

Abstract
Neurons in the lateral hypothalamus (LH) that contain hypocretin/orexin have been established as important promoters of arousal. Deficiencies in the hypocretin/orexin system lead to narcolepsy. The inhibition of hypocretin/orexin neurons by sleep-promoting neurotransmitters has been suggested as one part of the sleep regulation machinery. Adenosine has been identified as a sleep promoter and its role in sleep regulation in the basal forebrain has been well documented. However, the effect of adenosine on arousal-promoting hypocretin/orexin neurons has not been addressed, despite recent evidence that immunocytochemical visualization of adenosine receptors was detected in these neurons. In this study, we examined the hypothesis that adenosine inhibits the activity of hypocretin/orexin neurons by using electrophysiological methods in brain slices from mice expressing green fluorescent protein in hypocretin/orexin neurons. We found that adenosine significantly attenuated the frequency of action potentials without a change in membrane potential in hypocretin/orexin neurons. The adenosine-mediated inhibition is due to depression of excitatory synaptic transmission to hypocretin/orexin neurons, since adenosine depresses the amplitude of evoked excitatory postsynaptic potential and the frequency of spontaneous and miniature excitatory postsynaptic currents in these neurons. At the cell body of the hypocretin/orexin neurons, adenosine inhibits voltage-dependent calcium currents without the induction of GIRK current. The inhibitory effect of adenosine is dose-dependent, pertussis toxin-sensitive and mediated via A1 receptors. In summary, our data suggest that in addition to its effect in the basal forebrain, adenosine exerts its sleep-promoting effect in the LH via inhibition of hypocretin/orexin neurons.
INTRODUCTION
The neuropeptide hypocretin/orexin has received tremendous attention because of its role in sleep and arousal regulation (Kilduff and Peyron 2000; Mieda and Yanagisawa 2002; Siegel 2004; de Lecea and Sutcliffe 2005; Sakurai 2005). First, deficiencies in both peptide content and receptors of hypocrtin/orexin lead to narcolepsy in mice (Chemelli et al. 1999), dogs (Lin et al. 1999) and human patients (Nishino et al. 2000; Thannickal et al. 2000). Secondly, the concentration of hypocretin/orexin fluctuates in animals during the day. In rats, the hypocretin-1 level in cerebrospinal fluid is high when animals are active and reaches its lowest level at the end of the inactive period (Fujiki et al. 2001), while the concentration of hypocretin-1 in the LH and the medial thalamus elevates slowly during the active phase and decreases during the resting phase (Yoshida et al. 2001). In squirrel monkeys, the hypocretin-1 concentration in cerebrospinal fluid begins to increase after a few hours of wakefulness, reaches a plateau in the active phase and falls throughout the inactive phase (Zeitzer et al. 2003). Thirdly, the pattern of activity of hypocretin/orexin neurons changes along side the behavioral states of animals (Estabrooke et al. 2001). These neurons are generally active in the dark phase and basically silent during resting (sleep) in rats (Lee et al. 2005; Mileykovskiy et al. 2005). Thus, it is proposed that hypocretin/orexin neurons are in a unique position to consolidate wakefulness (Saper 2006). The modulation of the activity of these neurons is an essential part of the machinery responsible for sleep and arousal regulation.
Adenosine is an endogenous sleep-promoting neurotransmitter as demonstrated by a variety of studies (Basheer et al. 2004). First of all, local administration of adenosine and adenosine receptor agonists into the medial preoptic area, magnocellular cholinergic basal forebrain, stem cholinergic areas, the laterodorsal and pedenculopontine tegmental nuclei (LDT/PPT) and pontine reticular formation leads to sleep or reduction of wakefulness (Ticho and Radulovacki 1991; Portas et al. 1997; Marks and Birabil 1998). Secondly, the concentration of adenosine in the brain changes as the consequence of changes in the behavioral state of the animal. The extracellular concentration of adenosine decreases during spontaneous sleep in several sleep-related brain regions including the basal forebrain, cerebral cortex, thalamus, preoptic area of the hypothalamus, dorsal raphe nucleus and pedunculopontine tegmental nucleus (See review by Basheer et al. 2004). Adenosine content elevates in the basal forebrain following sleep deprivation (Porkka-Heiskanen et al. 1997) and remains at a high level throughout the period of recovery sleep (Porkka-Heiskanen et al. 2000). The sleep-promoting effect of adenosine in the brain has been demonstrated to be mediated by both A1 (Alam et al. 1999; Strecker et al. 2000; Thakkar et al. 2003) and A2 adenosine receptors (Satoh et al. 1996; 1998). At the cellular level, adenosine inhibits synaptic transmission, hyperpolarizes membrane potential, mobilizes intracellular stores of calcium, as well as activates PLC and then PKC via the ionsitol trisphosphate (IP3)-dependent pathways in the central nervous system (Yoon and Rothman 1991; Li and Perl 1994; Fisher 1995; Ulrich and Huguenard 1995; Lüscher et al. 1997; Biber et al. 1997; Basheer et al. 2002).
As a potent sleep-promoting neurotransmitter in the CNS, the role of adenosine in sleep regulation has been well established in brain areas such as the basal forebrain. However, it is still not known if adenosine exerts its effects on hypocretin/orexin neurons, an essential system for the maintenance of arousal and wakefulness in the CNS. A recent study by Thakkar and co-workers (2002) revealing the immunocytochemical visualization of adenosine receptor in hypocretin/orexin neurons in rats further exemplifies this lack of knowledge.
In the study presented here, the hypothesis that adenosine modulates the activity of hypocretin/orexin neurons in the LH was examined by using electrophysiological methods in acute brain slices. Our data suggest that adenosine inhibits the generation of action potentials, excitatory synaptic transmissions and ion channels via A1 receptors in the hypocretin/orexin neurons.
MATERIALS AND METHODS
Hypothalamic slice preparation
Coronal hypothalamic slices, 300 µm thick, were cut with a vibratome from the brains of mice postnatal days 14-21 (P14-P21) that expressed GFP under the control of a hypocretin/orexin promoter (Li et al. 2002; Yamanaka et al. 2003; Horvath and Gao 2005). Briefly, mice were anesthetized with ether and then decapitated. The brains were rapidly removed and immersed in cold (4° C) oxygenated cutting solution (containing (mM): sucrose 220, KCl 2.5, CaCl2 1, MgCl2 6, NaH2PO4 1.25, NaHCO3 26, and glucose 10, pH 7.3 with NaOH). After being trimmed to contain the hypothalamus and surrounding areas, slices were maintained at room temperature in a holding chamber with oxygenated ASCF (5% CO2 and 95% O2) containing (in mM): NaCl 124, KCl 3, CaCl2 2, MgCl2 2, NaH2PO4 1.23, NaHCO3 26, glucose 10, pH 7.4 with NaOH. Slices were then transferred to a recording chamber after at least 1hr recovery, and constantly perfused at 33° C with bath solution at a speed of 2 ml/min. All rodent use and methods are approved by the Yale University Committee on Animal Use.
Immunocytochemistry
The immunocytochemical experiments were performed to confirm the identity of GFP-expressing neurons as we previously described (Gao et al., 2003). Mice were anesthetized with ether and perfused transcardially with a fixative consisting of 4% paraformaldehyde in 0.1% sodium phosphate buffer, pH 7.4 (PB). Sections (30 μm thick) were then cut on a vibratome. After being washed in a buffer containing 0.1% lysine, 1% BSA, 0.1% Tris and 0.4% Triton X-100 and blocked with 2% normal goat serum, sections were incubated overnight at room temperature in a primary rabbit antiserum against hypocretin-2 (a gift from Dr. Anthony van den Pol, 1:2,000). After several washes with PB, sections were incubated in the secondary antibody (1:200) conjugated to Texas Red (red) (Vector Laboratories, Burlingame, CA) for 2 hours at room temperature. Specimens were examined with an FV 300 confocal laser scanning microscope from Olympus (Olympus America, Inc.).
Electrophysiology
Extracellular recording
Extracellular recordings were made in identified hypocretin-GFP neurons using a glass electrode filled with ACSF (resistance=2-5 MΩ) with a multiclamp 700A amplifier (Axon Instruments, Inc.). The recording electrode was propelled by a motorized micromanipulator (Sutter MP225, Sutter Instruments, Inc) to approach an identified hypocretin-GFP neuron. A loose seal was then formed (resistance= 10-20 MΩ) when the micropipette touched the surface of a neuron. After a 10-minute control was recorded, drugs were applied to the recording chamber via bath application. The band pass was 10-3000 Hz. All data were sampled at 500 Hz with an Apple Macintosh computer using Axograph 4.9 (Axon Instrument).
