

Abstract
In a search for nuclear genes that affect mutagenesis of mitochondrial DNA in Saccharomyces cerevisiae, an ATP-NAD (NADH) kinase, encoded by POS5, that functions exclusively in mitochondria was identified. The POS5 gene product was overproduced in Escherichia coli and purified without a mitochondrial targeting sequence. A direct biochemical assay demonstrated that the POS5 gene product utilizes ATP to phosphorylate both NADH and NAD+, with a twofold preference for NADH. Disruption of POS5 increased minus-one frameshift mutations in mitochondrial DNA 50-fold, as measured by the arg8m reversion assay, with no increase in nuclear mutations. Also, a dramatic increase in petite colony formation and slow growth on glycerol or limited glucose were observed. POS5 was previously described as a gene required for resistance to hydrogen peroxide. Consistent with a role in the mitochondrial response to oxidative stress, a pos5 deletion exhibited a 28-fold increase in oxidative damage to mitochondrial proteins and hypersensitivity to exogenous copper. Furthermore, disruption of POS5 induced mitochondrial biogenesis as a response to mitochondrial dysfunction. Thus, the POS5 NADH kinase is required for mitochondrial DNA stability with a critical role in detoxification of reactive oxygen species. These results predict a role for NADH kinase in human mitochondrial diseases.
In addition to their essential role in the intermediary metabolism of eukaryotic cells, mitochondria are also the principal site for the production of energy through electron transport and oxidative phosphorylation. In mammals, proper functioning of the mitochondria requires the coordinated function of over 1,500 nuclear gene products with the 37 gene products encoded by a discrete, circular chromosome located in the mitochondrial matrix. Heritable defects in nuclear and mitochondrial genes that disrupt cellular energy production cause devastating metabolic disorders that play a central role in many degenerative diseases, aging, and cancer. Because the vast majority of cellular energy is generated by the mitochondria, this class of energy-deficit diseases has been termed mitochondrial diseases (80). To date, over 100 autosomally inherited mitochondrial disorders have been linked to defects in nucleus-encoded mitochondrial proteins (6, 70). In 1988, Wallace and coworkers were the first to show that mutation of a mitochondrial gene, NADH dehydrogenase subunit 4, could cause Leber's hereditary optical neuropathy, a maternally inherited mitochondrial disease (81). Since that time, nearly 300 pathogenic point mutations and deletions in mitochondrial DNA (mtDNA) have been identified (http://www.mitomap.org).
Whereas most autosomally inherited mitochondrial diseases arise from defects in nuclear genes essential for energy metabolism, maternally inherited mitochondrial diseases result from enrichment of the mutant fraction of inherited mtDNA during development of the organism. An inherited predisposition to mutation of mtDNA helps to explain the delayed onset and progressive course of some mitochondrial diseases. Progressive external ophthalmoplegia (PEO), mitochondrial neurogastrointestinal encephalomyopathy, and Alpers syndrome belong to an interesting subset of autosomal mitochondrial diseases associated with depletion of mtDNA or the accumulation of mutations and deletions in mtDNA (21, 45, 78, 79). Over the last three years, nuclear genes responsible for these disorders have been identified, including genes for adenine nucleotide translocator 1 (26), thymidine phosphorylase (49), mitochondrial thymidine kinase (60), mitochondrial deoxyguanosine kinase (42), a putative mitochondrial helicase (67), and the catalytic subunit of the mtDNA polymerase, Pol γ (77). Because these enzymes function in either nucleotide metabolism or mtDNA replication, defects in their genes are expected to compromise mechanisms by which the cell maintains the integrity of its mtDNA. A previous study investigated one active-site mutation in Pol γ associated with PEO and demonstrated that the error-prone DNA synthesis catalyzed by the altered polymerase is consistent with the accumulation of mutations in tissues of PEO patients (57). Although much has been learned over the last several years about the protein factors that function to replicate and repair mtDNA, many genes that are putative candidates for mitochondrial diseases have not yet been discovered.
Defining the mechanisms affecting mutation and propagation of mtDNA is critical to understanding the origins of diseases stemming from mitochondrial genetic change. In an effort to identify nuclear genes that contribute to the stability of mtDNA, a genetic assay to measure the mutation rate of mtDNA in Saccharomyces cerevisiae was developed previously (72). In the present study, we utilized this assay to screen targeted disruptions of nuclear open reading frames with unknown functions to assess their role in mtDNA metabolism. A surprisingly high number of these gene disruptions exhibited a mitochondrial mutator phenotype. Disruption of the YPL188w gene, previously identified as the peroxide sensitivity allele POS5 (32), caused a 51-fold increase in the mitochondrial mutation rate. POS5 mutants also exhibit slow growth, enhanced frequency of petite formation, pronounced oxidative damage to mitochondrial proteins, and extreme sensitivity to exogenous copper ions. We demonstrated by direct biochemical assay that the cloned and overexpressed POS5 gene encodes a mitochondrial NADH kinase. The probable functions of the mitochondrial NADH kinase in mitochondrial mutagenesis and detoxification of reactive oxygen species (ROS) are discussed.
MATERIALS AND METHODS
Strains and plasmids.
TF235 and TF236 were generous gifts from Tom Fox (Cornell University). The genotype of TF235 is a ino1::HIS3 arg8::HISG pet9 (op1) ura3-52 lys2 cox3::ARG8m. The genotype of TF236 is α ino1::HIS3 arg8::HISG pet9 (op1) ura3-52 lys2 cox3::arg8m. Although the cox3 mutation renders TF235 and TF236 respiration deficient, these strains cannot lose their nonfunctional mitochondria because the pet9 mutation is lethal in the absence of mitochondria. The cox3::ARG8m construction has been described (69). YPH925 was obtained from Dmitry Gordenin (National Institute of Environmental Health Sciences), and the genotype is α ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 cyh2R kar1-Δ15 (68). The POS5 disruptions were made in both TF236 and YPH925 with a disruption plasmid provided by the Yale Genome Analysis Center. Disruptions were verified by PCR. All experiments were done in the YPH925 strain background unless otherwise indicated. The plasmid pFL39 has been described (3). The wild-type MIP1 gene was cloned into plasmid pFL39, and the gene was completely sequenced to confirm the cloned sequence. The exonuclease-deficient mip1-exo−(D171A, E173A) (40) mutant was constructed by using a Stratagene (La Jolla, Calif.) QuikChange site-directed mutagenesis kit, with the complementary oligonucleotide pair mip1-exo1 (5′-CTG GTG GTG TTT GCT GTA GCA ACA CTC TAT AAC G-3′) and mip1-exo2 (5′-GTT ATA GAG TGT TGC TAC AGC AAA CAC CAC CAG-3′) (the mutant nucleotides are underlined). The mip1-exo− gene was resequenced to confirm the mutagenic changes.
Growth curves.
Growth rates of cultures were measured in a spectrophotometer at an optical density at 600 nm (OD600). Cells were grown in 2-liter flasks with 400 ml of medium with vigorous shaking (250 rpm) at 30°C. YPD medium was made with 20 g of dextrose, 10 g of yeast extract, and 20 g of Bacto Peptone per liter. YPD low-sugar medium consists of 0.4 g of dextrose, 10 g of yeast extract, and 20 g of Bacto Peptone per liter. YPG medium was made with 14.2 ml of 1 M K2HPO4, 35.9 ml of 1 M KH2PO4, 30 ml of glycerol, 20 g of yeast extract, and 20 g of Bacto Peptone per liter. SD medium lacking adenine or lysine was prepared as described previously (64).
Mutation rate assays.
Mitochondrial mutation rates were measured by assaying reversion in the arg8m mutation assay (72). Initial screening consisted of 10 independent colonies for each disrupted open reading frame plated selectively on medium lacking arginine and plated nonselectively on YPD for a total cell count. Those that showed an elevated mitochondrial mutation frequency in the initial screen were assayed twice more, with 40 independent colonies per assay. Mutation rates were calculated by the method of Luria and Delbruck (41) or Lea and Coulson (34), as appropriate. Similarly, reversion of the lys2-801 and ade2-101 loci was assayed to measure nuclear mutation rates, and assays were performed at least four times.
Assay of mtDNA content.
Cells from a log-phase culture (200 ml) were harvested by centrifugation, washed with deionized water, and resuspended in extraction buffer (1 ml) containing 4 M guanidine thiocyanate, 25 mM sodium citrate (pH 7.1), and 0.56 M 2-mercaptoethanol. Cells were broken by vortexing for 5 min with glass beads. Following phenol-chloroform extraction and ethanol precipitation, total cellular DNA was resuspended in Tris-EDTA and spotted onto hybridization transfer membranes (DuPont NEN Research Products). Membranes were dried at 65°C for 1 h and then prehybridized for 1 to 3 h in 2% sodium dodecyl sulfate (SDS), 0.5% polyvinyl pyrrolidone, 0.2% heparin sulfate, 1 mM EDTA, 1 M NaCl, and 50 mM Tris-HCl (pH 7.5) at 37°C with constant agitation. Duplicate membranes were hybridized overnight with 32P-, 5′-end-labeled oligonucleotide probes corresponding to the mitochondrial origin of replication (5′-TTATTATGAAAATTATATCT TTTATTATATTCT-3′) or the nuclear Msh1 gene (5′-GACGATGATACATTGATTTGTTTGG-3′). Membranes were washed at room temperature to a final stringency of 0.1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-0.1% SDS, and blots were analyzed on a Molecular Dynamics PhosphorImager. Radioactivity was quantified with NIH Image 1.61 software, and mtDNA content was normalized to the nuclear signal.
