

Abstract
Background
Mucopolysaccharidosis type IIIA (MPS IIIA) is the most common of the mucopolysaccharidoses. The disease is caused by a deficiency of the lysosomal enzyme sulphamidase and results in the storage of the glycosaminoglycan (GAG), heparan sulphate. MPS IIIA is characterised by widespread storage and urinary excretion of heparan sulphate, and a progressive and eventually profound neurological course. Gene therapy is one of the few avenues of treatment that hold promise of a sustainable treatment for this disorder.
Methods
The murine sulphamidase gene cDNA was cloned into a lentiviral vector and high-titre virus produced. Human MPS IIIA fibroblast cultures were transduced with the sulphamidase vector and analysed using molecular, enzymatic and metabolic assays. High-titre virus was intravenously injected into six 5-week old MPS IIIA mice. Three of these mice were pre-treated with hyperosmotic mannitol. The weight of animals was monitored and GAG content in urine samples was analysed by polyacrylamide gel electrophoresis.
Results
Transduction of cultured MPS IIIA fibroblasts with the sulphamidase gene corrected both the enzymatic and metabolic defects. Sulphamidase secreted by gene-corrected cells was able to cross correct untransduced MPS IIIA cells. Urinary GAG was found to be greatly reduced in samples from mice receiving the vector compared to untreated MPS IIIA controls. In addition, the weight of treated mice became progressively normalised over the 6-months post-treatment.
Conclusion
Lentiviral vectors appear promising vehicles for the development of gene therapy for MPS IIIA.
Background
The mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders that arise from deficiencies in the catabolism of glycosaminoglycans (GAG) [1]. At present there are eleven known MPS, each resulting from the deficiency of a different lysosomal enzyme. Of the MPS, MPS IIIA (Sanfillipo A syndrome) is one of the most common, and as far as treatment goes, one of the most intractable, in that central nervous system (CNS) pathology is paramount [1]. Severely affected patients usually present by 2–3 years of age with a range of symptoms related to CNS pathology. These symptoms include delayed development, hyperactivity, aggressive behaviour, and sleep disturbances. Other symptoms include hirsutism and diarrhoea. The somatic manifestations of the disease, which include skeletal pathology, hepatosplenomegaly and joint stiffness, are generally milder and are more commonly found in older patients. MPS IIIA results from a genetically determined deficiency of sulphamidase, a lysosomal enzyme which normally catalyses the cleavage of N-linked sulphate from glucosamine residues at the non-reducing terminus of heparan sulphate. As this represents an obligatory step in the degradation of heparan sulphate, elevated levels of heparan sulphate fragments are found in tissues and in the urine. MPS IIIA also results in the secondary storage of GM2 and GM3 gangliosides in the CNS.
MPS IIIA represents a useful paradigm for therapies aimed at treating the widespread pathology which is found in many of the MPS. The availability of small (mouse) [2] and large (dog) [3,4] animal models of MPS IIIA provides a useful experimental resource for the preclinical development and testing of therapies.
The MPS IIIA mouse [2], the result of a spontaneous mutation, shows many of the progressive pathological features found in the human disease. By 6–7 months of age affected mice are noticeably less active, develop a scruffy appearance, hunched posture and abdominal distension, and lifespan is shortened. Affected animals show elevated levels of urinary GAG, which is predominantly heparan sulphate, greatly decreased sulphamidase activity in all tissues, normal or supranormal levels of other lysosomal enzymes and widely distributed storage. MPS IIIA mice are also significantly heavier than normals. The MPS IIIA mouse therefore provides an excellent model for the initial analysis of gene therapy strategies for the MPS in general, and MPS IIIA in particular.
Although intravenous enzyme replacement therapy has now been developed for a number of the MPS, it is obvious that this approach is not a viable option for treatment of CNS pathology due to its effective partitioning from the peripheral circulation by the blood-brain barrier [5]. Alternative therapies for MPS CNS pathology include small molecule therapies, aimed at preventing synthesis of storage material [6,7], and gene replacement therapy [7]. There is also accumulating evidence that the blood-brain barrier is not completely impermeable to lysosomal enzymes, and that high levels of enzyme in the peripheral circulation, delivered either by enzyme replacement therapy [8], or via gene therapy [9], result in delivery of significant amounts (i.e. high enough to affect pathology) of enzyme to the CNS.