Whole-cell recording
Whole-cell patch clamp recording was performed on identified hypocretin-GFP neurons with a Multiclamp 700A amplifier (Axon instrument, CA). The patch pipettes were made of borosilicate glass (World Precision Instruments) with a Sutter pipette puller (P-97). The tip resistance of the recording pipettes was 4-6 MΩ after filling with a pipette solution containing (mM): K-gluconate 135, MgCl2 2, HEPES 10, EGTA 1.1, Mg-ATP 2, Na2-phosphocreatine 10, and Na2-GTP 0.3, pH 7.3 with KOH. After a gigohm seal and whole-cell access were achieved, the series resistance was between 20 and 40 MΩ and partially compensated by the amplifier. Both input resistance and series resistance were monitored throughout the experiments. Only those recordings with stable series resistance and input resistance were accepted. Whole-cell voltage clamp was used to observe spontaneous and miniature excitatory postsynaptic currents (sEPSC and mEPSC) and membrane currents carried by ion channels. sEPSC were recorded in the presence of bicuculline (30 μM) and mEPSC were recorded in the presence of bicuculline (30 μM) and tetrodotoxin (TTX, 1 μM). Whole-cell voltage-dependent calcium currents were examined as reported previously (Gao and van den Pol 2001; 2002). To examine G-protein activated inwardly rectifying potassium (GIRK) current, a ramp protocol (from −140 mV to −20 mV, duration 600 ms) was used. Whole-cell current clamp was used to monitor the evoked postsynaptic potential (EPSP) and membrane potential in hypocretin/orexin neurons. To trigger evoked EPSPs in hypocretin/orexin neurons, a bipolar electrode was placed on the medial forebrain bundle (MFB) and a stimulator (Isostim A320, World Precision Instruments, Inc., Sarasota, Florida, USA) was used. Drugs were applied to the recording chamber via bath application after at least ten minutes of control recording.
All data were sampled at 3-10 kHz and filtered at 1-3 kHz with an Apple Macintosh computer using Axograph 4.9 (Axon Instruments). Electrophysiological data were analyzed with Axograph 4.9 and plotted with KaleidaGraph 3.6 (Synergy Software, Reading, PA, USA) and Igor Pro 5.04 (WaveMetrics, Lake Oswego, OR, USA). Spontaneous and miniature postsynaptic currents were detected and measured with an algorithm in Axograph 4.9 (Gao and van den Pol 2001). The frequency of sEPSC and mEPSC was normalized by comparing the average sEPSC or mEPSC frequency after drug application with 5-minute recordings just before drug application. Data from all recorded neurons were presented as mean ± S.E. An ANOVA test was used to examine statistical significance among three groups and the Kolmogorov-Smirnov test was used to examine significance between two distributions.
Adenosine and bicuculline were purchased from Sigma (St. Louis, MO, USA). Tetrodotoxin (TTX) and pertussis toxin (PTX) were obtained from Alomone Labs, Ltd. (Jerusalem, Israel). A1 receptor agonist 2-Chloro-N6-cyclopentyladenosine (CCPA), A1 receptor antagonist 8-Cyclopentyl-1, 3-dipropylxanthine (DPCPX), A2a receptor antagonist 2-(2-Furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine (SCH 58261), and A2b receptor antagonist N-(4-Acetylphenyl)-2-[-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]acetamide (MRS 1706) were obtained from Tocris Cookson, Inc (Ellisville, MO, USA).
RESULTS
Adenosine inhibits action potential generation in hypocretin/orexin neurons
The generation and propagation of action potentials (spikes) along axons of hypocretin/orexin neurons play a substantial role in the function of the LH (Li et al. 2002; Yamanaka et al. 2003; Lee et al. 2005; Mileykovskiy et al. 2005). We performed loose patch clamp recordings to monitor action potentials in visually identified hypocretin/orexin neurons under voltage clamp, through which the intracellular content of neurons could be kept intact for a long recording time. In slices from hypocretin-GFP mice, neurons containing hypocretin/orexin were easily identified under UV illumination and confirmed by immunocytochemical staining (van den Pol et al., 1998) (Figure 1 A-C). All GFP-expressing neurons are hypocretin/orexin-immunopositive. We did notice that about 30 to 40% of hypocrein/orexin-immunopositive neurons did not show green signals, which may be due to insufficient expression of GFP in those neurons. Action currents (spikes), as illustrated in Figure 1 D, were readily detected and verified by complete blockade with tetrodotoxin (TTX, 1μM) (data not shown). After at least 10 minutes of recording of control spikes (2.66 ± 0.48 Hz, n=14), adenosine (100 μM) applied via bath solution decreased the spike frequency to 66.5 ± 7.8% of control (n=14) in all tested neurons. The attenuation of the frequency of spikes was sustained as long as adenosine was present, as illustrated in Figure 1 E. The frequency of spikes recovered to 88.8 ± 9.7% of control (n=12) 10 minutes after withdrawal of adenosine. The ANOVA test suggested that the inhibition on action potential generation induced by adenosine in hypocretin/orexin neurons was significant (Degree of freedom= 39, F= 6.44, P<0.05). Recent data suggest that the firing rate of hypocretin/orexin neurons is sensitive to changes in the extracellular concentration of glucose (Burdakov et al. 2006). The extracellular concentration of glucose in the brain is 1-2 mM but not 10 mM which is used in most brain slice preparations including ours (McNay and Gold, 1999; De Vries et al. 2003). Therefore, the effect of adenosine on the firing rate of spikes was additionally examined using ACSF containing 2 mM glucose (Figure 1 G). The firing rate of spikes (5.8 ± 0.8 Hz, n=6, control) was 63.3 ± 3.3 % of control (n=6) during the application of adenosine and 104.4 ± 4.1 % of control (n=4) after its removal. The decrease in the firing rate of hypocretin/orexin neurons induced by adenosine in the presence of 2 mM glucose was significant (Degree of freedom= 15, F= 68.52, P<0.01, ANOVA test).
Fig.1.

Adenosine depresses the frequency of action potentials in hypocretin/orexin neurons. A-C, double labeling of hypocretin/orexin neurons in brain slices. A, expression of GFP in hypocretin/orexin neurons under the control of a specific hypocretin/orexin promoter. B, immunocytochemical staining of hypocretin/orexin immunoreactive neurons (red). C, green and red signals overlap in hypocretin/orexin neurons indicated by arrows. All GFP-expressing neurons are hypocretin/orexin positive. Scale bar: AC, 15 μm. D-G, loose patch recordings of spikes (action currents) in visually identified hypocretin/orexin neurons were performed in slices from P14-21 mice expressing GFP in hypocretin/orexin neurons. D, sample traces of action currents recorded at various stages of our experiments under voltage clamp with loose patch recordings are presented. E, the time course of a typical experiment shows that adenosine (100 μM) applied to the recorded neuron attenuated the frequency of spikes. This attenuation was reversible upon the removal of adenosine. Application of adenosine is indicated by the filled bar. F, pooled data for frequency of spikes from all hypocretin/orexin neurons examined in our experiments are presented. An ANOVA was used to examine the significance of difference among all three groups. An asterisk “*” indicates P<0.05. G, pooled data for frequency of spikes from all 6 hypocretin/orexin neurons examined in ACSF containing 2 mM glucose are presented. An ANOVA was used to examine the significance of difference among all three groups. A double asterisk “**” indicates P<0.01.
