formation. In: Paxinos G., editor. The human nervous system. Academic Press; San Diego, CA: 1990. pp. 711–755. [Google Scholar]
- Baddeley A. The concept of episodic memory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:1345–1350. doi: 10.1098/rstb.2001.0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin C., Linden M., Charnallet A., Denby C., Montaldi D., Roberts N., Mayes A.R. Dissociation between recall and recognition memory performance in an amnesic patient with hippocampal damage following carbon monoxide poisoning. Neurocase. 2004;10:330–344. doi: 10.1080/13554790490507650. [DOI] [PubMed] [Google Scholar]
- Bentivoglio M., Kultas-Ilinsky K., Illinsky I. Limbic thalamus: Structure, intrinsic organization, and connections. In: Vogt B.A., Gabriel M., editors. Neurobiology of cingulate cortex and limbic thalamus: A comprehensive handbook. Birkhäuser; Boston: 1993. pp. 71–122. [Google Scholar]
- Benton A.L., Hamsher K.D. Multilingual aphasia examination. University of Iowa Press; Iowa City: 1976. [Google Scholar]
- Benzing W.C., Squire L.R. Preserved learning and memory in amnesia: Intact adaptation-level effects and learning of stereoscopic depth. Behav. Neurosci. 1989;103:38–47. doi: 10.1037//0735-7044.103.3.538. [DOI] [PubMed] [Google Scholar]
- Butters N. Alcoholic Korsakoff’s syndrome: An update. Semin. Neurol. 1984;4:429–447. [Google Scholar]
- Caulo M., Van Hecke J., Toma L., Ferretti A., Tataro A., Colosimo C., Romani G.L., Uncini A. Functional MRI study of diencephalic amnesia in Wernicke-Korsakoff syndrome. Brain. 2005;128:1584–1594. doi: 10.1093/brain/awh496. [DOI] [PubMed] [Google Scholar]
- Cave C.B., Squire L.R. Intact and long-lasting repetition priming in amnesia. J. Exp. Psychol. Learn. Mem. Cogn. 1992;18:509–520. doi: 10.1037//0278-7393.18.3.509. [DOI] [PubMed] [Google Scholar]
- Corkin S., Amaral D.G., Gonzalez R.G., Johnson K.A., Hyman B.T. H.M.’s medial temporal lobe lesion: Findings from magnetic resonance imaging. J. Neurosci. 1997;17:3964–3979. doi: 10.1523/JNEUROSCI.17-10-03964.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crovitz H.F., Schiffman H. Frequency of episodic memories as a function of their age. Neuropsychologia. 1974;21:213–234. [Google Scholar]
- Delis D.C., Squire L.R., Bihrle A., Massman P. Componential analysis of problem-solving ability: Performance of patients with frontal lobe damage and amnesic patients on a new sorting test. Neuropsychologia. 1992;30:683–697. doi: 10.1016/0028-3932(92)90039-o. [DOI] [PubMed] [Google Scholar]
- Du F., Eid T., Lothman E.W., Kohler C., Schwarcz R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J. Neurosci. 1995;15:6301–6313. doi: 10.1523/JNEUROSCI.15-10-06301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H., Cohen N. From conditioning to conscious recollection: Memory systems of the brain. Oxford University Press; New York: 2001. [Google Scholar]
- Graff-Radford N.R., Tranel D., Van Hoesen G.W., Brandt J.P. Diencephalic amnesia. Brain. 1990;113:1–25. doi: 10.1093/brain/113.1.1. [DOI] [PubMed] [Google Scholar]
- Haist F., Musen G., Squire L.R. Intact priming of words and nonwords in amnesia. Psychobiology. 1991;19:275–285. [Google Scholar]
- Haist F., Shimamura A.P., Squire L.R. On the relationship between recall and recognition memory. J. Exp. Psychol. Learn. Mem. Cogn. 1992;18:691–702. doi: 10.1037//0278-7393.18.4.691. [DOI] [PubMed] [Google Scholar]
- Hanley J.R., Davies A.D., Downes J.J., Roberts J.N., Gong Q.Y., Mayes A.R. Remembering and knowing in a patient with preserved recognition and impaired recall. Neuropsychologia. 2001;39:1003–1010. doi: 10.1016/s0028-3932(01)00020-3. [DOI] [PubMed] [Google Scholar]