Cytochrome c oxidase assay.
Yeast cultures were grown in YPD medium with vigorous shaking at 30°C until log-phase growth reached an OD600 of 0.3. Cells were harvested by centrifugation, washed with deionized water, and resuspended in buffer containing 0.05 M KPO4 (pH 6.8), 25 mM NaCl, and 1.5% N-dodecyl-β-maltoside. Cells were lysed by vortexing at full speed for 5 min with ∼1 ml of acid-washed glass beads (425 to 600 μm). Beads were removed by centrifugation at 10,000 × g for 2 min at 4°C, and the supernatant was designated the whole-cell extract. The protein concentration of the whole-cell extracts was determined as described by Bradford (4). Cytochrome c oxidase activity of whole-cell extracts was measured as described previously (33). Briefly, horse heart cytochrome c (Sigma) was dissolved in 50 mM KPO4 (pH 6.8) and treated with 10 mM sodium dithionite on ice for 5 min. Reduced cytochrome c was purified by Sephadex G-25 chromatography and quantified by spectrophotometry at 550 nm. Reactions (1 ml) containing 50 mM KPO4 (pH 6.8), 0.08% maltoside, and 75 μM reduced cytochrome c were initiated by the addition of 0.1 ml of the specified whole-cell extract (0.1 to 0.2 mg of protein) prior to incubation at room temperature. Oxidation of cytochrome c was calculated from the decrease in absorbance at 550 nm over 10 to 60 min.
Expression and purification of POS5 gene product.
Analysis of the two S. cerevisiae POS5 sequences present in GenBank for mitochondrial target sequences using the computer program MitoProtII (7) predicted cleavage sites between amino acid residues 17 and 18. Chromosomal DNA was extracted from S. cerevisiae strain YPH925 (68), and the full-length POS5 gene and a POS5Δ17 version were amplified by PCR and cloned into the protein expression vector pQE30 (Qiagen, Chatsworth, Calif.) by using standard methods. The PCR primers used for amplification of the full-length POS5 gene were 5′-tat agg atc cAT GTT TGT CAG GGT TAA ATT GAA T-3′ and 5′-aat taa gct tTT AAT CAT TAT CAG TCT GTC TCT T-3′ (the POS5 start and stop codons are shown in bold, and the BamHI and HindIII restriction endonuclease sites that flank the POS5 gene are underlined; non-POS5 vector sequences are shown in lowercase). Primers for POS5Δ17 were 5′-tat agg atc cAG TAC GTT GGA TTC ACA TTC C-3′ and 5′-aat tgt cga cTT AAT CAT TAT CAG TCT GTC TCT T-3′ (the BamHI and SalI sites are underlined). The two pQE30yPOS5 constructs were verified by DNA sequencing, transformed into Escherichia coli strain XL1-Blue (Stratagene), and screened for expression of soluble protein. The POS5Δ17 clone expressed soluble protein in high yield, and this clone was selected for protein production. XL1-Blue pQE30/yPOS5Δ17 was grown at 37°C in 2× YT (pH 7.0) medium composed of 1.6% (wt/vol) Bacto-tryptone, 1.0% (wt/vol) yeast extract, 85.6 mM NaCl, and 0.1 mg of ampicillin per ml to an OD600 of ∼0.5 to 0.9. The temperature of the air shaker incubator was decreased to 30°C 30 min prior to induction with 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Following a 2-h induction period, the cells were harvested by centrifugation, flash-frozen in liquid nitrogen, and stored at −80°C.
Preparation of cell extracts and all chromatographic procedures were performed at 4°C. A cell pellet derived from 1 liter of induced XL1-Blue pQE30/yPOS5Δ17 was thawed on wet ice and resuspended in lysis buffer (20 ml) containing 0.1 M Tris-HCl (pH 7.5), 0.2 M NaCl, 8% glycerol, 2 mM 2-mercaptoethanol, 0.5% NP-40, 0.02 M imidazole, and 0.1 mM phenylmethylsulfonyl fluoride. This suspension was passed through a French pressure cell at 18,000 lb/in2 to produce the whole-cell extract. Following centrifugation for 15 min at 30,000 × g, material remaining in the supernatant constituted the soluble lysate. The soluble lysate (19.8 ml) was mixed end-over-end for 60 min with 2 ml of nickel-nitrilotriacetic acid (Ni-NTA) Sepharose (Qiagen) equilibrated in lysis buffer. Unbound material was separated by gentle centrifugation, and the resin was washed twice with a 30-ml solution containing 0.05 M Tris-HCl (pH 7.5), 0.5 M NaCl, 8% glycerol, 2 mM 2-mercaptoethanol, 0.1% NP-40, and 0.02 M imidazole and once with 30 ml of the same solution with the NaCl concentration reduced to 0.2 M. The resin was transferred to a disposable polypropylene column, washed with an additional 10 ml of the latter wash solution, and eluted with this solution containing 0.25 M imidazole. Fractions containing purified Pos5Δ17 protein were frozen with liquid nitrogen in small aliquots and stored at −80°C. Protein concentrations were determined as described by Bradford (4) with bovine serum albumin as the standard.
Enzymatic assay of NADH kinase activity.
NADH kinase activity was determined in reaction mixtures (20 μl) containing 0.02 M HEPES-KOH (pH 7.6), 4 mM MgCl2, 0.4 μg of acetylated bovine serum albumin, 1 mM [γ-32P]ATP (1.5 μCi), 2 mM NADH or NAD+, and various quantities of enzyme. Following incubation at 37°C for 20 min, reactions were terminated by the addition (1 μl) of 0.5 M EDTA. Samples (2 μl) of each reaction were spotted onto polyethyleneimine-cellulose-coated thin-layer chromatography sheets (Merck), dried, and developed with 0.5 M LiCl-1 M acetic acid as the ascending solvent. Reaction products were visualized with a Storm 860 PhosphorImager (Molecular Dynamics) and quantified with NIH Image 1.62 imaging software.
Preparation of yeast mitochondrial lysates.
Fresh YPD broth (0.4 liter) was inoculated with 50 ml of an overnight culture, and cells were grown with vigorous shaking (250 rpm) at room temperature for 90 min. Log-phase growth was verified by monitoring OD600. Cells were harvested by centrifugation, washed with deionized water, and resuspended in cold mannitol buffer (5 ml) consisting of 0.21 M mannitol, 0.07 M sucrose, 5 mM Tris-HCl (pH 7.5), and 5 mM EDTA. Cells were lysed by vortexing with ∼1 ml of acid-washed glass beads (425 to 600 μm) for 5 min, and beads were removed by centrifugation at 20 × g for 5 min. The supernatant was centrifuged at 6,400 × g for 15 min, the pellet was resuspended in 5 ml of cold mannitol buffer, and mitochondria were purified by sedimentation through a two-step discontinuous sucrose gradient, as described previously (59). Mitochondrial pellets (obtained from ∼5 g [wet weight] of YPH925 and YPH925 pos5 cells) were resuspended in 250 μl of lysis buffer (1% NP-40, 0.3 M NaCl, 10% glycerol, 20 mM Tris-HCl [pH 8.0], 14 mM 2-mercaptoethanol) and left to lyse on ice for 10 min. The lysates were then centrifuged for 2 h at 14,000 rpm in an Eppendorf microcentrifuge. The protein concentration of the supernatants was determined as described by Bradford (4) and was typically in the range of 2 to 2.5 mg/ml. Mitochondrial lysates were concentrated with Microcon 30 devices to approximately 10 mg of protein per ml, frozen in liquid nitrogen, and stored at −80°C.
Detection of oxidation of mitochondrial proteins.
Carbonyl groups can be formed in proteins by oxidation of lysine, arginine, proline, and threonine side chains. To detect such oxidized residues, samples of yeast mitochondrial lysates (50 μg of protein) were treated with 2,4-dinitrophenylhydrazine as directed in the OxyBlot kit (Intergen) to convert carbonyl groups to 2,4-dinitrophenylhydrazone derivatives. Proteins were resolved on denaturing 4-to-20% polyacrylamide gels and electrotransferred to Immobilon-P membranes (Millipore). Membranes were probed with a first antibody specific for 2,4-dinitrophenyl moieties and a horseradish peroxidase-conjugated second antibody. After treatment with SuperSignal West Pico chemiluminescent substrate (Pierce), oxidized proteins were visualized by autoradiography. Digital photography and NIH Image 1.61 software were utilized to quantify autoradiograms.
POS5 transcription levels.