Adenosine depresses glutamatergic synaptic transmission in hypocretin/orexin neurons
The excitatory inputs to hypocretin/orexin neurons make a remarkable contribution to the functions of these neurons (Li et al. 2002; Horvath and Gao 2005); particularly the glutamatergic synaptic transmission which governs the generation of action potentials in hypocretin/orexin neurons (Li et al. 2002). Here we examined the effect of adenosine on the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) mediated by glutamate in hypocretin/orexin neurons held at −60 mV under voltage clamp. sEPSCs were readily recorded in hypocretin/orexin neurons in the presence of the GABA-A receptor antagonist bicuculline (30 μM) after the establishment of whole cell configuration (Figure 2). The fact that sEPSCs were completely blocked in the presence of AP5 (50 μM) and CNQX (10 μM) in all tested hypocretin/orexin neurons (n=7) suggested that sEPSCs were mediated by glutamatergic synaptic transmission (Figure 2, inset). As shown in Figure 2 A-B, after a stable baseline of sEPSCs was recorded, adenosine (100 μM) applied to the recorded neurons via bath application induced a reduction in the frequency of sEPSCs. The frequency of sEPSCs (14.7 ± 1.8 Hz, n=9; control) was 61.2 ± 5.6 % of control (n=9) 5 minutes after the onset of application of adenosine and returned to 103.2 ± 3.4 % of control (n=9) shortly after withdrawal of adenosine (Figure 2 A and B). The reduction in the frequency of sEPSCs was significant as indicated by ANOVA (Degree of freedom = 25, F= 38.4, P<0.01). Next, the effect of different concentrations of adenosine on the frequency of sEPSCs was tested. In the presence of 3, 10 and 30 μM of adenosine, the frequency of sEPSCs was 91.7 ± 4.1 % (Degree of freedom=14, F= 2.70, P>0.05, ANOVA test), 86.0 ± 3.1% (Degree of freedom= 23, F= 9.04, P<0.01, ANOVA test), and 78.9 ± 6.8% of control (Degree of freedom=19, F=15.17, P<0.01, ANOVA test), respectively. In a parallel experiment, we examined the effect of adenosine on the frequency of sEPSCs in ACSF containing 2 mM glucose, which is not commonly used in brain slice preparations but is close to physiological levels of glucose in the brain. In this case, the frequency of sEPSCs (7.6 ± 1.6 Hz, n=8, control) decreased to 67.6 ± 6.3 % of control (n=8) during the application of adenosine and recovered to 102.7 ± 21.6 % of control (n=3) after its removal. This attenuation of the frequency of sEPSCs was significant as determined by ANOVA (Degree of freedom = 18, F=8.126, P<0.01). Altogether, our data suggest that adenosine induces a dose-dependent inhibitory effect on the frequency of sEPSCs in hypocretin/orexin neurons.
Fig.2.

Adenosine decreases the frequency of sEPSCs in hypocretin/orexin neurons. Experiments were performed in the presence of bicuculline (30 μM) in all solutions and sEPSCs were recorded in hypocretin/orexin neurons held at −60 mV under voltage clamp. The frequency of sEPSCs decreased rapidly in the presence of adenosine (100 μM). Sample traces recorded in our experiments at various stages are presented in A and the time course of a typical experiment is plotted in B. Application of adenosine is indicated by the filled bar in B. C, pooled data from all tested hypocretin/orexin neurons at different concentrations of adenosine show that the inhibitory effect of adenosine on the frequency of sEPSCs is dose-dependent. The symbol “**” indicates P<0.01 (ANOVA test). D, frequency of sEPSCs obtained from all hypocretin/orexin neurons (n=8) examined in ACSF containing 2 mM glucose demonstrated that adenosine depressed the frequency of sEPSCs in hypocretin/orexin neurons (**, P<0.01, ANOVA). Inset, sample traces demonstrated that sEPSCs were completely blocked by ionotropic glutamate receptor antagonists AP5 and CNQX (n=7), suggesting a tone of glutamatergic transmission.
Hypocretin/orexin neurons receive intensive innervation from other brain areas, which participate in the modulation of their activity (Sakurai et al. 2005). To examine the hypothesis that adenosine depresses excitatory inputs to hypocretin/orexin neurons from other brain areas, evoked excitatory postsynaptic potentials (eEPSPs) were recorded in these neurons under current clamp via stimulation of the medial forebrain bundle (MFB) with a bipolar electrode (Figure 3 A), since the MFB provides significant excitatory inputs to the LH (Henny and Jones 2006). To obtain a stable recording of eEPSPs, the membrane potential of hypocretin/orexin neurons was slightly hyperpolarized to a level between −60 and −70 mV by current injection. After a stable recording of eEPSPs was obtained, adenosine (100 μM) was applied to the recorded slices through bath application. As shown in Figure 3 B, adenosine induced a decline in the amplitude of eEPSPs upon application and attenuation of the amplitude of eEPSPs recovered after withdrawal of adenosine. The amplitude of eEPSPs (11.4 ±2.5 mV, n=6, control) was 48.1 ± 6.1 % of control (n=6) in the presence of adenosine and 82.2 ± 10.4 % of control (n=4) after its withdrawal (Figure 3 C). As suggested by the ANOVA test, this decrease in the amplitude of eEPSPs was significant (Degree of freedom= 15, F= 22.3, P<0.01). In the same experiment, the effect of adenosine on the membrane potential of hypocretin/orexin neurons was examined (Figure 3 D). The membrane potential was −69.9 ± 2.4 mV (n=7) before, −68.4 ± 2.5 mV (n=7) during and −68.7 ± 2.0 mV (n=7) after the application of adenosine, i.e., there was no significant change in membrane potential (Degree of freedom=18, F=0.826, P>0.05). To further test the direct effect of adenosine on the membrane potential in hypocretin/orexin neurons, TTX was applied to the recorded slices to functionally isolate these neurons (Figure 3 E and F). In the presence of TTX, the membrane potential was −72.3 ± 5.2 mV (n=6) before, −74.8 ± 4.8 mV (n=6) during and −75.0 ± 4.4 mV (n=6) after the application of adenosine. Again, there was no change in membrane potential (Degree of freedom=17, F=0.140, P>0.05). Overall, our results suggest that adenosine depresses the amplitude of evoked EPSP but has no direct effect on the membrane potential in hypocretin/orexin neurons.
Fig.3.

Adenosine attenuates the amplitude of evoked EPSPs in hypocretin/orexin neurons. Whole-cell patch clamp experiments were performed in the presence of bicuculline (30 μM) in all solutions and the stimulating electrode was placed on the medial forebrain bundle (MFB). Evoked EPSPs (eEPSPs) were recorded in hypocretin/orexin neurons under current clamp and presented in A. In order to obtain a stable recording of eEPSP without triggering action potentials, hyperpolarizing current was injected to the recorded neurons to maintain the membrane potential between −60 to −70 mV. The amplitude of eEPSPs declined in the presence of adenosine and recovered after its removal. The time course of a typical experiment is plotted in B. C, pooled data from all experiments show that the amplitude of eEPSPs is significantly decreased by the application of adenosine (**, p<0.01, ANOVA). D, pooled data from all experiments show that the membrane potential of hypocretin/orexin neurons does not change (P>0.05, ANOVA). E-F, the membrane potential of hypocretin/orexin neurons recorded in the presence of TTX (1 μM). E, a sample trace from our experiments is presented (M.P., −68.0 mV). The application of adenosine is indicated with the filled bar and the dotted line represents the baseline of the recording. F, pooled data from all experiments show that adenosine does not cause any changes in membrane potential in the presence of TTX (P>0.05, ANOVA).
Adenosine-mediated effect on glutamatergic synaptic transmission is pertussis toxin-sensitive
Next, we wanted to explore the mechanism underlying adenosine-induced inhibition of glutamatergic synaptic transmission. Previous evidence suggests that the adenosine receptor-mediated effect is coupled to the Gi/Go pathway (Dunwiddie and Masino 2001). Pertussis toxin (PTX) has been demonstrated to block this Gi/Go pathway-mediated effect. After being treated with PTX (500 ng/ml) for at least two hours, brain slices containing hypocretin-GFP neurons were transferred to the recording chamber and continuously perfused with ACSF containing PTX. After a stable recording of sEPSCs was obtained, adenosine (100 μM) was applied to the recorded slices (indicated by the filled bar in Figure 4 B). The frequency of sEPSCs (12.1 ±1.6 Hz, n=7, control) was 83.0 ± 5.5 % of control (n=7) during the application of adenosine and 99.7 ± 9.4% of control (n=6) after its withdrawal (Figure 4 C). Adenosine induced a modest inhibition in the frequency of sEPSCs in slices pre-treated with PTX, which was not significant as indicated by ANOVA (Degree of freedom = 19, F=2.77, P>0.05). Our data suggest that adenosine-mediated inhibition of the frequency of sEPSCs was PTX-sensitive and probably mediated via the Gi/Go pathway.
Fig.4.

Adenosine-mediated inhibition of frequency of sEPSCs was abolished in the presence of pertussis toxin (PTX). All experiments were performed in slices treated with PTX (500 ng/ml) for two hours in advance. sEPSCs were recorded in hypocretin/orexin neurons under voltage clamp in the presence of bicuculline (30 μM) in all solutions. A, sample traces of sEPSCs recorded at various stages of our experiment. B, the time course of a typical experiment is shown. Application of adenosine is indicated by the filled bar. C, pooled data from all tested neurons showing that the frequency of sEPSCs does not significantly decrease in the presence of adenosine in slices pre-treated with PTX (P>0.05, ANOVA).