Gene replacement therapy holds obvious potential for the treatment of the MPS [7,10,11], including MPS IIIA. We have previously demonstrated retroviral-mediated gene correction of cultured MPS IIIA fibroblasts [12]. However, retroviruses have serious limitations [13] that preclude their use in gene delivery to the CNS, and make them of limited utility in the transduction of any tissue in vivo. In order to further the development of gene therapy for MPS IIIA we have developed a lentiviral vector that expresses the murine sulphamidase gene and shown that it can be used to correct MPS IIIA cells in vitro. After intravenous administration of the vector to MPS IIIA mice urinary GAG and the weight of treated mice became progressively normalised over the 6-month period following vector administration.
Materials and methods
PCR
The primers used for amplification of the murine sulphamidase gene were msulatg (GGGCCCATCGATGCCACC ATGCACTGCCCGGGACTGGCCTG); msulbglr (GAGGGTCGTAGATCTGGGGTGTCC); msulbglf (GGACACCCCAGATCTACGACCCTC) and msultga (GGGCCCGAATTC TCAGAGTTCATTGTGAAGCGGTC). Sequences homologous to the murine sulphamidase gene sequence are shown in italics. The reaction conditions for all PCRs were 94°C, 30 seconds, 60°C, 20 seconds and 68°C, 1 minute for 20 cycles and used the Expand High Fidelity system (Roche). First strand cDNA from NIH3T3 cells (ATCC CRL-1658) was used as template. A 648 bp fragment corresponding to the 5' part of the murine sulphamidase cDNA sequence was amplified with the primers msulatg and msulbglr, and cloned as a ClaI/BglII fragment in pSP70 (Progen). A 872 bp fragment corresponding to the 3' part of the sequence was amplified with the primers msulbglf and msultga and cloned as an BglII/EcoRI fragment, again in pSP70.
Lentiviral vector
The lentiviral vector used in this study was essentially the same as the pAF2Δ-SE vector [14] except that the SV40 early promoter was replaced with the murine phosphoglycerate kinase gene promoter from MSCVpac [15] to give pHIV-1pgkEYFP
Virus production
The production and purification of the virus used in this work has been described elsewhere [16]. The virus was resuspended in 0.9% (w/v) NaCl and quantified by p24 ELISA (NEN-Dupont). Virus for in vivo administration was shown to be negative for replication competent virus [14].
Cell culture, transduction, and enzymatic and metabolic analysis
Normal and MPS IIIA human skin fibroblasts were plated in 6-well plates and grown till confluent in DMEM/10% (v/v) FCS (2 mL per well). The medium was then aspirated and the cells fed with 1.5 mL of DMEM/10% (v/v) FCS containing 8 μg/mL polybrene. MPS IIIA cells were then transduced with vector for 24 hours. The medium was then exchanged for growth medium. For analysis, medium was exchanged for Ham's F12/10% (v/v) FCS, and after 4 hours the medium was exchanged again for Ham's F12/10% (v/v) FCS containing 10 μCi/ml 35SO4; a further 24 hours later the label was removed and the cells fed with DMEM/10% (v/v) FCS. To assess enzymatic cross-correction, labelled MPS IIIA cells were exposed to medium collected from lentivirus-transduced cells for 24 hours. For analysis of storage, cells were harvested 72 hours after labelling and cell lysates prepared by freeze/thaw in 20 mM Tris-HCl, pH 7.0, 500 mM NaCl. The cell lysates were then clarified by microcentrifugation (13,000 g, 5 minutes) and the supernatants assayed for sulphamidase [17] and β-hexosaminidase [18] activity, total protein and 35S cpm. The pellets resulting from the microcentrifugation of the freeze/thaw cell lysates were used to prepare genomic DNA using the Promega Wizard SV Genomic DNA kit.
Real time PCR analysis
Vector sequences were detected in genomic DNA using a TaqMan assay for gag gene sequences present in the vector (Forward primer 5' AGCTAGAACGATTCGCAGTTGAT 3', reverse primer 5' CCAGTATTTGTCTACAGCCTTCTGA 3', probe 5' CCTGGCCTGTTAGAAAC 3' with FAM/NFQ reporter). Results were normalized using a single copy sequence in the transferrin gene (Forward primer 5' AAGCAGCCAAATTAGCATGTTGAC 3', reverse primer 5' GGTCTGATTCTCTGTTTAGCTGACA 3', probe 5' CTGGCCTGAGCTCCT 3' with FAM/NFQ reporter). The assays were run under standard conditions and using Applied biosystems TaqMan Universal PCR Master Mix. DNA from an A549 derived cell line, and containing a single copy of the lentiviral vector, was used to provide an absolute standard for copy number. Real time PCR was performed on an ABI 7300 cycler and analyzed using Sequence Detection Software v1.2.2 (Applied Biosystems). All samples were analyzed in triplicate.