- Harding A., Halliday G., Caine D., Kril J. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123:141–154. doi: 10.1093/brain/123.1.141. [DOI] [PubMed] [Google Scholar]
- Heaton R.K. Wisconsin card sorting test manual. Psychology Assessment Resources; Odessa, FL: 1981. [Google Scholar]
- Holdstock J.S., Mayes A.R., Roberts N., Cezayirli E., Isaac C.L., O’Reilly R.C., Norman K.A. Under what conditions is recognition spared relative to recall after selective hippocampal damage in humans? Hippocampus. 2002;12:341–351. doi: 10.1002/hipo.10011. [DOI] [PubMed] [Google Scholar]
- Huppert F.A., Piercy M. Normal and abnormal forgetting in organic amnesia: Effect of locus of lesion. Cortex. 1979;15:385–390. doi: 10.1016/s0010-9452(79)80065-9. [DOI] [PubMed] [Google Scholar]
- Janowsky J.S., Shimamura A.P., Squire L.R. Source memory impairment in patients with frontal lobe lesions. Neuropsychologia. 1989;27:1043–1056. doi: 10.1016/0028-3932(89)90184-x. [DOI] [PubMed] [Google Scholar]
- Jones E.G. The thalamus. Plenum Press; New York: 1985. [Google Scholar]
- Kaplan E.F., Goodglass H., Weintraub S. The Boston naming test. Lea and Febiger; Philadelphia: 1983. [Google Scholar]
- Kishiyama M.M., Yonelinas A.P., Kroll N.E., Lazzara M.M., Nolan E.C., Jones E.G., Jagust W.J. Bilateral thalamic lesions affect recollection- and familiarity-based recognition memory judgments. Cortex. 2005;41:778–788. doi: 10.1016/s0010-9452(08)70296-x. [DOI] [PubMed] [Google Scholar]
- Knowlton B.J., Ramus S.J., Squire L.R. Intact artificial grammar learning in amnesia: Dissociation of category-level knowledge and explicit memory for specific instances. Psychol. Sci. 1992;3:172–179. [Google Scholar]
- Kopelman M.D., Stanhope N. Recall and recognition memory in patients with focal frontal, temporal lobe and diencephalic lesions. Neuropsychologia. 1998;36:785–795. doi: 10.1016/s0028-3932(97)00167-x. [DOI] [PubMed] [Google Scholar]
- MacKinnon D.F., Squire L.R. Autobiographical memory and amnesia. Psychobiology. 1989;17:247–256. [Google Scholar]
- Mair W.G., Warrington E.K., Weiskrantz L. Memory disorder in Korsakoff’s psychosis: A neuropathological and neuropsychological investigation of two cases. Brain. 1979;102:749–783. doi: 10.1093/brain/102.4.749. [DOI] [PubMed] [Google Scholar]
- Manns J.R., Hopkins R.O., Reed J.M., Kitchener E.G., Squire L.R. Recognition memory and the human hippocampus. Neuron. 2003;37:171–180. doi: 10.1016/s0896-6273(02)01147-9. [DOI] [PubMed] [Google Scholar]
- Markowitsch H.J. Diencephalic amnesia: A reorientation toward tracts? Brain. Res. Rev. 1988;13:351–370. doi: 10.1016/0006-8993(88)91226-7. [DOI] [PubMed] [Google Scholar]
- Mattis S. Dementia rating scale. In: Bellack R., Karasu B., editors. Geriatric psychiatry. Vol. 1. Grune and Stratton; New York: 1976. pp. 77–121. [Google Scholar]
- Mayes A.R., Meudell P.R., Mann D., Pickering A. Location of lesions in Korsakoff’s syndrome: Neuropsychological and neuropathological data on two patients. Cortex. 1988;24:367–388. doi: 10.1016/s0010-9452(88)80001-7. [DOI] [PubMed] [Google Scholar]
- McKee R.D., Squire L.R. Equivalent forgetting rates in long-term memory for diencephalic and medial temporal lobe amnesia. J. Neurosci. 1992;12:3765–3772. doi: 10.1523/JNEUROSCI.12-10-03765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner B., Squire L.R., Kandel E.R. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- Mishkin M. A memory system in the monkey. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1982;298:83–95. doi: 10.1098/rstb.1982.0074. [DOI] [PubMed] [Google Scholar]