Competitive PCR was utilized to measure transcription of the POS5 gene. Competitor DNA was amplified from chromosomal DNA by PCR using the oligonucleotides 5′-AGGATAGCAACTCATCTATTGTGTGCTTGCACCATCCCATTTAACTGTA-3′ and 5′-GAGGAATACCATCAACAGAAAGTTGATACCTTGGTGTCTTGGTCTAC-3′. The 23 nucleotides at each 5′ end of this primer pair are homologous to the POS5 gene, whereas the 3′ portions of these primers amplify the actin gene. mRNA was isolated from 400-ml liquid cultures having an OD600 of <1.0 with the Promega AT1000 system. Based on absorbance at 260 nm, mRNA concentrations were equalized, and cDNA was synthesized by reverse transcription using Clontech's Advantage RT-for-PCR kit. In quantitative PCRs, fixed quantities of competitor DNA and variable POS5 cDNA competed for the POS5 primers 5′-AGGATAGCAACTCATCTATTGTG-3′ and 5′-GAGGAATACCATCAACAGAAAGT-3′ to generate distinct products of similar size. PCR products were resolved on a 1.8% agarose gel and were quantified with a Marathon Central densitometer and NIH Image 1.61 software. The quantitative PCR was repeated twice with different batches of cDNA. As indicated in Results, some cultures were supplemented with 2.5 mM CuSO4 and grown an additional 10 to 12 min at room temperature with shaking at 250 rpm prior to harvesting of the cells.
RESULTS
Identification of mitochondrial mutators.
To hasten the discovery of nuclear genes that affect the integrity of the mitochondrial genome, a new genetic assay for measuring mitochondrial mutation rates in S. cerevisiae was developed previously (72). The nuclear ARG8 gene was removed from the nuclear DNA, recoded for mitochondrial translation, and inserted into the mitochondrial genome. The mitochondrial location of the ARG8m gene supports growth of the strain TF235 on media lacking arginine (69). The mitochondrial mutation rate assay exploits a spontaneous mutation in the TF235 ARG8m gene that renders the resulting strain, TF236 arg8m, auxotrophic for arginine (69). Sequence analysis of the arg8m gene in strains TF236 and TF236 pos5 revealed three changes from the original ARG8m gene (69) in which reversion (i.e., a −1 frameshift) of the mitochondrial mutation at position 1592 results in an ARG+ phenotype (72). Revertants of arg8m can be selected by growth on media lacking arginine. Other ARG8m constructs have recently been used to measure the stability of repetitive DNA sequences in mtDNA in vivo (65).
Over 80 nuclear gene disruptions were screened in the arg8m reversion assay, and rates of reversion were scored on minimal plates lacking arginine. This approach identified a number of unknown genes as mitochondrial mutators, as well as several known genes not previously associated with mitochondrial mutagenesis (Table 1). For example, disruption of the YBL018c gene, which encodes the mitochondrial MRP (mitochondrial RNA processing) RNase that processes heavy-strand mitochondrial transcripts for use as a primer in DNA replication, increased the mitochondrial mutation rate by a factor of 5. Hap2 (for “heme activation protein 2”) is an oxygen-responsive transcription factor known to modulate expression of several cytochrome subunits, and we note that the gene for the yeast mitochondrial single-stranded DNA binding protein (RIM1) contains a Hap2 binding site. Whether the sixfold mitochondrial mutator effect due to Hap2 disruption is mediated through altered expression of RIM1 is not known. The YPL188w was originally identified as POS5 by Krems et al. in a screen for sensitivity to hydrogen peroxide (32). The pronounced mutator effect in the arg8m reversion assay, together with the close connection between mitochondria and the cellular response to oxidative stress, prompted us to select the POS5 gene for further study (below). One important gene that scored as a mitochondrial mutator was the Can1 gene, which encodes arginine permease. The inability of can1 mutants to import arginine gives a huge selective advantage to arg8m revertants, even when growing on rich medium, thus giving the false appearance of an elevated mitochondrial mutation rate. Comparisons of growth rate on medium limiting for arginine easily identify Can1 and other genes affecting arginine import, and such tests verify that pos5 yeasts are not defective in uptake of arginine (data not shown).
TABLE 1.
Mitochondrial mutator genes identified in the arg8m reversion assay
| Gene | Relative arg8m reversion ratea | Phenotype and/or functionb |
|---|---|---|
| YJR046w | 17 | Molecular function unknown, DNA replication licensing |
| YNL242w | 42 | Molecular function unknown, autophagy, peroxisome degradation |
| YOR279c | 34 | Peripheral membrane protein required for sporulation, molecular function unknown |
| YBL018c | 5 | Mitochondrial ribonuclease MRP |
| YPL188w (pos5) | 50 | Peroxide sensitive (this study) |
| YDL186w | 6 | Molecular and biological function unknown |
| Ngr1 | 6 | Negative growth regulatory protein |
| YDR295c | 11 | Molecular and biological function unknown |
| Hap2 | 6 | Heme-regulated transcription factor |
| YJL076w | 105 | Net1, Sir2-associated nucleolar protein required for rDNA silencing |
| Ecm10 | 509 | hsp70 homolog in the mitochondrial matrix |
| Can1 | 13,762 | Canavanine resistance, arginine permease |
Relative to the reversion rate of TF236.
During the course of this work, putative functions were assigned to several mitochondrial mutator genes by the Saccharomyces Genome Database (http://genome-www.stanford.edu/Saccharomyces/).
The pos5 mutation impairs mitochondrial function and increases mitochondrial but not nuclear mutations.
The wild-type TF236 strain had a mutation rate of 0.02 × 10−6 in the arg8m reversion assay, whereas the rate for TF236pos5 was 1.0 × 10−6. Thus, disruption of POS5 increased the average mitochondrial mutation rate by a factor of 50 (Table 2). The magnitude of this effect becomes apparent when it is compared to the effect of a relevant control strain. Elimination of the 3′-5′ exonuclease activity of the mtDNA polymerase decreases the fidelity of mtDNA replication by preventing excision, or proofreading, of DNA replication errors. Strain TF236 carrying a plasmid that overexpresses the exonuclease-deficient mtDNA polymerase (pFL39::mip1-exo−) had an arg8m reversion rate of 0.12 × 10−6, which is sixfold above the background (Table 2). Given such a large effect on the mitochondrial mutation rate, pos5 mutants were also expected to display a significant increase in the frequency of formation of petite colonies. TF236 is respiration deficient because construction of the mutational target disrupted the mitochondrial COX3 gene (cox3::arg8m). Accordingly, experiments to determine the frequency of formation of petite colonies utilized the strain YPH925. After growth on glycerol to eliminate preexisting petites, the petite frequency was 4.9% in the wild type and 27% in the pos5 mutant, indicating a sixfold increase upon disruption of POS5 (Table 2). The mip1-exo− control exhibits a fourfold-elevated rate of petite formation, and the double mutant has a 15-fold elevation in the mutation rate. This synergistic effect indicates that Mip1 and Pos5 act in different pathways, both of which may affect mitochondrial mutagenesis. The YPH925 strain contains two nuclear mutations, lys2-801 and ade2-101, permitting nuclear mutation rates to be estimated by measuring reversion to Lys+ and Ade+ prototrophy. When cells were grown on YPD, the POS5 disruption reduced the nuclear mutation rate roughly twofold (Table 2); however, this difference was not significant (P = 0.11). The differences between the wild type and the mutant grown on YPG were also insignificant, demonstrating that the POS5 disruption does not increase the mutation rate at these two nuclear alleles. Since reversion of arg8m requires a −1 frameshift event, the rate of reverting a nuclear frameshift mutation was also measured in wild-type and pos5 strains containing nuclear homomononucleotide runs of 5 and 14 in the lys2 gene (75). With the lys2-A5 (predominantly measuring a +1 addition) and lys2-A14 (predominantly measuring a −1 deletion) markers, no difference was observed between the wild type and the pos5 mutant. Thus, disruption of POS5 specifically enhances mutation of mtDNA without affecting the mutation of nuclear DNA.
TABLE 2.
Mutation frequencies of the wild type and pos5 mutants
| Strain genotype | arg8m reversion ratea | Petite frequency (%)a,b | Nuclear mutation rate
|
|
|---|---|---|---|---|
| YPD | YPG | |||
| Wild type | 0.02 × 10−6 (1) | 4.9 (1) | 0.6 × 10−8 | 5.4 × 10−8 |
| pos5 | 1.00 × 10−6 (50) | 27 (6) | 0.25 × 10−8 | 6.4 × 10−8 |
| pFL39:mip1-exo− | 0.12 × 10−6 (6) | 20 (4) | NDc | ND |
| pos5/pFL39:mip1-exo− | ND | 72 (15) | ND | ND |
Reversion rate per cell per generation. Values in parentheses are normalized to the wild-type value.
Petite frequency is the combined frequencies of white versus total (red and white) colonies from three independent experiments done with the YPH925 strain with the pFL39 plasmid present.
ND, not done.