A1 adenosine receptor is responsible for the effect of adenosine on glutamatergic synaptic transmission
To date, four types of adenosine receptors (A1, A2a, A2b and A3) have been reported. Both A1 and A2 receptors have been implied to participate in the modulation of sleep (see review by Basheer et al. 2004). We have already shown that adenosine inhibits action potential generation and glutamatergic synaptic transmission in hypocretin/orexin neurons. Here, we asked which receptor types are responsible for this effect. First, the effect of DPCPX, a selective A1 receptor antagonist, was tested on adenosine-mediated inhibition of the frequency of sEPSCs. It is noteworthy that the application of DPCPX alone induced only a negligible change in the frequency of sEPSCs (data not shown); the frequency of sEPSCs in DPCPX serves as the control for subsequent experiments. In the presence of DPCPX (3 μM) in all solutions, adenosine (100 μM) did not induce significant change in the frequency of sEPSCs (Figure 5 A-B). The frequency of sEPSCs (14.3 ± 3.0 Hz, n=8, control) was 100.3 ± 1.4 % of control (n=8) in the presence of adenosine and 104.8 ± 2.2 % of control (n=4) after its withdrawal (Figure 5 C) (Degree of freedom=19, F=3.556, P>0.05, ANOVA test). To confirm the involvement of A1 receptor in achieving the effect of adenosine, a selective A1 receptor agonist, CCPA (1 μM) was used to mimic the effect of adenosine. In the presence of bicuculline (30 μM), sEPSCs were recorded from identified hypocretin/orexin neurons under voltage clamp. The selective A1 receptor agonist CCPA was then applied to the recorded slices after a stable recording of sEPSCs was achieved (Figure 5 D). The frequency of sEPSCs (16.6 ± 2.0 Hz, n=6, control) was 55.6 ± 9.1 % of control (n=6) in the presence of CCPA and was significantly lower than that in control solution (Degree of freedom=16, F=8.29, P<0.01) as indicated by ANOVA. After withdrawal of CCPA the frequency of sEPSCs was 69.6 ± 11.6% of control (n=5) and was significantly lower than the control level (P<0.05, Post Hoc Test).
Fig.5.

A1 adenosine receptor is responsible for the effect of adenosine in hypocretin/orexin neurons. Whole-cell recordings were performed in hypocretin/orexin neurons held at −60 mV under voltage clamp. Bicuculline (30 μM) was present in all solutions. In the presence of DPCPX (3 μM), a selective A1 receptor antagonist, adenosine (100 μM) was applied to recorded neurons after a stable recording of sEPSCs was obtained and its effect on the frequency of sEPSCs was monitored. Sample traces are presented in A and the time course of a typical experiment is shown in B. Application of adenosine is indicated by the filled bar above the time course plotting. C, pooled data from all neurons examined in our experiments show that adenosine does not inhibit the frequency of sEPSCs in the presence of DPCPX (P>0.05, ANOVA). D, experiments were performed in ACSF containing bicuculline (30 μM). After a stable recording of sEPSCs was obtained, a selective A1 receptor agonist, CCPA was applied to the recorded neurons. Data from all tested hypocretin/orexin neurons are pooled and presented here. The frequency of sEPSCs significantly declined in the presence of CCPA and after its removal (**, p<0.01; *, P<0.05, ANOVA).
Next, we examined whether A2 adenosine receptors participate in the adenosine-mediated inhibition in hypocretin/orexin neurons by using A2 receptor selective antagonists (Figure 6 A-C). Since two subtypes (A2a and A2b) of A2 receptors have been reported (Dunwiddie and Masino 2001), the effect of the combined A2 receptor antagonists SCH58261 (1 μM) and MRS1706 (0.1 μM) on adenosine-mediated inhibition was tested. Whole cell recording was performed in hypocretin/orexin neurons under voltage clamp at −60 mV. After a stable recording of sEPSCs was obtained, adenosine (100 μM) was applied to the recorded slices in the presence of A2 receptor antagonists. The frequency of sEPSCs (12.3 ± 1.9 Hz, n=6, control) was 68.9 ± 6.0 % of control (n=6) during and 99.9 ± 14.6 % of control (n=6) after application of adenosine (Figure 6 C) which represents a significant inhibition (Degree of freedom= 17, F= 3.86, P<0.05, ANOVA test). Our results indicate that in the presence of A2 receptor antagonists, adenosine still depresses the frequency of sEPSCs.
Fig.6.

A2 adenosine receptors are not responsible for the effect of adenosine in hypocretin/orexin neurons. Whole-cell recordings were performed in hypocretin/orexin neurons held at −60 mV under voltage clamp. Bicuculline (30 μM) was present in all solutions. Selective A2 receptor antagonists, MRS 1706 and SCH 58261, were applied to the recorded neurons. After a stable recording of sEPSCs were obtained, in the presence of MRS 1706 and SCH 58261, adenosine (100 μM) was applied and its effect on the frequency of sEPSCs was monitored. Sample traces are presented in A and the time course of a typical experiment is shown in B. In the presence of MRS 1706 and SCH 58261, the frequency of sEPSCs declined in the presence of adenosine (100 μM) and recovered after its withdrawal. Application of adenosine is indicated by the filled bar. C, pooled data from all neurons examined in our experiments show that the inhibitory effect of adenosine was intact in the presence of selective A2 receptor antagonists (**, P<0.01, ANOVA).
In conclusion, our data suggest that the adenosine-induced inhibitory effect on glutamatergic synaptic transmission is mediated by the A1 adenosine receptor.
Adenosine inhibits glutamatergic synaptic transmission at presynaptic terminals
Next we determined whether the attenuation in frequency of sEPSCs induced by adenosine occurred pre- or postsynaptically; this was accomplished through the measurement of miniature excitatory postsynaptic currents (mEPSCs). In the presence of bicuculline (30 μM) and TTX (1 μM), mEPSCs were recorded in hypocretin/orexin neurons under voltage clamp at −60 mV. A change in the frequency of mEPSCs is generally due to modulations at presynaptic terminals, while a change in the amplitude is normally due to modulations at postsynaptic components (Bekkers and Stevens 1995; Gao and van den Pol, 2001). After a stable baseline of mEPSCs was recorded in hypocretin/orexin neurons, adenosine (100 μM) was applied to recorded neurons via bath application in the presence of bicuculline and TTX (Figure 7 A-B). The frequency of mEPSCs (12.2 ± 0.8 Hz, n=6, control) was 72.2 ± 3.0 % of control (n=6) in the presence of adenosine and 90.7 ± 3.4 % of control (n=6) after its removal representing a significant decrease (Degree of freedom= 17, F=29.89, P<0.01, ANOVA test) (Figure 7 C). The effect of adenosine on the amplitude of mEPSCs was examined in our study as well. The cumulative probability curve was plotted for the amplitude of mEPSC events detected before and during the application of adenosine (Figure 7 D) and no significant difference was seen in any of the recorded hypocretin/orexin neurons, as indicated by the Kolmogorov-Smirnov test (P>0.05, Figure 7 D).
Fig.7.