Treatment of MPS IIIA mice
The MPS IIIA mouse colony was originally established from mice provided by Dr. P. Stanley (Albert Einstein Institute College Medicine, New York). The mice were housed in the Women's and Children's Hospital Animal Care Facility where general maintenance was provided by trained animal care staff. MPS IIIA and normal mice were genotyped by PCR using previously established methods [19]. Six 5-week old male MPS IIIA mice were injected with 50 μg p24 equivalent of the lentiviral vector via injection into the tail vein. Three mice were pre-treated by intravenous injection of hyperosmotic mannitol (200 μl of 25% (w/v) mannitol in saline) 5 minutes prior to the administration of the vector in an attempt to achieve vector delivery to the CNS.
Analysis of urinary GAG
Samples of mouse urine were incubated for one hour at 37°C with two volumes of 0.1% cetylpyridinium chloride in 0.054 M Na3 citrate (pH 4.8). Samples were centrifuged for 10 minutes at 3000 rpm and pellets were resuspended in 150 μL 2 M LiCl. Following addition of 800 μL absolute ethanol, samples were incubated at -20°C for one hour and then centrifuged for 10 minutes at 3000 rpm. Pellets were resuspended in 200 μL of water, lyophilised, and then resuspended in 20 μL water.
Purified glycosaminoglycan samples (0.2 μmol creatinine equivalents) were analysed on 40–50% linear gradient polyacrylamide gels as previously described [20].
Statistical analysis
Except for weight data, results were analysed using one way ANOVA/SNK [21].
Mouse weights were analysed by comparison of non-linear trends for each group. Firstly, the relationship between body weight and age for each group was modelled using a cubic smoothing spline. Modelling using different non-linear trends for each group was then compared with modelling using the same non-linear trend for all groups and the log-likelihood values compared to determine significance. Where significance was found prediction intervals were calculated for the splines to determine the ages at which the groups were significantly different. In age intervals where predicted values for a group did not overlap the predicted interval for another group the treatment groups being compared were taken as being significantly different (p < 0.001).
Results
Isolation of the murine sulphamidase cDNA sequence and construction of lentiviral vectors transducing murine sulphamidase
The murine sulphamidase cDNA sequence was PCR amplified in two parts as described in Materials and methods. The 5' primer used to amplify the 5' part of the sequence was designed to introduce a Kozak consensus sequence immediately prior to the initiation codon. DNA sequencing was used to confirm the absence of PCR induced errors. The two fragments were then joined via the common BglII site to generate a full length sequence. The full length sequence was then cloned into the pHIV-1pgk vector placing it under the transcriptional control of the pgk promoter (Fig. 1). The resulting construct was designated pHIV-1pgkmsulp
Figure 1.
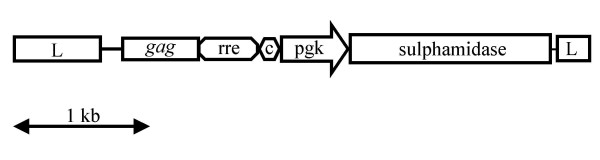
Schematic representation of pHIV-1pgkmsulp lentiviral vector. Schematic of pHIV-1pgkmsulp lentiviral vector showing pertinent elements. L, long terminal repeat, gag, gag gene sequence; rre, Rev-response element; c, central polypurine tract; pgk, murine phosphoglycerate kinase gene promoter; sulphamidase, murine sulphamidase cDNA sequence.
Correction of MPS IIIA skin fibroblasts, metabolic analysis
MPS IIIA skin fibroblasts were transduced with 68, 167 or 508 ng p24 equivalent of the pHIV-1pgkmsulph vector per well (6-well plate) as described in Materials and methods. Labelling with 35SO4 demonstrated that all vector transduced cells were metabolically normalised (Fig. 2), as were cells exposed to medium collected from cells transduced with 508 ng p24 equivalent of vector (Fig. 2). This medium contained 16 pmol/min/ml of sulphamidase activity. Conditioned medium from control MPS IIIA cells did not contain detectable sulphamidase activity. In all cases storage was significantly different from control MPS IIIA cells (P < 0.01) and not significantly different from control normal cells (P > 0.05).
Figure 2.