Interestingly, even though the pos5 mutation had no significant effect, nuclear mutation rates were affected dramatically when cells were grown on a nonfermentable carbon source (Table 2). Growth on YPG medium resulted in a 9-fold increase for the wild-type strain (P < 0.0001) and a 25-fold increase for the mutant (P < 0.0001). Since growth on YPG requires cells to perform electron transport and oxidative phosphorylation to generate ATP, we speculate that a high level of mitochondrial activity is linked to the increased nuclear mutation rates. Mitochondrial mutation rates may also be elevated on YPG, but mitochondrial mutation rates could not be measured in glycerol media because the mitochondrial COX3 gene was disrupted by insertion of arg8m.
Yeast cells with mutations in ade2 accumulate a red pigment due to disruption of the adenine pathway. Yeasts without functioning mitochondria do not get to this step in the adenine synthesis pathway and thus are white. YPH925 cells with functional mitochondria have red grande colonies due to their ade2-101 mutation, whereas respiration-deficient YPH925 colonies are easily scored as white petite colonies. On solid media there is an obvious visual increase in the number of petite colonies of YPH925 pos5 (Fig. 1). The petite colonies in the pos5 strain were smaller than either spontaneous petites or petites induced by mip1-exo−, suggesting a specific metabolic defect that is exacerbated by the acquired respiratory dysfunction. The irregular borders and distinct morphology of pos5 colonies also suggest a general growth defect (Fig. 1, insets). To better define the function of POS5 as it relates to respiration, growth rates of YPH925 and YPH925 pos5 in YPD, low-sugar YPD, and YPG media were monitored (Fig. 2). Growth in YPD medium did not reveal significant differences between the two strains. However, growth of YPH925 pos5 was substantially inhibited in low-sugar YPD and YPG media. Since growth on glycerol requires functional electron transport and oxidative phosphorylation, POS5 may function directly to support respiration. On the other hand, because the pos5 phenotype is most pronounced when the cell is deprived of glucose, POS5 may function in metabolism of fermentable carbon sources.
FIG. 1.

Appearance on solid media in a red-white colony color assay of wild-type and mip1-exo− and pos5 mutant strains. Growth of the wild-type (POS5) and mutant (pos5) strains on YPD plates was assessed. YPH925 contains the ade2-101 mutation, which gives red colonies on glucose-based media, in respiration-competent ([rho]+) cells. Petite cells in this strain are phenotypically white. (A) Wild-type YPH925 (MIP1) carrying an empty pFL39 vector, showing normal levels of petite formation. (B) YPH925 containing the wild-type chromosomal MIP1 gene plus a mip1-exo− copy (as pFL39::mip1-exo−), showing an increased severity of petite sectors within grande colonies (inset). These growth-impaired sectors are rapidly overgrown by the nonpetite cells, giving rise to the highly irregular shape of the borders of the colonies. (C) YPH925 pos5 pFL39 mutant, showing an increased frequency of petite formation as well as an increased severity of petite sectors within grande colonies (inset). (D) YPH925 (MIP1) pos5 pFL39::mip1-exo− double mutant, demonstrating significantly increased petite frequency and increased growth defect.
FIG.2.

Effect of pos5 mutation on growth rates in YPG or YPD. Growth rates of wild-type YPH925 (closed squares) and YPH925 pos5 (open circles) strains in YPD (A), low-sugar YPD (B), or YPG (C) were measured spectroscopically at OD600. Cells were grown as described in Materials and Methods. Data points are averages of four independent experiments.
Mutation of POS5 stimulates mitochondrial biogenesis.
The elevated frequency of petite formation and the slow growth on glycerol could both be caused by the loss of mtDNA. To test this idea, we measured the amount of mtDNA relative to a nuclear marker by competitive PCR, as described in Materials and Methods. Total cellular DNA was isolated from YPH925 and YPH925 pos5 cells grown in YPD. Although the absolute numbers produced by this method cannot be compared from one experiment to the next, the relative values are very precise. The mitochondrial/nuclear DNA ratio was 81 in YPH925, whereas the mitochondrial/nuclear DNA ratio was 264 for YPH925 pos5. Thus, the pos5 mutant maintains 3.3-fold more mtDNA than the wild type, clearly indicating that the pos5 phenotype does not result from a loss of mtDNA. However, the elevated mitochondrial mutation rate may compromise mitochondrial function to the point that the cell synthesizes more mtDNA to attempt to compensate for the reduced dose of functional mitochondrial genes.
Cytochrome c oxidase is an enzymatic marker of mitochondrial function. Whole-cell extracts were prepared from YPH925 and YPH925 pos5 cultures, and cytochrome c oxidase activity of the extracts was measured in duplicate as described in Materials and Methods. Wild-type YPH925 extracts (0.1 mg of protein) oxidized cytochrome c at 7.9 nmol/h, and YPH925 pos5 extracts oxidized cytochrome c at 18.9 nmol/h. Thus, mutant cells possess 2.4-fold more cytochrome c oxidase than wild-type cells. When the concomitant increase in mtDNA is taken into consideration, the cytochrome c oxidase activity per mitochondrial genome is not significantly different from that of the wild type. This proportional increase in mtDNA content and cytochrome c oxidase activity suggests that pos5 cells make more mitochondria than wild-type cells. We believe that enhanced biogenesis of mitochondria may be an indirect phenotype of the POS5 disruption, caused by the cell's attempting to compensate for loss of mitochondrial function.
The POS5 gene belongs to a highly conserved NAD kinase family.
A BLAST search revealed that the predicted amino acid sequence of the POS5 gene has strong homology to a number of known NADH kinase genes from a variety of different species. The NAD kinase genes UTR1 from S. cerevisiae, FLJ13052 from Homo sapiens, and the genes from E. coli, Salmonella enterica serovar Typhimurium, and Micrococcus flavus have been expressed and demonstrated by biochemical assay to have ATP-dependent NAD kinase activity (5, 27-29, 38).
Cloning, overexpression, and purification of Pos5.
We undertook cloning and overexpression of the POS5 gene to confirm the biochemical activity of this putative NADH kinase. We cloned the coding sequence of POS5 from chromosomal DNA by PCR, as described in Materials and Methods. The full-length POS5 coding sequence was transferred into the pQE30 expression vector and overexpressed in E. coli as a His6-affinity-tagged protein. Expression of the full-length protein was very high, but the vast majority of the protein was insoluble (data not shown). Since the phenotype of the pos5 mutant indicated a mitochondrial function, we suspected that the Pos5 protein might have been targeted for import into mitochondria. Subcellular localization by immunofluorescence of an overexpressed POS5-V5 C-terminal fusion protein indicates that the POS5 gene product accumulates in mitochondria (http://ygac.med.yale.edu/). Intracellular trafficking is controlled by the sequence of amino acids at the N terminus of nascent proteins. Analysis of the amino-terminal sequence of the full-length Pos5 protein by the target peptide prediction programs TargetP, version 1.0 (11), and MitoProtII (7) predicted cleavage sites between amino acid residues 17 and 18. A POS5 gene construct lacking the coding sequence for the first 17 amino acid residues, designated POS5Δ17, was overexpressed in E. coli XL1-Blue as described in Materials and Methods. Overall expression of POS5Δ17 was comparable to expression of POS5, and the solubility of the Pos5Δ17 protein was significantly higher than that of full-length Pos5 protein (Fig. 3). Recombinant Pos5Δ17 protein was purified to >95% homogeneity from soluble lysates by affinity chromatography over Ni-NTA Sepharose (Fig. 3). Approximately 12 mg of soluble Pos5Δ17 protein could be obtained from 1 liter of induced XL1-Blue.
FIG. 3.