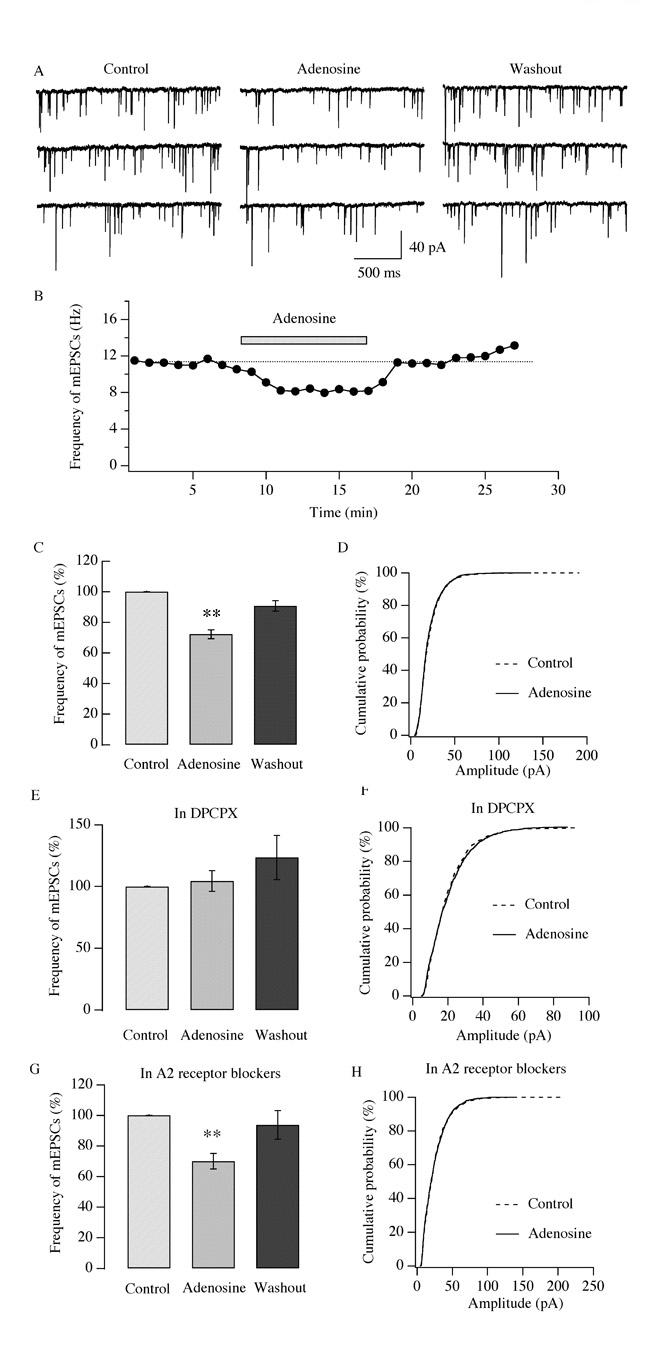
Adenosine inhibits the frequency but not the amplitude of miniature EPSCs. mEPSCs were recorded in hypocretin/orexin neurons held at −60mV under voltage clamp in the presence of bicuculline and TTX in all solutions. A-C, after a stable recording of mEPSCs was obtained, adenosine (100 μM) was applied to the recorded neuron via bath solution. Sample traces of mEPSCs recorded at various stages of our experiments are shown in A and the time course of a typical experiment is presented in B. The filled bar above the trace in B indicates the application of adenosine. C, pooled data of frequency of mEPSCs from all tested neurons indicate that the frequency of mEPSCs significantly declines in the presence of adenosine and recovers after its withdrawal (**, P<0.01, ANOVA). D, accumulative probability curves for the amplitude of mEPSC events detected before and during application of adenosine are generated and plotted. There is no significant difference between events in control and adenosine groups as indicated by the Kolmogorov-Smirnov test. Solid line: control, 2770 events; dotted line: plus adenosine, 1937 events. E-F, the effect of adenosine on the frequency and amplitude of mEPSCs was examined in the presence of DPCPX, a selective A1 receptor antagonist. Adenosine does not inhibit the frequency and amplitude of mEPSCs in the presence of DPCPX as suggested by ANOVA (P>0.05, E) and the Kolmogorov-Smirnov test (P>0.05, F). In F, solid line: control, 1228 events; dotted line: plus adenosine, 1170 events. G-H, experiments were performed as described above in the presence of A2 receptor antagonists (SCH 58261 and MRS 1706). In their presence, adenosine significantly depresses the frequency (G, **, P<0.01, ANOVA) but not the amplitude of mEPSCs (H, P>0.05, Kolmogorov-Smirnov test). In H, solid line: control, 1450 events; dotted line: plus adenosine, 974 events.
In a parallel experiment, the adenosine-mediated effect on the frequency and amplitude of mEPSCs was tested in the presence of DPCPX (Figure 7 E-F). The application of DPCPX alone induced only a negligible change in the frequency of mEPSCs (data not shown); the frequency of mEPSCs in DPCPX serves as the control for subsequent experiments. The frequency of mEPSCs (5.7 ± 1.9 Hz, n=6, control) was 104.4 ± 8.3 % of control (n=6) in the presence of adenosine and 123.4 ± 17.8 % of control (n=6) after its withdrawal when DPCPX was in all solutions (Figure 7 E); this change was not significant as determined by ANOVA (Degree of freedom=17, F=1.203, P>0.05). As shown in Figure 7 F, cumulative probability curves were plotted for the amplitude of mEPSC events detected before and during the application of adenosine and no change in amplitude was observed as suggested by the Kolmgorov-Smirnov test (P>0.05). Our experiments indicate that the selective A1 receptor antagonist DPCPX could eliminate the adenosine-induced inhibitory effect on the frequency of mEPSCs. Consistent with the results reported above, the inhibitory effect of adenosine on the frequency of mEPSCs was intact in the presence of A2 receptor antagonists (Figure 7 G). The frequency of mEPSCs (6.23 ± 1.1 Hz, n=7, control) was 70.1 ± 5.1 % of control (n=7) in the presence of adenosine (100 μM) and 93.8± 9.4 % of control (n=7) after its removal when SCH58261 (1 μM) and MRS1706 (0.1 μM) were present in all solutions; this attenuation was significant (Degree of freedom=20, F=6.53, P<0.01, ANOVA test). As shown in Figure 7 H, cumulative probability curves were plotted for the amplitude of mEPSC events detected before and during the application of adenosine in the presence of A2 receptor antagonists SCH58261 (1 μM) and MRS1706 (0.1 μM); there was no change in amplitude as revealed by the Kolmogorov-Smirnov test (P>0.05).
In summary, our results indicate that adenosine inhibits glutamatergic synaptic transmission in hypocretin/orexin neurons presynaptically.
Inhibition of voltage-dependent calcium currents by adenosine
We have demonstrated how adenosine modulates glutamatergic transmission presynaptically in hypocretin/orexin neurons. It is intriguing to ask if adenosine directly modulates the cell bodies of these neurons, since there was evidence that these soma possess adenosine receptors in rats (Thakkar et al. 2002). Previous reports demonstrated that adenosine directly hyperpolarizes the membrane potential of the soma of nerve cells and inhibits the influx of calcium ions at neuronal cell bodies by modulating the G-protein coupled inwardly rectifying potassium (GIRK) currents and voltage-dependent calcium currents (Dunwiddie and Masino 2001). Therefore, we tested whether adenosine modulates ion channels carrying calcium and GIRK currents at the cell bodies of hypocretin/orexin neurons. To monitor whole-cell calcium currents a voltage step from −80 mV to −20 mV (100 ms duration) was applied to the recorded neurons in the presence of TTX (1 μM) plus TEA (40 mM) in the bath solution and Cs+ in the pipette solution (Figure 8 A-C). After a stable recording of whole cell calcium currents was obtained in hypocretin/orexin neurons under voltage clamp (clamped at −80 mV), adenosine (30 μM) was applied to recorded neurons. The amplitude of whole-cell calcium currents (758.05 ± 92.35 pA, n=5, control) was 77.94 ± 4.02% of control (n=5) during the application of adenosine and 104.11 ± 6.53 % of control (n=4) after its removal (Figure 8 C), indicating a significant depression of the whole-cell calcium currents (Degree of freedom=13, F=12.69, P<0.01, ANOVA test). In addition, since it has been reported that the Gi/Go pathway couples to the GIRK current in hypocretin/orexin neurons (Fu et al. 2004; Xie et al. 2006), we tested whether adenosine induces the GIRK current in hypocretin/orexin neurons (Figure 8 D). A ramp (from −140 mV to −20 mV, 600 ms duration) was applied to the recorded hypocretin/orexin neurons under voltage clamp in the presence of TTX and Cd2+. After recording a stable baseline of membrane current response to the ramp protocol, adenosine (100 μM) was applied to the recorded neurons and membrane currents were recorded. A typical I-V relationship of membrane currents responding to the test ramp is shown in Figure 8 D. The amplitude of membrane currents was 109.5 ± 14.9 % of control (n=8) in the presence of adenosine and 86.3 ± 8.0% of control (n=4) after its withdrawal, which was not significantly different from that of control (Degree of freedom=19, F=0.929, P>0.05, ANOVA test). It appears that adenosine did not induce a detectable GIRK current in hypocretin/orexin neurons.
Fig.8.