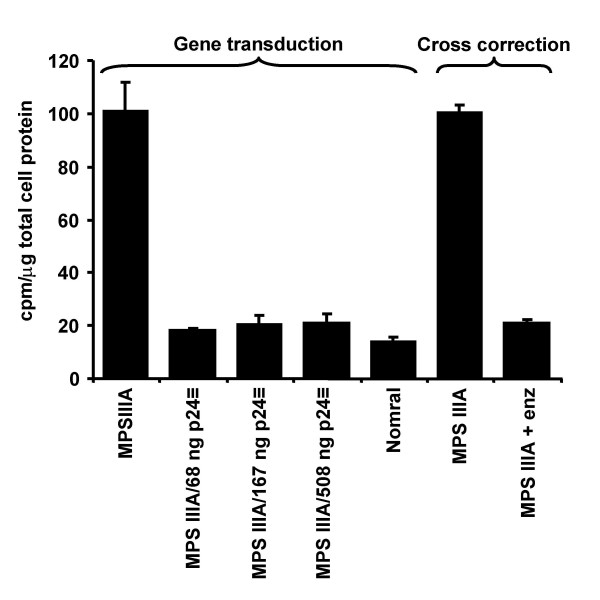
Correction of MPS IIIA storage phenotype in vitro. MPS IIIA cells were transduced with 68, 167 or 508 ng p24 equivalent of the pHIV-1pgkmsulp vector and cell lysates assayed for incorporation of 35SO4 as described in Materials and methods (Gene transduction). Conditioned medium from the cells transduced with 508 ng p24 equivalent of vector was added to untransduced cells (cross correction), and again, cell lysates assayed for incorporation of 35SO4. Normal and untransduced MPS IIIA cells were used as controls.
Correction of MPS IIIA skin fibroblasts, enzymatic analysis
Analysis of the levels of sulphamidase activity (Fig. 3) showed that the level of enzyme replacement increased with increasing amounts of vector added, with the two larger amounts effectively normalizing enzyme levels. In comparison to untransduced MPS IIIA cells, the increase in activity with the smallest amount of virus (68 ng p24) was insignificant, while the increase in sulphamidase activity with the two larger amounts of virus (167 and 508 ng p24) was significant (P < 0.01). The correction in enzyme activity in the cultures transduced with 167 ng p24 of vector was not significantly different from the level found in normal cells (P > 0.05), while enzyme activity resulting from transduction with 508 ng p24 of vector was significantly higher (P < 0.05) than in the normal control cells. β-hexosaminidase was not significantly different in any of the samples (P > 0.05). In the cross-correction experiments, sulphamidase activity in cells exposed to conditioned medium from transduced cells was not detectable above background.
Figure 3.
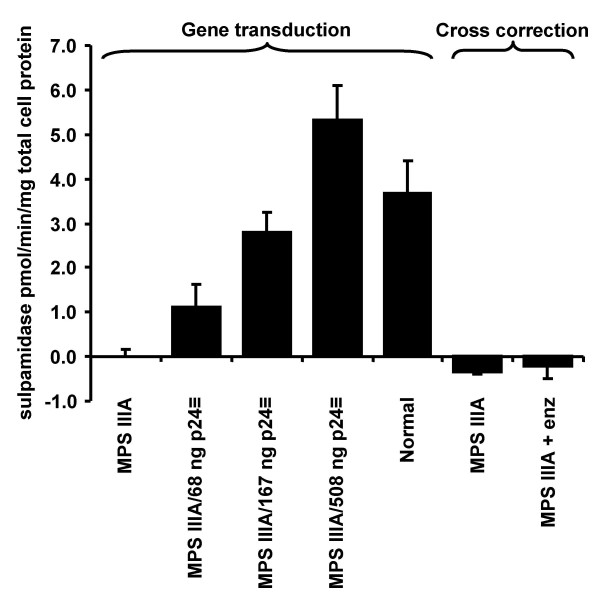
Correction of MPS IIIA enzymatic phenotype in vitro. MPS IIIA cells were transduced with 68, 167 or 508 ng p24 equivalent of the pHIV-1pgkmsulp vector and cell lysates assayed for sulphamidase activity as described in Materials and methods (Gene transduction). Conditioned medium from the cells transduced with 508 ng p24 equivalent of vector was added to untransduced cells (cross correction), and again, cell lysates assayed for sulphamidase activity. Normal and untransduced MPS IIIA cells were used as controls.