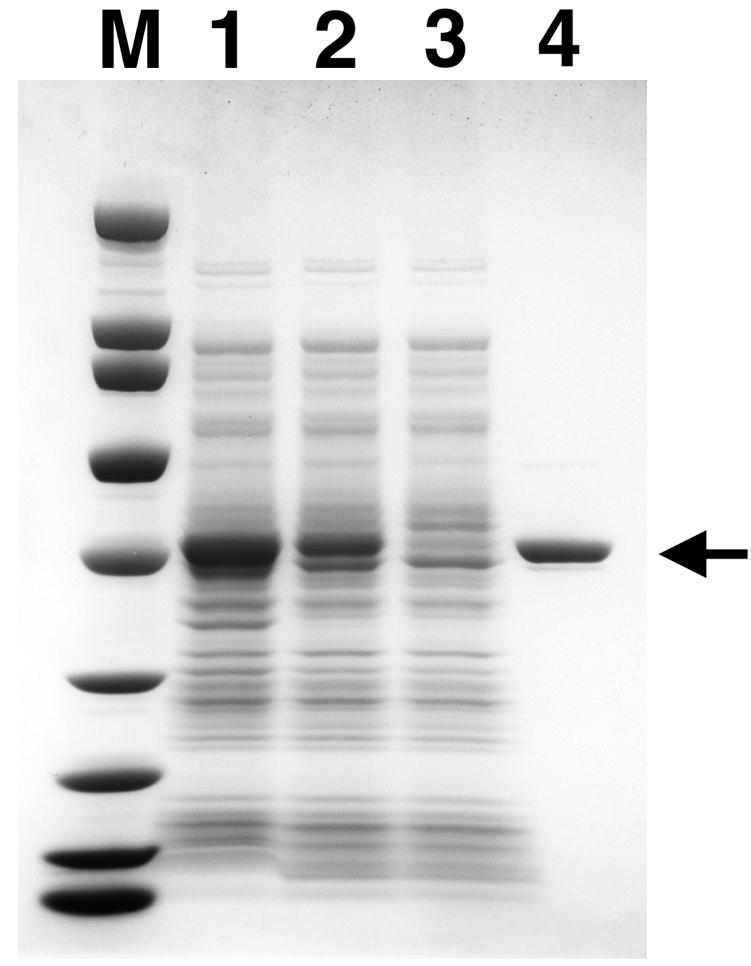
Purification of recombinant Pos5Δ17. Recombinant Pos5Δ17 protein was expressed in E. coli and purified as described in Materials and Methods. Samples throughout purification were resolved by electrophoresis on a 4-to-20% polyacrylamide gradient gel (Zaxis) in the presence of 0.1% SDS and stained with Coomassie brilliant blue. Lane 1, whole-cell extract (2 μl); lane 2, soluble lysate (2 μl); lane 3, unbound Ni-NTA Sepharose fraction (2 μl); lane 4, purified Pos5Δ17 protein (2.3 μg). Broad-range molecular mass markers (Bio-Rad) were 200, 116, 97, 66, 45, 31, 21.5, 14.4, and 6.5 kDa (lane M). The arrow indicates the position of recombinant Pos5Δ17 protein.
Pos5 protein has NADH kinase activity.
Existing biochemical assays of NAD+ kinase activity rely upon the enzymatic reduction of freshly formed NADP+ to NADPH by glucose-6-phosphate dehydrogenase. For reasons of sensitivity and simplicity, we developed a new assay to measure the direct phosphorylation of NAD+ to NADP+. Because this assay is independent of the oxidation state of the substrates, phosphorylation of NADH to NADPH can also be monitored. The transfer of the γ-phosphate group from [γ-32P]ATP to either NAD+ or NADH generates [32P]NADP+ or [32P]NADPH. We selected a solvent system capable of resolving the three radiolabeled reactants and products by thin-layer chromatography, as described in Materials and Methods. Steady-state kinetic analysis of enzymes that utilize two diffusible substrates requires one substrate to be maintained at a saturating concentration while the other is varied. Accordingly, a biochemical assay of purified Pos5Δ17 was performed at several fixed concentrations of ATP with variable concentrations of NAD+ or NADH. This assay revealed that the enzyme possesses bona fide NADH kinase activity and that the enzyme is capable of phosphorylating either NAD+ or NADH in vitro (Fig. 4). Higher concentrations of ATP inhibited the reaction due to titration of Mg2+ ions (data not shown), which confounds a rigorous kinetics analysis. Nevertheless, a comparison of the rates of generating NADP+ or NADPH at 0.25, 1.0, and 5 mM ATP indicates as much as a twofold preference for NADH over NAD+ throughout the full range of ATP concentrations tested (Fig. 5). Preliminary kinetics analysis suggests a sequential reaction mechanism, although confirmation of this would require a comprehensive study of product inhibition. Previously, two forms of NAD+ kinase were detected in S. cerevisiae: a NADH-specific form was localized exclusively in mitochondria, and a NAD+-specific form was distributed in microsomal and cytosolic fractions (24). Our observation of the same substrate specificity for the mitochondrial NADH kinase Pos5 implies that we have cloned and characterized the mitochondrial form of the enzyme originally described and partially purified by Iwahashi et al. (24).
FIG. 4.

Direct in vitro assay for NADH kinase activity. NADH kinase reactions were performed as described in Materials and Methods. Reaction mixtures contained 1 mM ATP, 4 μg of Pos5Δ17 and 0, 0.05, 0.1, 0.2, 0.5, 1.0, or 2.0 mM NAD+ (lanes 2 to 8, respectively), or 0, 0.05, 0.1, 0.2, 0.5, 1.0, or 2.0 mM NADH (lanes 9 to 15, respectively). The mixture in lane 1 did not contain enzyme. The origin of the thin-layer chromatogram and the relative mobilities of ATP, NADPH, and NADP+ are indicated with arrows.
FIG. 5.
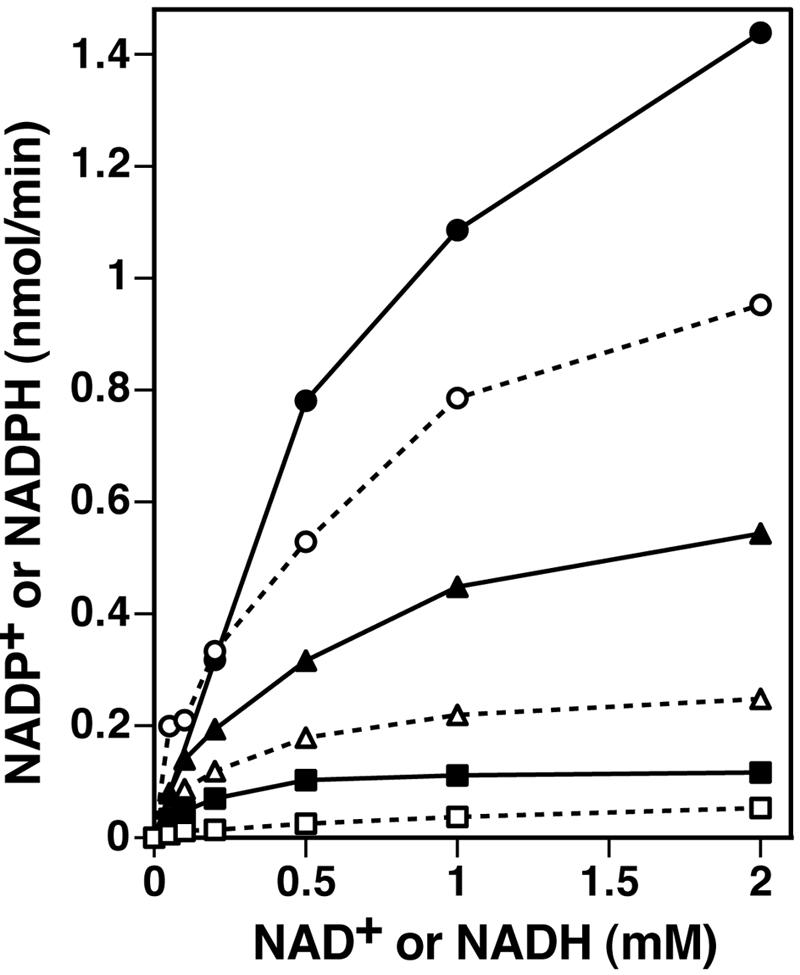
The Pos5 NAD kinase prefers NADH over NAD+. NADH kinase reactions included the indicated concentrations of NADH or NAD+, while ATP concentrations were maintained at 0.25 mM (squares), 1.0 mM (triangles), or 5.0 mM (circles). Production of NADPH (filled symbols) or NADP+ (open symbols) was determined as described in Materials and Methods. Reaction mixtures contained 4 μg of Pos5Δ17.
POS5 participates in detoxification of ROS.
Analysis of the POS5 promoter sequence predicts that gene expression may be regulated by the transcription factors Yap1, Skn7, and Ace1. Yap1 and Skn7 regulate the expression of at least 32 genes, many of which are involved in the cellular response to oxidative stress (37), and Yap1 can be activated by oxidation of reactive protein thiols with hydrogen peroxide (15). Ace1 is a copper-responsive positive transcription factor that regulates expression of Cu,Zn-superoxide dismutase in addition to Cup1 and Crs5, two copper metallothioneins involved in the cellular response to oxidative stress (18, 23, 71, 74). The presence of binding sites for these three transcription factors strongly suggests that POS5 functions to counter both metal ion toxicity and oxidative stress. Oxidative stress is a natural consequence of living in an aerobic environment. Molecular oxygen liberates electrons from the electron transport chain to produce highly reactive superoxide anions. Most superoxide is converted to hydrogen peroxide by superoxide dismutase, but free superoxide can destroy iron-sulfur centers in proteins, causing the release of Fe(II) (39). Through the Fenton reaction, transition metals such as Fe(II) and Cu(I) can reduce hydrogen peroxide to generate highly reactive hydroxyl radicals that damage cellular components (39).