Adenosine inhibits voltage-dependent calcium currents in hypocretin/orexin neurons. A, sample traces of whole-cell voltage-dependent calcium currents recorded at various stages in our experiments are shown. Voltage-dependent calcium currents were induced in hypocretin/orexin neurons held at −80 mV under voltage clamp with a voltage step from −80 mV to −20 mV. B, the time course of a typical experiment is shown. The application of adenosine (30 μM) is indicated as the filled bar above the time course curve. Letters a, b and c indicate time points when sample traces were recorded. C, pooled data from all tested neurons were analyzed and plotted, which demonstrate that the amplitude of voltage-dependent calcium currents significantly decreases in the presence of adenosine (30 μM) (**, P<0.01, ANOVA). D, the I-V relationship of membrane currents induced by a ramp pulse (from −140 mV to −20 mV, duration=600 ms) before, during and after the application of adenosine is shown. There is no significant GIRK current in the presence of adenosine.
In conclusion, adenosine inhibits voltage-dependent calcium currents and does not induce GIRK current in hypocretin/orexin neurons.
DISCUSSION
The role of adenosine in promoting sleep has been well documented in cholinergic neurons in the basal forebrain (Basheer et al. 2004). However, the effect of adenosine on the LH, particularly on the arousal/wakefulness promoting hypocretin/orexin neurons has not been examined despite the fact that adenosine receptors exist in these neurons in rats (Thakkar et al. 2002). In this study, we report the novel finding that adenosine exerts an inhibitory effect on hypocretin/orexin neurons in the LH. Activation of the A1 adenosine receptor attenuated the generation of action potentials and inhibited voltage-dependent calcium currents in these neurons. The inhibition in the generation of action potentials might be due to the inhibitory effect of adenosine on the excitatory synaptic transmission to the hypocretin/orexin neurons since adenosine did not have direct effects on membrane potential or conductance in these cells.
Adenosine inhibits the activity of hypocretin/orexin neurons
Adenosine has been reported to exert both inhibitory and stimulatory effects in the CNS. Its inhibitory effect is usually mediated via the A1 receptor, while its stimulatory effect is usually mediated by the A2 receptor (Dunwiddie and Masino 2001). In the LH, our data showed that adenosine caused a dose-dependent inhibition in hypocretin/orexin neurons. Adenosine was able to significantly decrease the frequency of sEPSCs at a low concentration of 10 μM and the maximum inhibition was obtained when the concentration of adenosine reached 100 μM (Figure 2 C). Although the concentration of adenosine used in our experiments is high compared to reports that its extracellular concentration in the basal forebrain, thalamus, striatum and cerebellar cortex ranges from 40 nM to 270 nM (Ballarin et al. 1991; Pazzagli et al. 1994, 1995; Porkka-Heiskanen et al. 2000), we believe that the nanomolar levels of extracellular adenosine in vivo may be the residue of intensively released adenosine, which maintains a tonic modulatory role. In our experiments, a tonic inhibition of synaptic transmission by endogenous release of adenosine was not observed, since application of the A1 receptor antagonist DPCPX induced little change in the frequency of sEPSCs and mEPSCs (data not shown). We believe that this is probably due to a relatively quiescent state of the neurons in our slice preparations and washout of adenosine during slice preparation, storage and perfusion in the recording chamber. We do not exclude the possibility of intensive release of endogenous adenosine under other conditions in brain slices.
Four types (A1, A2A, A2B and A3) of adenosine receptors exist in the central nervous system (Dunwiddie and Masino 2001; Ribeiro et al. 2003). The distribution of these subtypes has been examined in brain regions such as the amygdala, cortex, cerebellum, hippocampus, nucleus tractus solitarius, olfactory bulb, spinal cord, and thalamus (Review by Ribeiro et al. 2003). Immunocytochemical staining for the A1 adenosine receptor has revealed its existence in the hypocretin/orexin neurons of the LH in rats (Thakkar et al. 2002). In this study, the expression of functional A1 adenosine receptor in the LH of mice was demonstrated (Figure 5 and 6). The A1 receptor exists not only on the cell bodies of hypocretin/orexin neurons but also on presynaptic terminals innervating these neurons as suggested by our data (Figures 7 and 8), which is similar to previous reports on other neuronal types in the CNS (Chen and van den Pol 1997; Wetherington and Lambert 2002).
Mechanism of adenosine-mediated inhibition in hypocretin/orexin neurons
The inhibitory effect of adenosine was blocked in slices pre-treated with PTX, suggesting the involvement of the Gi/Go pathway in signaling triggered by the A1 adenosine receptor (Figure 4). This finding is in line with previous reports (Park et al. 2001). Based on our current results, we believe that adenosine exerts its inhibitory effect on the arousal-promoting hypocretin/orexin neurons in the LH at multiple sites as illustrated in Figure 9.
Fig.9.
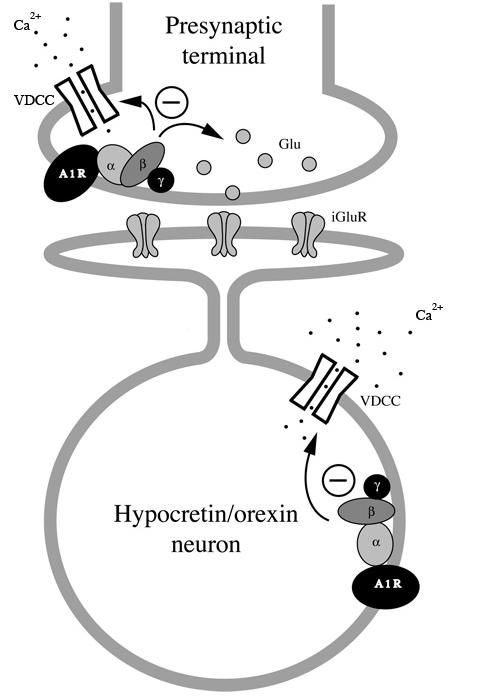
A schematic graph illustrates pathways mediating the effect of adenosine in hypocretin/orexin neurons. Activation of A1 receptor occurs both at presynaptic terminals innervating hypocretin/orexin neurons and at cell bodies of these neurons. At presynaptic terminals, adenosine might depress voltage-dependent calcium channels which leads to a reduction in calcium-dependent glutamate release. Adenosine might also directly inhibit calcium-independent exocytosis of vesicles containing glutamate. At the soma of hypocretin/orexin neurons, adenosine inhibits voltage-dependent calcium channels but does not induce a GIRK current. A1R: A1 adenosine receptor; VDCC: voltage-dependent calcium channel; iGluR: ionotropic glutamate receptor.
Presynaptic terminals
Adenosine inhibits synaptic transmission via the depression of neurotransmitter release at presynaptic terminals in many brain areas including the hippocampus (Yoon and Rothman 1991; Manzoni et al. 1994), cerebellar cortex (Kocsis et al. 1984), thalamus (Ulrich and Huguenard 1995), SCN (Chen and van den Pol 1997) and spinal cord (Li and Perl 1994). Consistent with previous reports, our data suggest that adenosine inhibits glutamatergic synaptic transmission to the hypocretin/orexin neurons by two possible mechanisms. The fact that adenosine decreased the frequency of sEPSCs and the amplitude of evoked EPSPs suggests that it could reduce the release of glutamate by inhibition of calcium channels at presynaptic terminals as reported by Wu and Saggau (1997), since both sEPSCs and evoked EPSPs are due to calcium-dependent release of glutamate driven by the propagation of action potentials to nerve terminals. The other mechanism may involve the direct inhibition of the release apparatus, which is supported by the fact that adenosine decreased the frequency of mEPSCs, i.e. the quantal release of glutamate form nerve terminals (Figure 7). The direct interaction of presynaptic adenosine receptor with the machinery responsible for synaptic vesicle exocytosis has been reported (Scholz and Miller 1992). The origin of the excitatory presynaptic terminals that are modulated by adenosine could be from both local circuitry such as the glutamatergic circuits proposed by Li and co-workers (2002) and afferent fibers from other brain areas such as the MFB as suggested in this study (Figure 3).