- Mori E., Yamadori A., Mitani Y. Left thalamic infarction and disturbance of verbal memory: A clinicoanatomical study with a new method of computed tomographic stereotaxic lesion localization. Ann. Neurol. 1986;20:671–676. doi: 10.1002/ana.410200604. [DOI] [PubMed] [Google Scholar]
- Musen G., Shimamura A.P., Squire L.R. Intact text-specific reading skill in amnesia. J. Exp. Psychol. Learn. Mem. Cogn. 1990;16:1068–1076. doi: 10.1037//0278-7393.16.6.1068. [DOI] [PubMed] [Google Scholar]
- Osterrieth P.A. Le test de copie d’une figure complexe [The test of copying a complex figure] Arch. Psychol. (Frankf.) 1944;30:306–356. [Google Scholar]
- Parkin A.J. Amnesic syndrome: A lesion-specific disorder. Cortex. 1984;20:479–508. doi: 10.1016/s0010-9452(84)80053-2. [DOI] [PubMed] [Google Scholar]
- Phaf H.R., Geurts H., Eling P.A. Word frequency and word stem completion in Korsakoff patients. J. Clin. Exp. Neuropsychol. 2000;22:817–829. doi: 10.1076/jcen.22.6.817.956. [DOI] [PubMed] [Google Scholar]
- Reber P.J., Squire L.R. Parallel brain systems for learning with and without awareness. Learn. Mem. 1994;1:217–229. [PubMed] [Google Scholar]
- Reber P.J., Knowlton B.J., Squire L.R. Dissociable properties of memory systems: Differences in the flexibility of declarative and nondeclarative knowledge. Behav. Neurosci. 1996;110:861–871. doi: 10.1037//0735-7044.110.5.861. [DOI] [PubMed] [Google Scholar]
- Reed J.M., Squire L.R. Impaired recognition memory in patients with lesions limited to the hippocampal formation. Behav. Neurosci. 1997;111:667–675. doi: 10.1037//0735-7044.111.4.667. [DOI] [PubMed] [Google Scholar]
- Rempel-Clower N.L., Zola S.M., Squire L.R., Amaral D.G. Three cases of enduring memory impairment after bilateral damage limited to the hippocampal formation. J. Neurosci. 1996;16:5233–5255. doi: 10.1523/JNEUROSCI.16-16-05233.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey A. L’examinen clinique en psychologie [The clinical exam in psychology] Universitaires de France; Paris: 1964. [Google Scholar]
- Ribot T. Les Maladies de la Memoire [Diseases of memory] Appleton-Century-Crofts; New York: 1881. [Google Scholar]
- Russchen F.T., Amaral D.G., Price J.L. The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J. Comp. Neurol. 1987;256:175–210. doi: 10.1002/cne.902560202. [DOI] [PubMed] [Google Scholar]
- Sanders H.I., Warrington E.K. Memory for remote events in amnesic patients. Brain. 1971;94:661–668. doi: 10.1093/brain/94.4.661. [DOI] [PubMed] [Google Scholar]
- Schacter D.L. The seven sins of memory: How the mind forgets and remembers. Houghton Mifflin; Boston: 2001. [Google Scholar]
- Schwarcz R., Eid T., Du F. Neurons in layer III of the entorhinal cortex. A role in epileptogenesis and epilepsy? Ann. N.Y. Acad. Sci. 2000;911:328–342. doi: 10.1111/j.1749-6632.2000.tb06735.x. [DOI] [PubMed] [Google Scholar]
- Scoville W.B., Milner B. Loss of recent memory after bilateral hippocampal lesions. J. Neurol. Neurosurg. Psychiatr. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A.P., Jernigan T.L., Squire L.R. Korsakoff’s syndrome: Radiological (CT) findings and neuropsychological correlates. J. Neurosci. 1988;8:4400–4410. doi: 10.1523/JNEUROSCI.08-11-04400.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire L.R. Memory and the hippocampus: A synthesis from findings with rats, monkeys, and humans. Psychol. Rev. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- Squire L.R., Alvarez P. Retrograde amnesia and memory consolidation: A neurobiological perspective. Curr. Opin. Neurobiol. 1995;5:169–177. doi: 10.1016/0959-4388(95)80023-9. [DOI] [PubMed] [Google Scholar]
- Squire L.R., Frambach M. Cognitive skill learning in amnesia. Psychobiology. 1990;18:109–117. [Google Scholar]