The sensitivity of POS5 mutants to exogenous hydrogen peroxide (32) suggests that Pos5 participates in detoxification of ROS. Accordingly, pos5 strains should suffer more oxidative damage to their macromolecules than wild-type strains. Oxidation of lysine, arginine, proline, and threonine side chains in proteins produces carbonyl groups. Since carbonyl groups do not normally exist in polypeptides, their presence is an indication of oxidative damage. Mitochondrial lysates were prepared from YPH925 and YPH925 pos5 yeast strains, and equal quantities of mitochondrial protein from each lysate were resolved by SDS-polyacrylamide gel electrophoresis. Staining of protein with Coomassie blue indicated a nearly even distribution of mitochondrial proteins over the 20- to 200-kDa range (data not shown). Protein oxidation was estimated by immunoblot analysis with antibodies specific to oxidized side chains (Fig. 6). With the exception of a particularly strong signal for a ∼100-kDa protein, the YPH925 mitochondrial lysate displayed relatively minor protein oxidation (Fig. 6, lane 1). By comparison, the YPH925 pos5 lysate exhibited 28-fold-higher protein oxidation, and proteins of all sizes were damaged (Fig. 6, lane 2). The enhanced oxidative damage to mitochondrial proteins in the pos5 strain supports the hypothesis that pos5 mutants are sensitive to exogenous peroxides because Pos5 participates in the detoxification of ROS.
FIG. 6.
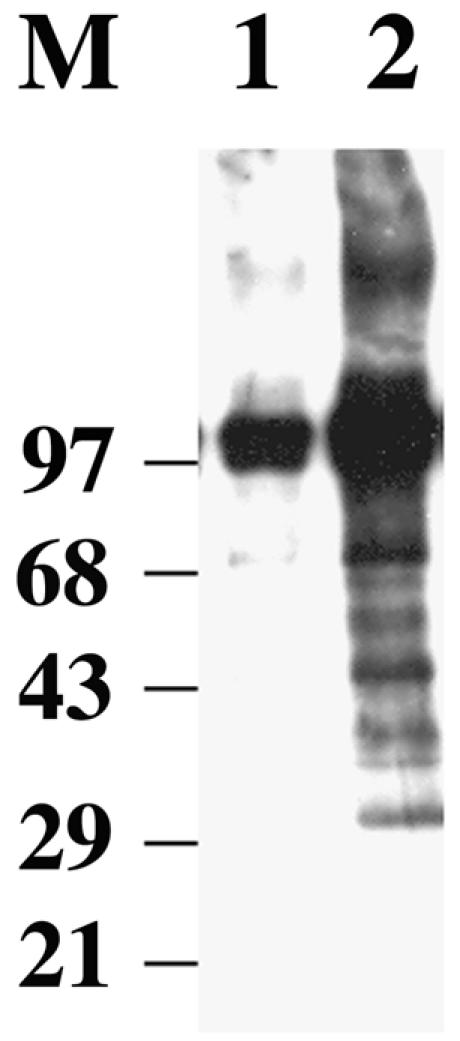
Disruption of POS5 enhances oxidation of mitochondrial proteins. Preparation of yeast mitochondrial lysates and detection of oxidized protein residues were as described in Materials and Methods. Oxidation of mitochondrial proteins (50 μg) derived from wild-type YPH925 and YPH925 pos5 lysates is displayed in lanes 1 and 2, respectively. The positions and molecular masses (in kilodaltons) of standard proteins are indicated.
Exogenous metal ions are toxic because they accentuate oxidative stress and shift the redox balance of the cell. Elimination of exogenous metal ions requires a reductant, such as reduced glutathione (16), and treatment of S. cerevisiae with 0.5 mM CuSO4 significantly depletes cellular stores of reduced glutathione (13). Limitation of reduced glutathione (or reduced thioredoxin) depresses the enzymatic reduction of hydrogen peroxide by glutathione peroxidases (or thioredoxin peroxidases). Together with a fresh supply of reduced transition metal ions, this persistence of hydrogen peroxide increases production of hydroxyl radicals through the Fenton reaction. Thus, metal ions simultaneously impair detoxification and enhance production of ROS. The ability of exogenous copper to stimulate the formation of free radicals in vivo has been demonstrated experimentally (25), and free radical scavengers have been shown to reduce copper-induced neuronal cell death (63). Additionally, disruption of the yeast thioredoxin peroxidase gene Ahp1 induced extreme sensitivity to organic peroxides, a variety of metal ions, and the glutathione-depleting agent diethyl maleate (47). To further define the role of POS5 in the oxidative stress response, we tested the effects of exogenous copper ions on the growth of wild-type and pos5 cultures. YPH925 and YPH925 pos5 were grown in YPD supplemented with 25 μM CuSO4, and growth was monitored by OD600. The addition of copper ions had no effect on growth of YPH925, but growth of YPH925 pos5 was completely inhibited by only 25 μM CuSO4 (Fig. 7). This pronounced copper toxicity, combined with the peroxide sensitivity, mitochondrial mutagenesis, and enhanced oxidative damage to mitochondrial proteins, firmly establishes a role for POS5 in the mitochondrial response to oxidative stress.
FIG. 7.

Exogenous copper inhibits growth of YPH925 pos5. Growth rates of wild-type YPH925 (black squares) and YPH925 pos5 (open circles) cultures were measured spectroscopically at OD600. Cells were grown in YPD supplemented with 25 μM CuSO4, as described in Materials and Methods. Data points are averages of two independent experiments.
The transcription factor Yap1 regulates expression of many oxidative response genes, possibly including POS5. Deletion of YAP1 causes hypersensitivity both to hydrogen peroxide and to cadmium ions (15). Since disruption of POS5 also induces peroxide sensitivity and metal toxicity, it is possible that reduced expression of POS5 is sufficient to explain the phenotype of the YAP1 deletion strain. The ability of copper ions to accentuate oxidative stress suggests that the cell may respond to exogenous copper by up-regulating expression of oxidative response genes. The presence of two Ace1 binding sites in the promoter region of the POS5 gene prompted us to measure the effects of copper on the steady-state level of wild-type POS5 transcripts. Log-phase cultures of YPH925 were supplemented with 2.5 mM CuSO4 and grown for an additional 10 to 12 min. POS5 transcripts were measured by competitive PCR, as described in Materials and Methods. In the first experiment, POS5 transcripts increased from 460 U (arbitrary units) without Cu2+ to 1,390 U with 2.5 mM Cu2+. In the second experiment, the values were 348 U without Cu2+ and 1,300 U with Cu2+. Averaging these results indicates a 3.4-fold increase in the abundance of POS5 transcripts upon exposure to copper ions, which is consistent with the critical role of POS5 in oxidative stress and metal ion toxicity.
DISCUSSION
The vast majority of oxygen consumed by eukaryotic cells is utilized for oxidative phosphorylation within the mitochondria. Although cells balance the stream of reducing equivalents and oxygen to avoid the injurious effects of oxygen deficiency and oxygen surplus, as much as 1 to 5% of the oxygen consumed by the mitochondria can be released as superoxide, hydrogen peroxide, or hydroxyl radicals (76, 82). Consequently, mitochondria are both the main source of ROS and a principal target for oxidative damage, and these organelles are expected to play a central role in the cellular response to oxidative stress. Indeed, several enzymes involved in the detoxification of ROS have been localized to the mitochondria, including glutathione peroxidase, thioredoxin peroxidase (12, 44, 53, 54), and manganese superoxide dismutase (14). The POS5 gene was originally identified as a hydrogen peroxide resistance allele (32), and a subcellular localization study by the Yale Genome Analysis Center has determined that Pos5-V5 C-terminal fusion proteins accumulate in mitochondria. Thus, a mitochondrial function for the POS5 gene product was expected, and our identification of the Pos5 protein as a mitochondrial NADH kinase helps to reveal the central role of NADPH in the mitochondrial response to oxidative stress. Nieminen and coworkers observed that application of exogenous t-butyl hydroperoxide to cultured hepatocytes induced the rapid oxidative depletion of mitochondrial NAD(P)H, followed by generation of mitochondrial ROS, the mitochondrial permeability transition, and death of the cells (48). More recently, photoactivation of rhodamine dyes was utilized to generate in situ singlet oxygen within the mitochondrial matrix of cultured hepatocytes. Depletion of NAD(P)H was dependent on the dose of this endogenous ROS, and higher doses induced the mitochondrial permeability transition and apoptotic cell death (55). Both studies strongly suggest that the reducing equivalents of NAD(P)H function as an endogenous antioxidant to protect mitochondria from the injurious effects of ROS. In addition to this direct chemical reduction of ROS, NADPH is also an essential cofactor for the enzymatic detoxification of ROS. For example, one molecule of NADPH is bound tightly to each subunit of the catalase tetramer (30). Through an interaction with heme in each active site, NADPH exerts a protective effect to maintain catalase in an active state (31). The ability of glutathione peroxidases and thioredoxin peroxidases to eliminate hydrogen peroxide depends on a continuous supply of reduced glutathione and thioredoxin. The mitochondrial enzymes that replenish these cofactors, glutathione reductase and thioredoxin reductase, both exclusively utilize NADPH as the reductant (22, 73). As the control point for the conversion of NADH to NADPH, POS5 function supports these enzymatic systems of ROS detoxification by preventing depletion of the mitochondrial NADP+/NADPH pool.