Postsynaptic neurons
In addition to modulation of neurotransmitter release by adenosine in CNS neurons, ion channels at cell bodies of central neurons are targets of adenosine-mediated modulation as well. Calcium channels responsible for Ca2+ influx during action potentials are modulated directly or indirectly via adenosine receptors. For instance, adenosine inhibits calcium currents via the A1 receptor in sensory neurons (Kasai and Aosaki 1989), pyramidal neurons of the hippocampus (Scholz and Miller 1991; Mogul et al. 1993; Wu and Saggau 1994), sympathetic neurons (Zhu and Ikeda 1993), and GABAergic neurons in the SCN (Chen and van den Pol 1997). The activation of A2 receptor leads to potentiation of the P-type calcium channel in neurons of the brainstem (Umemiya and Berger 1994). In this study, we demonstrate that adenosine inhibits whole-cell voltage-dependent calcium currents in hypocretin/orexin neurons (Figure 8A-C), which is in line with the inhibitory effect mediated by A1 receptor mentioned above and suggests that functional A1 receptor is expressed in hypocretin/orexin neurons in mice. As an important regulator of membrane excitability, the GIRK current is triggered by activation of G-protein coupled adenosine A1 receptor in relay neurons in the thalamus (Pape 1992), pyramidal neurons of the hippocampus (Lüscher et al. 1997; Takigawa and Alzheimer 2002; Wetherington and Lambert 2002), cholinergic neurons in the magnocellular preoptic nucleus and substantia innominata (Arrigoni et al. 2006) and neurons of the locus coeruleus (Pan et al. 1995). In contrast, our results demonstrate that activation of adenosine receptor does not induce GIRK current in hypocretin/orexin neurons (Figure 8), suggesting that the A1 receptor does not couple to GIRK channels in these neurons. This result is consistent with our finding that adenosine does not directly modulate membrane potential in hypocretin/orexin neurons (Figure 4), which is a distinct property of the GIRK current in these cells (Fu et al. 2004; Xie et al. 2006).
Functional Implications
It is widely accepted that adenosine participates in sleep induction via the A1 receptor in several brain areas. On the one hand, adenosine directly inhibits neurons promoting wakefulness in many brain regions (Arrigoni et al. 2001; 2006; Kohlmeier and Leonard 2006). On the other hand, adenosine disinhibits neurons that directly depress neurons in wakefulness-maintaining brain regions such as the ventrolateral preoptic nucleus (VLPO), which is a key regulator of the behavioral state that promotes sleep (Chamberlin et al. 2003). In this study, our data revealed a new potential site of action of adenosine in light of its function in sleep regulation. We find that adenosine depresses action potentials in hypocretin/orexin neurons via the A1 adenosine receptor, which is achieved by a possible attenuation of excitatory inputs to these neurons within (local) and outside (afferent fibers from the MFB) of the LH. The excitatory inputs onto the hypocretin/orexin neurons are of considerable importance to the functioning of this system (Li et al. 2002; Horvath and Gao 2005); a reduction in the excitatory afferents to the hypocretin/orexin system by adenosine greatly attenuates its excitability and limits its capability to control its targets. Adenosine may also restrain the function of the hypocretin/orexin neurons by depressing the calcium influx via voltage-dependent calcium channels during action potentials. Voltage-dependent calcium channels at cell bodies play a critical role in excitation-transcription coupling in neurons (Catterall 1998), since the calcium influx via calcium channels is required for activation of the cAMP- and calcium-dependent transcription factor CREB (Bading et al. 1993; Abel et al. 1997; Rajadhyaksha et al. 1999) and transcription of immediate-early genes triggered by neuronal activity (Murphy et al. 1991; Rajadhyaksha et al. 1999). In our case, the inhibition of calcium currents in the hypocretin/orexin neurons by adenosine would decrease excitation-dependent synthesis and release of hypocretin/orexin. Altogether, we propose that it is highly possible for adenosine to reduce wakefulness by attenuating the output of the hypocretin/orexin system in the LH in addition to its actions in other brain areas, which could be the subject of a follow-up study. A recent report just emerged that application of A1 adenosine receptor agonist, N6 cyclopentyladenosine (CPA), into the perifornical lateral hypothalamic area promotes sleep in rats (Kumar et al. 2006, SFN Abstrsct).
In summary, our results showing that adenosine decreases the generation of action potentials, excitatory synaptic transmission and calcium currents in hypocretin/orexin neurons reveal a new role for adenosine in sleep regulation, which is complementary to the current framework of sleep research.
ACKNOWLEGEMENTS
We'd like to thank Dr. Anthony van den Pol for antiserum against hypocretin-2, Ms. Marya Shanabrough and Susan Andranovich for helpful suggestions on the manuscript.
Footnotes
GRANTS
This work is supported by NIH grants DK 061478 and DK 070723.
REFERENCES
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourthouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based longterm memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Alam MN, Szymusiak R, Gong H, King J, McGinty D. Adenosinergic modulation of rat basal forebrain neurons during sleep and waking: neuronal recording with microdialysis. J Physiol. 1999;521:679–690. doi: 10.1111/j.1469-7793.1999.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni E, Rainnie DG, McCarley RW, Greene RW. Adenosine-mediated presynaptic modulation of glutamatergic transmission in the laterodorsal tegmentum. J Neurosci. 2001;21:1076–1085. doi: 10.1523/JNEUROSCI.21-03-01076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni E, Chamberlin NL, Saper CB, McCarley RW. Adenosine inhibits basal forebrain cholinergic and noncholinergic neurons in vitro. Neuroscience. 2006;140:403–413. doi: 10.1016/j.neuroscience.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Ballarin M, Fredholm BB, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand. 1991;142:97–103. doi: 10.1111/j.1748-1716.1991.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Basheer R, Arrigoni E, Thatte HS, Greene RW, Ambudkar IS, McCarley RW. Adenosine induces inositol 1,4,5-trisphosphate receptor-mediated mobilization of intracellular calcium stores in basal forebrain cholinergic neurons. J Neurosci. 2002;22:7680–6. doi: 10.1523/JNEUROSCI.22-17-07680.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep–wake regulation. Progress in Neurobio. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Quantal analysis of EPSCs recorded from small numbers of synapses in hippocampal culture. J Neurophysiol. 1995;73:1145–1156. doi: 10.1152/jn.1995.73.3.1145. [DOI] [PubMed] [Google Scholar]
- Biber K, Klotz KN, Berger M, Gebicke-Harter PJ, van Calker D. Adenosine A1 receptor-mediated activation of phospholipase C in cultured astrocytes depends on the level of receptor expression. J Neurosci. 1997;17:4956–4964. doi: 10.1523/JNEUROSCI.17-13-04956.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O'Kelly I, Gerasimenko O, Fugger L, Verkhratsky A. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. doi: 10.1016/j.neuron.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24:307–323. doi: 10.1016/s0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- Chamberlin NL, Arrigoni E, Chou TC, Scammell TE, Greene RW, Saper CB. Effects of adenosine on gabaergic synaptic inputs to identified ventrolateral preoptic neurons. Neuroscience. 2003;119:913–918. doi: 10.1016/s0306-4522(03)00246-x. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Adenosine modulation of calcium currents and presynaptic inhibition of GABA release in suprachiasmatic and arcuate nucleus neurons. J Neurophysiol. 1997;77:3035–3047. doi: 10.1152/jn.1997.77.6.3035. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Sutcliffe JG. The hypocretins and sleep. FEBS J. 2005;272:5675–5688. doi: 10.1111/j.1742-4658.2005.04981.x. [DOI] [PubMed] [Google Scholar]
- de Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes. 2003;52:2767–2773. doi: 10.2337/diabetes.52.11.2767. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–1662. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu LY, Acuna-Goycolea C, van den Pol AN. Neuropeptide Y inhibits hypocretin/orexin neurons by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci. 2004;24:8741–8751. doi: 10.1523/JNEUROSCI.2268-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki N, Yoshida Y, Ripley B, Honda K, Mignot E, Nishino S. Changes in CSF hypocretin-1 (orexin A) levels in rats across 24 hours and in response to food deprivation. Neuroreport. 2001;12:993–997. doi: 10.1097/00001756-200104170-00026. [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Melanin-concentrating hormone depresses L-, N-, and P/Q-type voltage-dependent calcium channels in rat lateral hypothalamic neurons. J Physiol. 2002;542:273–286. doi: 10.1113/jphysiol.2002.019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol. 2001;533:237–252. doi: 10.1111/j.1469-7793.2001.0237b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henny P, Jones BE. Vesicular glutamate (VGlut), GABA (VGAT), and acetylcholine (VACht) transporters in basal forebrain axon terminals innervating the lateral hypothalamus. J Comp Neurol. 2006;496:453–467. doi: 10.1002/cne.20928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity's association with insomnia. Cell Metab. 2005;1:279–286. doi: 10.1016/j.cmet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Kasai H, Aosaki T. Modulation of Ca-channel current by an adenosine analog mediated by a GTP-binding protein in chick sensory neurons. Pflugers Arch. 1989;414:145–149. doi: 10.1007/BF00580956. [DOI] [PubMed] [Google Scholar]