Correction of MPS IIIA skin fibroblasts, real time PCR analysis
Real time PCR analysis of DNA samples from transduced fibroblasts revealed the vector copy number to be proportional (R2 = 0.99) to the dose of virus used (Fig. 4). The copy number varied from 0.3 copies/cell (68 ng p24 dose) to 1.7 copies/cell (508 ng p24 dose). No vector was detected in un-transduced cells or cells exposed to conditioned medium collected from transduced cells. Enzyme expression was proportional (R2 = 0.90) to virus dose (Fig. 4), and hence also to vector copy number (R2 = 0.95).
Figure 4.
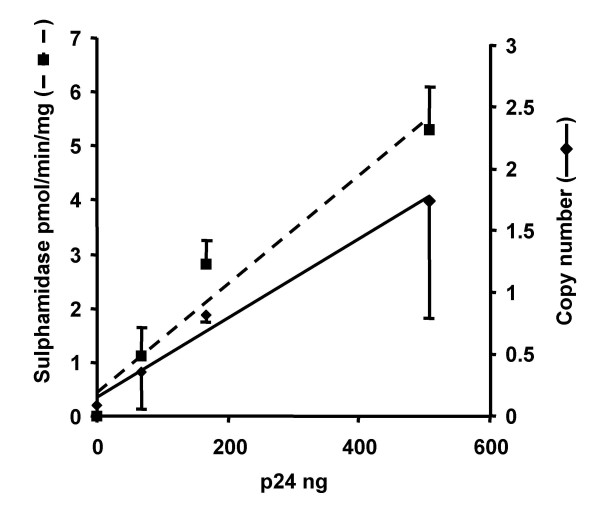
Vector copy number in transduced MPS IIIA cells. MPS IIIA cells were transduced with 68, 167 or 508 ng p24 equivalent of the pHIV-1pgkmsulp vector and both vector copy number, and sulphamidase enzyme activity, were determined as described in Materials and methods. Both vector copy number and enzyme levels are proportional to vector dose.
In vivo administration of vector, analysis of urinary GAG and body weight
Six 5 week old MPS IIIA mice (animal #s 2, 3, 7, 94, 98 and 99) were intravenously injected with vector as described in Materials and methods. Three of these mice (#s 2, 3 and 7) also received hyperosmotic mannitol immediately prior to administration of the vector. At various times post treatment urine was collected, GAG purified by CPC precipitation and analysed by gradient-PAGE as described in Materials and methods. The results of this analysis show that there is a large and consistent reduction in urinary GAG to normal, or near normal levels, in the treated animals (Fig 5). Comparison of samples taken from individual animals at different time points suggest that the reduction in urinary GAG becomes more marked after a longer period of treatment (e.g. animal 2, 122 days versus 54 days after treatment).
Figure 5.

Urine analysis. Urine from selected mice was analysed by gradient PAGE as described in Materials and methods. Lane M, octasaccharide size standard; lane 1, empty; lane 2, normal; lane 3, normal; lane 4, MPS IIIA; lane 5, MPS IIIA; lane 6, treated #2, 54 days post-treatment; lane 7, treated #2, 122 days post-treatment; lane 8, treated #3, 67 days post-treatment; lane 9, treated #7, 61 days post-treatment; lane 10, treated #7, 196 days post-treatment; lane 11, treated #94, 106 days post-treatment; lane 12, treated #98, 98 days post-treatment.
At the time of treatment all animals showed the weight gain typical of MPS IIIA affected mice. However, over the 6-month treatment period their weight progressively trended towards normal and became closer to the normal range than to the range seen in untreated mice (Fig 6). No difference in growth was seen between the mice given hyperosmotic mannitol prior to treatment and those that were not (data not shown). Accordingly, the treated mice were analysed as one group. Statistical analysis showed that the weight of treated mice was significantly different from that of untreated mice after age 54 days (17–22 days post-treatment), and not significantly different from normal after age 166 days (18.5–19 weeks post-treatment).
Figure 6.
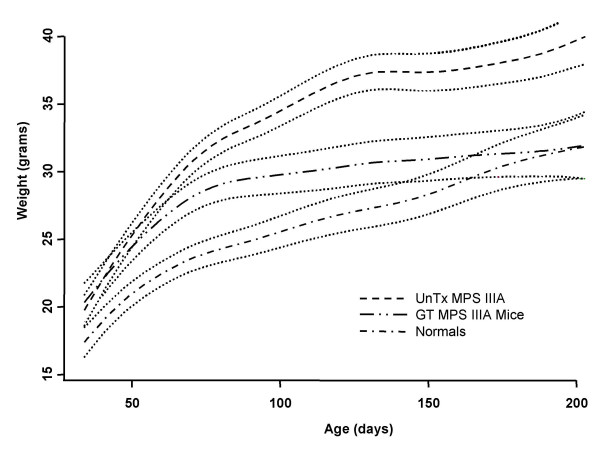
Growth analysis. Mouse weights were analysed by comparison of non-linear trends for each group as described in Materials and methods. The cubic smoothing spline and p < 0.001 prediction interval (dotted line) is shown for each group. ---, untreated MPS IIIA; -••-, treated MPS IIIA; -•-, normal.