The predicted surplus of ROS in cells with compromised POS5 function is expected to enhance oxidative damage of macromolecules. Proteins in mitochondrial lysates derived from a POS5 disruption strain exhibited substantially higher levels of protein oxidation than did proteins from wild-type mitochondrial lysates (Fig. 6). This accumulation of oxidized proteins occurred without the application of exogenous agents to induce oxidative stress, supporting our hypothesis that pos5 strains suffer elevated concentrations of endogenous ROS. In addition to attacking proteins and lipids, ROS are a significant source of chemical damage to DNA. ROS oxidize both the sugar and base components of DNA, causing a variety of adducts as well as abasic sites and single- and double-stranded breaks (1, 9). Such premutagenic oxidative lesions to DNA, if left unrepaired, have been shown to cause point mutations and frameshift mutations (20).
Our observation of enhanced mutagenesis in mtDNA also is consistent with our theory of elevated endogenous ROS in the pos5 strain. However, the specific mutagenesis of mtDNA in the pos5 strain, compared to mutagenesis of nuclear markers, requires explanation. S. cerevisiae possesses two additional genes homologous to POS5: UTR1 and Yel041w. Pos5 protein is clearly targeted to mitochondria, whereas UTR1 encodes a functional cytosolic NADH kinase (29). Yel041w does not contain a mitochondrial targeting sequence and has no known function, even though UTR1 and Yel041w likely arose from an 8-kb chromosomal duplication (43). Strict subcellular compartmentalization of NADH kinase function would predict the mitochondrial phenotype. Alternatively, mtDNA has been shown to be more susceptible to oxidative damage than nuclear DNA (2, 58, 61, 62, 66, 83), perhaps due to the closer proximity of mtDNA to the endogenous source of ROS. This susceptibility would likely be exacerbated in a pos5 background. The selective vulnerability of mtDNA to chemical damage has also been explained by the absence of protective histones in mitochondria. Mammalian and yeast systems for the repair of oxidative damage to mtDNA have been described (8, 35, 36, 51, 56), but the possibility that these mtDNA repair systems are saturated in a pos5 background has not been tested. It has been demonstrated that the catalytic subunit of human DNA Pol γ is sensitive to oxidation by exogenous H2O2 both in vivo and in vitro (19). Although oxidation inhibits the overall activity of the polymerase, the effects on fidelity of replication were not tested, and extension of this thinking to the yeast enzyme is risky. Wallace has proposed that oxidative damage to mtDNA could lead to mutation of the mitochondrially encoded genes needed for electron transport and that dysfunctional electron transport would aggravate the production of mitochondrial ROS (80). Although the specific mutagenesis of mtDNA relative to nuclear DNA in pos5 strains is likely caused by strict functional compartmentalization of this NADH kinase activity, we cannot discount the possible contributions of enhanced DNA damage, reduced repair capacity, compromised fidelity of DNA replication, or complex features of mitotic segregation to the specificity.
In addition to their role in energy production, mitochondria are also the site of many essential reactions in the intermediary metabolism of eukaryotic cells, including steps in the biosynthesis and catabolism of pyrimidines and amino acids, β- and ω-oxidation of fatty acids, and a number of steps in the tricarboxylic acid and urea cycles. Oxidation-reduction reactions in mitochondria that exclusively utilize NADPH as the electron donor could be compromised in pos5 strains, and several such biochemical pathways can be linked indirectly to mitochondrial mutagenesis. As mentioned before, mitochondrial thioredoxin and glutathione are maintained in a reduced state by NADPH-dependent reductases (22, 73). Cytosolic ribonucleotide reductase exclusively utilizes the NADPH-dependent thioredoxin-thioredoxin reductase system as the electron source for the reduction of ribonucleotides to deoxyribonucleotides. Young and coworkers have partially purified a mitochondrion-specific form of ribonucleotide reductase (84), suggesting that a metabolically distinct deoxynucleoside triphosphate (dNTP) pool exists within mitochondria. Although the finer details remain unknown, such a system in pos5 yeasts could exhibit depleted mitochondrial dNTP pools. Similarly, a defect in thymidine phosphorylase, the enzyme that catalyzes the first committed step in the pathway for degradation of thymidine, causes the human disease mitochondrial neurogastrointestinal encephalomyopathy and is associated with deletions in mtDNA. The working hypothesis for this pathogenesis invokes an imbalance in mitochondrial dNTP pools (49). The second reaction in this catabolic pathway, the reduction of thymine to dihydrothymine, is catalyzed by the NADPH-dependent enzyme dihydropyrimidine dehydrogenase. The predicted dysfunction of this pathway in pos5 strains may also lead to mutagenic imbalances in mitochondrial dNTP pools. Similarly, dihydrofolate reductase requires NADPH to replenish tetrahydrofolate. Restriction of methylene-tetrahydrofolate inhibits thymidylate synthase and may lead to unbalanced pools of pyrimidine nucleotides. The pentose phosphate pathway is the source of the sugar components needed to synthesize RNA, DNA, and nucleotide coenzymes, and it is also a principle mechanism for regenerating NADPH from NADP+. Perturbation of cellular NADP+/NADPH concentrations in pos5 yeast could compromise metabolism that depends on the pentose phosphate pathway, including biosynthesis of the nucleotide precursors needed for synthesis of mitochondrial and nuclear DNA. NADP+/NADPH-dependent mitochondrial pathways other than nucleotide metabolism could also affect mitochondrial mutation rates. For example, exogenous mutagens can be detoxified by the mitochondrial monooxygenase cytochrome P450 (46). Each catalytic cycle requires the contribution of one electron from the NADPH-dependent flavoprotein cytochrome P450 reductase, suggesting that a lack of mitochondrial NADPH could interfere with detoxification of mutagens. Other important oxygenases are metalloenzymes that cycle between reduced and oxidized states. Reducing equivalents needed to regenerate the active, reduced enzymes are ultimately derived from mitochondrial NADPH by way of thioredoxin and ascorbate redox pairs.
Recent research predicts that nitric oxide probably plays an important role in mitochondrial mutagenesis by modulating production of ROS and reactive nitrogen species in mitochondria (17). A mitochondrial form of NADPH-dependent nitric oxide synthase was described recently (10). Since nitric oxide stimulates the biogenesis of new mitochondria (50), the consequences of mitochondrial NADH kinase deficiency for these roles for nitric oxide are unknown.
The central role of mitochondrial NADPH in metabolism predicts pleiotropic phenotypic effects for NADH kinase-deficient cells. We have demonstrated an elevated mitochondrial mutation rate and impaired mitochondrial function, as shown by both the increase in petite frequency and the slow-growth phenotypes on reduced glucose or glycerol. Although there are a number of plausible mechanisms by which a deficiency in mitochondrial NADH kinase increases the mitochondrial mutation rate, the most direct model is impaired detoxification of mitochondrial ROS. The enhanced sensitivity to exogenous copper and hydrogen peroxide and a higher level of oxidative damage to mitochondrial proteins in pos5 yeasts support this model. S. cerevisiae contains three NADH kinases by genetic analysis, two of which have been shown biochemically to encode NADH kinases. However, all larger eukaryotes have only one NADH kinase gene. The results described in this paper predict an elevated frequency of mitochondrial mutations and mitochondrial diseases in higher eukaryotes with a NADH kinase deficiency.
ADDENDUM
After this paper was submitted, Outten and Culotta published a paper revealing that POS5 encodes a mitochondrial NADH kinase that functions in iron homeostasis and protects against hyperoxia (52). Because these investigators did not remove the mitochondrial targeting presequence, POS5 was overexpressed in E. coli, which produced the insoluble pre-Pos5 protein. The enzymatic activity of the renatured pre-Pos5p was rather low (specific activity ∼ 6 nmol of NADPH/min/mg of pre-Pos5p), and the authors describe an apparent 50-fold preference for NADH over NAD+ in a two-stage cycling assay linked to the function of glucose-6-phosphate dehydrogenase (52). In contrast, our direct-phosphorylation assay (Fig. 5) clearly shows that both NAD+ and NADH are phosphorylated efficiently by mature Pos5p (specific activity > 300 nmol/min/mg of Pos5p). Nevertheless, several complementary observations are made in the two studies. The dramatically reduced activities of the mitochondrial Fe-S proteins aconitase and succinate dehydrogenase in pos5Δ yeast (52) are consistent with the mutagenesis of mtDNA (Table 2) and oxidative damage to mitochondrial proteins (Fig. 6) demonstrated in the present work. Together with the known sensitivity to hydrogen peroxide (32) and hyperoxia (52), these collected observations strongly support a model in which POS5 supports detoxification of ROS within mitochondria by maintaining the mitochondrial supply of NADP+ and NADPH.
Acknowledgments
We thank Tom Fox, Dmitry Gordenin, and the Yale Genome Analysis Center for strains and plasmids.
G.R.S. was partially supported by a grant from the U.S. Army.