- Kilduff TS, Peyron C. The hypocretin/orexin ligand-receptor system: implications for sleep and sleep disorders. Trends Neurosci. 2000;23:359–365. doi: 10.1016/s0166-2236(00)01594-0. [DOI] [PubMed] [Google Scholar]
- Kocsis JD, Eng DL, Bhisitkul RB. Adenosine selectively blocks parallel-fiber-mediated synaptic potentials in rat cerebellar cortex. Proc. Natl. Acad. Sci. USA. 1984;81:6531–6534. doi: 10.1073/pnas.81.20.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier KA, Leonard CS. Transmitter modulation of spike-evoked calcium transients in arousal related neurons: muscarinic inhibition of SNX-482-sensitive calcium influx. Eur J Neurosci. 2006;23:1151–62. doi: 10.1111/j.1460-9568.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Rai S, Szymusiak R, McGinty D, Alam N. Effects of adenosine A1 receptor agonist into the perifornical lateral hypothalamic area on sleep. Program No. 458.12. 2006 Neuroscience Meeting Planner. Society for Neuroscience; Atlanta, GA: 2006. Online. [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Perl ER. Adenosine inhibition of synaptic transmission in the substantia gelatinosa. J Neurophysiol. 1994;72:1611–1621. doi: 10.1152/jn.1994.72.4.1611. [DOI] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN. Hypocretin/Orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- Marks GA, Birabil CG. Enhancement of rapid eye movement sleep in the rat by cholinergic and adenosinergic agonists infused into the pontine reticular formation. Neuroscience. 1998;86:29–37. doi: 10.1016/s0306-4522(98)00005-0. [DOI] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem. 1999;72:785–790. doi: 10.1046/j.1471-4159.1999.720785.x. [DOI] [PubMed] [Google Scholar]
- Mieda M, Yanagisawa M. Sleep, feeding, and neuropeptides: roles of orexins and orexin receptors. Curr Opin Neurobiol. 2002;12:339–345. doi: 10.1016/s0959-4388(02)00331-8. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–98. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogul DJ, Adams ME, Fox AP. Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca2+ current in hippocampal CA3 neurons. Neuron. 1993;10:327–334. doi: 10.1016/0896-6273(93)90322-i. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- Pan WJ, Osmanovic SS, Shefner SA. Characterization of the adenosine A1 receptor-activated potassium current in rat locus ceruleus neurons. J Pharmacol Exp Ther. 1995;273:537–544. [PubMed] [Google Scholar]
- Pape HC. Adenosine promotes burst activity in guinea-pig geniculocortical neurons through two different ionic mechanisms. J Physiol. 1992;447:729–753. doi: 10.1113/jphysiol.1992.sp019026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Jeong SW, Cha SK, Lee BS, Kong ID, Ikeda SR, Lee JW. Modulation of N-type Ca2+ currents by A1-adenosine receptor activation in male rat pelvic ganglion neurons. J Pharmacol Exp Ther. 2001;299:501–508. [PubMed] [Google Scholar]
- Pazzagli M, Corsi C, Latini S, Pedata F, Pepeu G. In vivo regulation of extracellular adenosine levels in the cerebral cortex by NMDA and muscarinic receptors. Eur J Pharmacol. 1994;254:277–282. doi: 10.1016/0014-2999(94)90465-0. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, McCarley RW. Brain sitespecificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience. 2000;99:507–517. doi: 10.1016/s0306-4522(00)00220-7. [DOI] [PubMed] [Google Scholar]
- Portas CM, Thakkar M, Rainnie DG, Greene RW, McCarley RW. Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neuroscience. 1997;79:225–235. doi: 10.1016/s0306-4522(96)00640-9. [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha A, Barczak A, Macias W, Leveque JC, Lewis SE, Konradi C. L-Type Ca2+ channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. Journal of Neuroscience. 1999;19:6348–6359. doi: 10.1523/JNEUROSCI.19-15-06348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro JA, Sebastiao AM, de Mendonca A. Adenosine receptors in the nervous system: pathophysiological implications. Prog Neurobiol. 2002;68:377–392. doi: 10.1016/s0301-0082(02)00155-7. [DOI] [PubMed] [Google Scholar]
- Sakurai T. Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev. 2005;9:231–241. doi: 10.1016/j.smrv.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, Kageyama H, Kunita S, akahashi S, Goto K, Koyama Y, Shioda S, Yanagisawa M. Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron. 2005;46:297–308. doi: 10.1016/j.neuron.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Saper CB. Chapter 14: Staying awake for dinner: hypothalamic integration of sleep, feeding, and circadian rhythms. Prog Brain Res. 2006;153:243–252. doi: 10.1016/S0079-6123(06)53014-6. [DOI] [PubMed] [Google Scholar]
- Satoh S, Matsumura H, Suzuki F, Hayaishi O. Promotion of sleep mediated by the A2a-adenosine receptor and possible involvement of this receptor in the sleep induced by prostaglandin D2 in rats. Proc Natl Acad Sci U S A. 1996;93:5980–5984. doi: 10.1073/pnas.93.12.5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Matsumura H, Hayaishi O. Involvement of adenosine A2A receptor in sleep promotion. Eur J Pharmacol. 1998;351:155–162. doi: 10.1016/s0014-2999(98)00302-1. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Analysis of adenosine actions on Ca2+ currents and synaptic transmission in cultured rat hippocampal pyramidal neurones. J Physiol. 1991;435:373–393. doi: 10.1113/jphysiol.1991.sp018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- Siegel JM. The neurotransmitters of sleep. J Clin Psychiatry. 2004;65(Suppl 16):4–7. [PMC free article] [PubMed] [Google Scholar]
- Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW. Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res. 2000;115:183–204. doi: 10.1016/s0166-4328(00)00258-8. [DOI] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C. Phasic and tonic attenuation of EPSPs by inward rectifier K+ channels in rat hippocampal pyramidal cells. J Physiol. 2002;539:67–75. doi: 10.1113/jphysiol.2001.012883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar MM, Winston S, McCarley RW. Orexin neurons of the hypothalamus express adenosine A1 receptors. Brain Res. 2002;944:190–194. doi: 10.1016/s0006-8993(02)02873-1. [DOI] [PubMed] [Google Scholar]
- Thakkar MM, Winston S, McCarley RW. A1 receptor and adenosinergic homeostatic regulation of sleep-wakefulness: effects of antisense to the A1 receptor in the cholinergic basal forebrain. J Neurosci. 2003;23:4278–4287. doi: 10.1523/JNEUROSCI.23-10-04278.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticho SR, Radulovacki M. Role of adenosine in sleep and temperature regulation in the preoptic area of rats. Pharmacol Biochem Behav. 1991:33–40. doi: 10.1016/0091-3057(91)90317-u. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. Purinergic inhibition of GABA and glutamate release in the thalamus: implications for thalamic network Activity-dependent activity. Neuron. 1995;15:909–918. doi: 10.1016/0896-6273(95)90181-7. [DOI] [PubMed] [Google Scholar]
- Umemiya M, Berger AJ. Activation of adenosine A1 and A2 receptors differentially modulates calcium channels and glycinergic synaptic transmission in rat brainstem. Neuron. 1994;13:1439–1446. doi: 10.1016/0896-6273(94)90429-4. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB. Pre- and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J Neurosci. 1998;18:7962–7971. doi: 10.1523/JNEUROSCI.18-19-07962.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington JP, Lambert NA. Differential desensitization of responses mediated by presynaptic and postsynaptic A1 adenosine receptors. J Neurosci. 2002;22:1248–1255. doi: 10.1523/JNEUROSCI.22-04-01248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Xie X, Crowder TL, Yamanaka A, Morairty SR, Lewinter RD, Sakurai T, Kilduff TS. GABA(B) receptor-mediated modulation of hypocretin/orexin neurones in mouse hypothalamus. J Physiol. 2006;574:399–414. doi: 10.1113/jphysiol.2006.108266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, Mignot E, Nishino S. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–1081. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]
- Yoon KW, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Adenosine modulates voltage-gated Ca2+ channels in adult rat sympathetic neurons. J Neurophysiol. 1993;70:610–620. doi: 10.1152/jn.1993.70.2.610. [DOI] [PubMed] [Google Scholar]
- Zeitzer JM, Buckmaster CL, Parker KJ, Hauck CM, Lyons DM, Mignot E. Circadian and homeostatic regulation of hypocretin in a primate model: implications for the consolidation of wakefulness. J Neurosci. 2003;23:3555–3560. doi: 10.1523/JNEUROSCI.23-08-03555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]