- Squire L.R., Shimamura A.P. Characterizing amnesia patients for neurobehavioral study. Behav. Neurosci. 1986;100:866–877. doi: 10.1037//0735-7044.100.6.866. [DOI] [PubMed] [Google Scholar]
- Squire L.R., Amaral D.G., Zola-Morgan S., Kritchevsky M., Press G. Description of brain injury in the amnesic patient N.A. based on magnetic resonance imaging. Exp. Neurol. 1989a;105:23–35. doi: 10.1016/0014-4886(89)90168-4. [DOI] [PubMed] [Google Scholar]
- Squire L.R., Haist F., Shimamura A.P. The neurology of memory: Quantitative assessment of retrograde amnesia in two groups of amnesic patients. J. Neurosci. 1989b;9:828–839. doi: 10.1523/JNEUROSCI.09-03-00828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire L.R., Amaral D.G., Press G.A. Magnetic resonance imaging of the hippocampal formation and mammillary nuclei distinguish medial temporal lobe and diencephalic amnesia. J. Neurosci. 1990;10:3106–3117. doi: 10.1523/JNEUROSCI.10-09-03106.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire L.R., Stark C.E.L., Clark R.E. The medial temporal lobe. Annu. Rev. Neurosci. 2004;27:279–306. doi: 10.1146/annurev.neuro.27.070203.144130. [DOI] [PubMed] [Google Scholar]
- Stefanacci L., Buffalo E.A., Schmolck H., Squire L.R. Profound amnesia after damage to the medial temporal lobe: A neuroanatomical and neuropsychological profile of patient E.P. J. Neurosci. 2000;20:7024–7036. doi: 10.1523/JNEUROSCI.20-18-07024.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen S.P., Puttemans V., Lamote S. Procedural memory in Korsakoff’s disease under different movement feedback conditions. Behav. Brain Res. 2005;159:127–133. doi: 10.1016/j.bbr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Talland G.A. Deranged memory: A psychonomic study of the amnesic syndrome. Academic Press; New York: 1965. [Google Scholar]
- Turriziani P., Fadda L., Caltagirone C., Carlesimo G.A. Recognition memory for single items and for associations in amnesic patients. Neuropsychologia. 2004;42:426–433. doi: 10.1016/j.neuropsychologia.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Van der Werf Y.D., Witter M.P., Uylings H.B., Jolles J. Neuropsychology of infarctions in the thalamus: A review. Neuropsychologia. 2000;38:613–627. doi: 10.1016/s0028-3932(99)00104-9. [DOI] [PubMed] [Google Scholar]
- Victor M., Adams R.D., Collins G.H. The Wernicke-Korsakoff syndrome and related neurologic disorders due to alcoholism and malnutrition. F.A. Davis Company; New York: 1989. 2nd ed. [Google Scholar]
- von Cramon D.Y., Hebel N., Schuri U. A contribution to the anatomical basis of thalamic amnesia. Brain. 1985;108:993–1008. doi: 10.1093/brain/108.4.993. [DOI] [PubMed] [Google Scholar]
- Wais P., Wixted J.T., Hopkins R.O., Squire L.R. The hippocampus supports both the recollection and the familiarity components of recognition memory. Neuron. 2006;49:459–466. doi: 10.1016/j.neuron.2005.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington E.K. Recognition memory test. NFER-Nelson; Windsor, UK: 1984. [Google Scholar]
- Zola-Morgan S., Squire L.R., Amaral D.G. Human amnesia and the medial temporal region: Enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J. Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zola-Morgan S., Squire L.R., Ramus S.J. Severity of memory impairment in monkeys as a function of locus and extent of damage within the medial temporal lobe memory system. Hippocampus. 1994;4:483–495. doi: 10.1002/hipo.450040410. [DOI] [PubMed] [Google Scholar]
- Zoppelt D., Koch B., Schwarz M., Daum I. Involvement of the mediodorsal thalamic nucleus in mediating recollection and familiarity. Neuropsychologia. 2003;41:1160–1170. doi: 10.1016/s0028-3932(03)00019-8. [DOI] [PubMed] [Google Scholar]