Discussion
Gene replacement therapy for the MPS has several potential advantages over enzyme replacement therapy, the current gold-standard for treatment where it is available. These include a reduced frequency of treatment, better efficacy and the prospect of being able to treat CNS disease by the introduction of gene vectors directly into the CNS. Until recently, the development of gene therapy for the MPS has foundered on the lack of suitable gene delivery vehicles. Generally, integrative vectors would seem to be preferable for inherited metabolic disorders such as the MPS as they confer genetic stability on the transduced gene and hence the potential for long-term effects. Because of this, retroviral vectors have long been the gene delivery vehicle of choice. However, vectors derived from oncogenic retroviral vectors are unable to transduce non-cycling cells [13], severely limiting their usefulness. For this reason we, and others, have developed lentiviral vectors [14,22-25]. These have the general positive attributes of retroviral vectors with the additional feature of being able to transduce non-cycling cells, meaning they have great utility for in vivo gene delivery. This has led to the use of lentiviral vectors in the development of gene therapy for a range of disorders, including the MPS [26-29].
In this study we have constructed a lentiviral vector and demonstrated proof of principle experiments in vitro. Human cells were used for these experiments simply for convenience; they were immediately available while cultures of control and MPS IIIA murine fibroblasts were not. Lentiviral-mediated gene delivery to human MPS IIIA skin fibroblasts resulted in correction of the metabolic and enzymatic defects exhibited by these cells, even at the lowest dose of virus used. In addition, the complete correction of the metabolic defect in cultured MPS IIIA cells with the lowest copy number of vector, and the fact that medium secreted from gene corrected cells was able to cross-correct the metabolic defect in non-transduced cells, demonstrates the potential of gene therapy to affect multiple cells in addition to those directly transduced by vector. In the cross-correction experiment it was not possible to assess whether enzyme uptake was via the mannose-6-phosphate (M6P) receptor, as metabolic correction was seen when enzyme was added in the presence or absence of M6P (data not shown). In addition, sulphamidase activity was too low to be detected in all samples from the cross correction experiment. This, at least in part, reflects the relatively low sensitivity of the enzyme assay, and in part the relatively low levels of enzyme activity in the conditioned medium used (towards the lower level of activity that can be easily detected).
In the in vitro study the pgk promoter appears to be relatively weak, as the level of expression obtained with the vector was not greater than that found in normal cells, suggesting it may be useful to assess the level of expression from other promoters. However, the use of strong promoters must be developed with caution as they increase the risk of insertional mutagenesis via oncogene activation [30].
The development of a real time PCR assay for our vector, and control cell line containing a single copy of our vector, will prove useful in further studies, for example determination of vector copy number in tissues after in vivo administration. By careful selection of the vector sequences that the real time PCR detects we have made this assay generic so that it will detect all versions of our vector, whatever the transgene or promoter sequence the vector carries.
Vector was administered to MPS IIIA mice either with or in the absence of a hyperosmotic mannitol pre-treatment. In the studies presented in this paper these two sets of animals could not be distinguished, therefore, all animals were grouped together for analysis. Administration of the vector to MPS IIIA mice resulted in partial normalisation of urinary GAG as evidenced by gradient PAGE analysis, giving an early indication of in vivo efficacy. The power of the gradient PAGE system [20] is that it allows the specific assessment of the small to large size free GAG molecules (i.e. four to thirty saccharide residues in length) typical of lysosomal storage material, rather than small free GAG (di- to tetra-saccharides) which can be analysed by mass spectrometry, or the total (free and conjugated) GAG measured by analysis of uronic acid.
The weight of the treated animals was also progressively normalised, suggesting that the treatment is having a widespread effect on the disease pathology even though enzyme activity could not be detected in blood samples from treated mice (data not shown). Further analysis of these animals is ongoing and will be published elsewhere.
In conclusion, lentiviral vectors appear to be promising reagents for the development of effective therapy for MPS IIIA. Future work will involve in vivo delivery of the vector to somatic and CNS cells and detailed analysis of the disease phenotype in treated animals.
Acknowledgments
Acknowledgements
This work was supported by funding from the Australian National Health and Medical Research Council, the National (US) MPS Society, and Adelaide University. We would like to thank Kate Dowling, Biometrics SA, University of Adelaide, for help with the statistical analysis of body weights.
Contributor Information
Donald S Anson, Email: donald.anson@adelaide.edu.au.
Chantelle McIntyre, Email: chantelle.mcintyre@adelaide.edu.au.
Belinda Thomas, Email: belinda.thomas@southernhealth.org.au.
Rachel Koldej, Email: rachel.koldej@adelaide.edu.au.
Enzo Ranieri, Email: enzo.ranieri@adelaide.edu.au.
Ainslie Roberts, Email: ainslie.roberts@adelaide.edu.au.
Peter R Clements, Email: peter.clements@adelaide.edu.au.
Kylie Dunning, Email: kylie.dunning@adelaide.edu.au.
Sharon Byers, Email: sharon.byers@adelaide.edu.au.
References
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editor. The metabolic and molecular bases of inherited disease. 8. McGraw Hill, NY; 2001. [Google Scholar]
- Bhaumik M, Muller VJ, Rozaklis T, Johnson L, Dobrenis K, Bhattacharyya R, Wurzelmann S, Finamore P, Hopwood JJ, Walkley SU, Stanley P. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome) Glycobiol. 1999;9:1389–1396. doi: 10.1093/glycob/9.12.1389. [DOI] [PubMed] [Google Scholar]
- Yogalingam G, Pollard T, Gliddon B, Jolly RD, Hopwood JJ. Identification of a mutation causing mucopolysaccharidosis type IIIA in New Zealand Huntaway dogs. Genomics. 2002;79:150–3. doi: 10.1006/geno.2002.6699. [DOI] [PubMed] [Google Scholar]
- Fischer A, Carmichael KP, Munnell JF, Jhabvala P, Thompson JN, Matalon R, Jezyk PF, Wang P, Giger U. Sulfamidase deficiency in a family of Dachshunds: a canine model of mucopolysaccharidosis IIIA (Sanfilippo A) Pediatr Res. 1998;44:74–82. doi: 10.1203/00006450-199807000-00012. [DOI] [PubMed] [Google Scholar]
- Sands MS, Vogler C, Kyle JW, Grubb JH, Levy B, Galvin N, Sly WS, Birkenmeier EH. Enzyme replacement therapy for murine mucopolysaccharidosis type VII. J Clin Invest. 1994;93:2324–2331. doi: 10.1172/JCI117237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AL, Thomas BJ, Wilkinson AS, Fletcher JM, Byers S. Inhibition of glycosaminoglycan synthesis using rhodamine B in a mouse model of mucopolysaccharidosis type IIIA. Pediatr Res. 2006;60:309–314. doi: 10.1203/01.pdr.0000233037.00707.da. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Mayatepek E. Neurological manifestations in lysosomal storage disorders – from pathology to first therapeutic possibilities. Neuropediatrics. 2005;36:285–289. doi: 10.1055/s-2005-872810. [DOI] [PubMed] [Google Scholar]
- Vogler C, Levy B, Grubb JH, Galvin N, Tan Y, Kakkis E, Pavloff N, Sly WS. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA. 2005;102:14777–14782. doi: 10.1073/pnas.0506892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sferra TJ, Backstrom K, Wang C, Rennard R, Miller M, Hu Y. Widespread correction of lysosomal storage following intrahepatic injection of a recombinant adeno-associated virus in the adult MPS VII mouse. Mol Ther. 2004;10:478–491. doi: 10.1016/j.ymthe.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Winchester B, Vellodi A, Young E. The molecular basis of lysosomal storage diseases and their treatment. Biochem Soc Trans. 2000;28:150–154. doi: 10.1042/bst0280150. [DOI] [PubMed] [Google Scholar]
- Desnick RJ. Enzyme replacement and beyond. J Inher Met Dis. 2001;24:251–265. doi: 10.1023/A:1010331404448. [DOI] [PubMed] [Google Scholar]
- Bielicki J, Hopwood JJ, Anson DS. Correction of Sanfilippo A skin fibroblasts by retroviral mediated gene transfer. Hum Gene Ther. 1996;7:1965–1970. doi: 10.1089/hum.1996.7.16-1965. [DOI] [PubMed] [Google Scholar]
- Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson DS, Fuller M. Rational development of a HIV-1 gene therapy vector. J Gene Med. 2003;5:829–838. doi: 10.1002/jgm.415. [DOI] [PubMed] [Google Scholar]
- Lieu FH, Hawley TS, Fong AZ, Hawley RG. Transmissibility of murine stem cell virus-based retroviral vectors carrying both interleukin-12 cDNAs and a third gene: implications for immune gene therapy. Cancer Gene Ther. 1997;4:167–75. [PubMed] [Google Scholar]
- Koldej R, Cmielewski P, Stocker A, Parsons DW, Anson DS. Optimisation of a Multipartite Human Immunodeficiency Virus Based Vector System; Control of Virus Infectivity and Large Scale Production. J Gene Med. 2005;7:1390–1399. doi: 10.1002/jgm.803. [DOI] [PubMed] [Google Scholar]
- Hopwood JJ, Elliott H. Diagnosis of Sanfilippo A syndrome by estimation of sulfamidase activity using a radiolabelled tetrasaccharide substrate. Clin Chim Acta. 1982;123:241–250. doi: 10.1016/0009-8981(82)90168-1. [DOI] [PubMed] [Google Scholar]
- Leaback DH, Walker PG. Studies on glucosaminidase. 4. The fluorimetric assay of N-acetyl-beta-glucosaminidase. Biochem J. 1961;78:151–6. doi: 10.1042/bj0780151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliddon BL, Hopwood JJ. Enzyme-Replacement Therapy from Birth Delays the Development of Behavior and Learning Problems in Mucopolysaccharidosis Type IIIA Mice. Pediatr Res. 2004;56:65–72. doi: 10.1203/01.PDR.0000129661.40499.12. [DOI] [PubMed] [Google Scholar]
- Byers S, Rozaklis T, Brumfield LK, Ranieri E, Hopwood JJ. Glycosaminoglycan accumulation and excretion in the mucopolysaccharidoses: characterization and basis of a diagnostic test for MPS. Mol Genet Metab. 1998;65:282–290. doi: 10.1006/mgme.1998.2761. [DOI] [PubMed] [Google Scholar]
- Vassar Stats http://faculty.vassar.edu/lowry/VassarStats.html
- Fuller M, Anson DS. Helper plasmids for production of HIV-1 derived vectors. Hum Gene Ther. 2001;12:2085–2097. doi: 10.1089/10430340152677421. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Kotsopoulou E, Kim VN, Kingsman AJ, Kingsman SM, Mitrophanous KA. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon optimised HIV-1 gag-pol gene. J Virol. 2000;74:4839–4852. doi: 10.1128/JVI.74.10.4839-4852.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasakumar N, Schuening FG. A lentivirus packaging system based on alternative RNA transport mechanisms to express helper and gene transfer vector RNAs and its use to study the requirement of accessory proteins for particle formation and gene delivery. J Virol. 1999;73:9589–98. doi: 10.1128/jvi.73.11.9589-9598.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CS, Kang Y, Sauter SL, Townsend K, Staber P, Derksen TA, Martins I, Qian J, Davidson BL, McCray PB., Jr In vivo treatment of hemophilia A and mucopolysaccharidosis type VII using nonprimate lentiviral vectors. Mol Ther. 2001;3:850–6. doi: 10.1006/mthe.2001.0325. [DOI] [PubMed] [Google Scholar]
- Di Domenico C, Villani GR, Di Napoli D, Reyero EG, Lombardo A, Naldini L, Di Natale P. Gene therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum Gene Ther. 2005;16:81–90. doi: 10.1089/hum.2005.16.81. [DOI] [PubMed] [Google Scholar]
- Di Natale P, Di Domenico C, Gargiulo N, Castaldo S, Gonzalez Y, Reyero E, Mithbaokar P, De Felice M, Follenzi A, Naldini L, Villani GR. Treatment of the mouse model of mucopolysaccharideosis type IIIB with lentiviral-naglu vector. Biochem J. 2005;388:639–646. doi: 10.1042/BJ20041702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AI, Stein CS, Hughes SM, Heth J, McCray PM, Jr, Sauter SL, Johnston JC, Cory-Slechta DA, Federoff HJ, Davidson BL. Functional correction of established central nervous system deficits in an animal model of lysosomal storage disease with feline immunodeficiency virus-based vectors. Proc Natl Acad Sci USA. 2002;99:6216–21. doi: 10.1073/pnas.082011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2 -associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]