REFERENCES
- 1.Aust, A. E., and J. F. Eveleigh. 1999. Mechanisms of DNA oxidation. Proc. Soc. Exp. Biol. Med. 222:246-252. [DOI] [PubMed] [Google Scholar]
- 2.Ballinger, S. W., B. Van Houten, G. F. Jin, C. A. Conklin, and B. F. Godley. 1999. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res. 68:765-772. [DOI] [PubMed] [Google Scholar]
- 3.Bonneaud, N., O. Ozier-Kalogeropoulos, G. Y. Li, M. Labouesse, L. Minvielle-Sebastia, and F. Lacroute. 1991. A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors. Yeast 7:609-615. [DOI] [PubMed] [Google Scholar]
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Cheng, W., and J. R. Roth. 1994. Evidence for two NAD kinases in Salmonella typhimurium. J. Bacteriol. 176:4260-4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinnery, P. F. 2003. Searching for nuclear-mitochondrial genes. Trends Genet. 19:60-62. [DOI] [PubMed] [Google Scholar]
- 7.Claros, M. G. 1995. MitoProt, a Macintosh application for studying mitochondrial proteins. Comput. Appl. Biosci. 11:441-447. [DOI] [PubMed] [Google Scholar]
- 8.Croteau, D. L., R. H. Stierum, and V. A. Bohr. 1999. Mitochondrial DNA repair pathways. Mutat. Res. 434:137-148. [DOI] [PubMed] [Google Scholar]
- 9.Dizdaroglu, M., P. Jaruga, M. Birincioglu, and H. Rodriguez. 2002. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic. Biol. Med. 32:1102-1115. [DOI] [PubMed] [Google Scholar]
- 10.Elfering, S. L., T. M. Sarkela, and C. Giulivi. 2002. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem. 277:38079-38086. [DOI] [PubMed] [Google Scholar]
- 11.Emanuelsson, O., H. Nielsen, S. Brunak, and G. von Heijne. 2000. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300:1005-1016. [DOI] [PubMed] [Google Scholar]
- 12.Esworthy, R. S., Y. S. Ho, and F. F. Chu. 1997. The Gpx1 gene encodes mitochondrial glutathione peroxidase in the mouse liver. Arch. Biochem. Biophys. 340:59-63. [DOI] [PubMed] [Google Scholar]
- 13.Fortuniak, A., R. Zadzinski, T. Bilinski, and G. Bartosz. 1996. Glutathione depletion in the yeast Saccharomyces cerevisiae. Biochem. Mol. Biol. Int. 38:901-910. [PubMed] [Google Scholar]
- 14.Fridovich, I. 1995. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 64:97-112. [DOI] [PubMed] [Google Scholar]
- 15.Georgiou, G. 2002. How to flip the (redox) switch. Cell 111:607-610. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh, M., J. Shen, and B. P. Rosen. 1999. Pathways of As(III) detoxification in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 96:5001-5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giulivi, C. 2003. Characterization and function of mitochondrial nitric-oxide synthase. Free Radic. Biol. Med. 34:397-408. [DOI] [PubMed] [Google Scholar]
- 18.Gralla, E. B., D. J. Thiele, P. Silar, and J. S. Valentine. 1991. ACE1, a copper-dependent transcription factor, activates expression of the yeast copper, zinc superoxide dismutase gene. Proc. Natl. Acad. Sci. USA 88:8558-8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graziewicz, M. A., B. J. Day, and W. C. Copeland. 2002. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 30:2817-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graziewicz, M. A., T. H. Zastawny, R. Olinski, and B. Tudek. 1999. SOS-dependent A→G transitions induced by hydroxyl radical generating system hypoxanthine/xanthine oxidase/Fe3+/EDTA are accompanied by the increase of Fapy-adenine content in M13 mp18 phage DNA. Mutat. Res. 434:41-52. [DOI] [PubMed] [Google Scholar]
- 21.Hirano, M., R. Marti, C. Ferreiro-Barros, M. R. Vila, S. Tadesse, Y. Nishigaki, I. Nishino, and T. H. Vu. 2001. Defects of intergenomic communication: autosomal disorders that cause multiple deletions and depletion of mitochondrial DNA. Semin. Cell Dev. Biol. 12:417-427. [DOI] [PubMed] [Google Scholar]
- 22.Holmgren, A., and M. Bjornstedt. 1995. Thioredoxin and thioredoxin reductase. Methods Enzymol. 252:199-208. [DOI] [PubMed] [Google Scholar]
- 23.Hottiger, T., P. Furst, G. Pohlig, and J. Heim. 1994. Physiological characterization of the yeast metallothionein (CUP1) promoter, and consequences of overexpressing its transcriptional activator, ACE1. Yeast 10:283-296. [DOI] [PubMed] [Google Scholar]
- 24.Iwahashi, Y., A. Hitoshio, N. Tajima, and T. Nakamura. 1989. Characterization of NADH kinase from Saccharomyces cerevisiae. J. Biochem. (Tokyo) 105:588-593. [DOI] [PubMed] [Google Scholar]
- 25.Kadiiska, M. B., P. M. Hanna, L. Hernandez, and R. P. Mason. 1992. In vivo evidence of hydroxyl radical formation after acute copper and ascorbic acid intake: electron spin resonance spin-trapping investigation. Mol. Pharmacol. 42:723-729. [PubMed] [Google Scholar]
- 26.Kaukonen, J., J. K. Juselius, V. Tiranti, A. Kyttala, M. Zeviani, G. P. Comi, S. Keranen, L. Peltonen, and A. Suomalainen. 2000. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289:782-785. [DOI] [PubMed] [Google Scholar]
- 27.Kawai, S., S. Mori, T. Mukai, W. Hashimoto, and K. Murata. 2001. Molecular characterization of Escherichia coli NAD kinase. Eur. J. Biochem. 268:4359-4365. [DOI] [PubMed] [Google Scholar]
- 28.Kawai, S., S. Mori, T. Mukai, S. Suzuki, T. Yamada, W. Hashimoto, and K. Murata. 2000. Inorganic polyphosphate/ATP-NAD kinase of Micrococcus flavus and Mycobacterium tuberculosis H37Rv. Biochem. Biophys. Res. Commun. 276:57-63. [DOI] [PubMed] [Google Scholar]
- 29.Kawai, S., S. Suzuki, S. Mori, and K. Murata. 2001. Molecular cloning and identification of UTR1 of a yeast Saccharomyces cerevisiae as a gene encoding an NAD kinase. FEMS Microbiol. Lett. 200:181-184. [DOI] [PubMed] [Google Scholar]
- 30.Kirkman, H. N., and G. F. Gaetani. 1984. Catalase: a tetrameric enzyme with four tightly bound molecules of NADPH. Proc. Natl. Acad. Sci. USA 81:4343-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirkman, H. N., M. Rolfo, A. M. Ferraris, and G. F. Gaetani. 1999. Mechanisms of protection of catalase by NADPH. Kinetics and stoichiometry. J. Biol. Chem. 274:13908-13914. [DOI] [PubMed] [Google Scholar]
- 32.Krems, B., C. Charizanis, and K. D. Entian. 1995. Mutants of Saccharomyces cerevisiae sensitive to oxidative and osmotic stress. Curr. Genet. 27:427-434. [DOI] [PubMed] [Google Scholar]
- 33.Law, R. H., S. Manon, R. J. Devenish, and P. Nagley. 1995. ATP synthase from Saccharomyces cerevisiae. Methods Enzymol. 260:133-163. [DOI] [PubMed] [Google Scholar]
- 34.Lea, D. E., and C. A. Coulson. 1949. The distribution of the number of mutants in bacterial populations. J. Genet. 49:264-285. [DOI] [PubMed] [Google Scholar]
- 35.LeDoux, S. P., W. J. Driggers, B. S. Hollensworth, and G. L. Wilson. 1999. Repair of alkylation and oxidative damage in mitochondrial DNA. Mutat. Res. 434:149-159. [DOI] [PubMed] [Google Scholar]
- 36.LeDoux, S. P., G. L. Wilson, E. J. Beecham, T. Stevnsner, K. Wassermann, and V. A. Bohr. 1992. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 13:1967-1973. [DOI] [PubMed] [Google Scholar]
- 37.Lee, J., C. Godon, G. Lagniel, D. Spector, J. Garin, J. Labarre, and M. B. Toledano. 1999. Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J. Biol. Chem. 274:16040-16046. [DOI] [PubMed] [Google Scholar]
- 38.Lerner, F., M. Niere, A. Ludwig, and M. Ziegler. 2001. Structural and functional characterization of human NAD kinase. Biochem. Biophys. Res. Commun. 288:69-74. [DOI] [PubMed] [Google Scholar]
- 39.Liochev, S. I., and I. Fridovich. 1994. The role of O2·− in the production of HO·: in vitro and in vivo. Free Radic. Biol. Med. 16:29-33. [DOI] [PubMed] [Google Scholar]
- 40.Longley, M. J., P. A. Ropp, S. E. Lim, and W. C. Copeland. 1998. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry 37:10529-10539. [DOI] [PubMed] [Google Scholar]
- 41.Luria, S. E., and M. Delbruck. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandel, H., R. Szargel, V. Labay, O. Elpeleg, A. Saada, A. Shalata, Y. Anbinder, D. Berkowitz, C. Hartman, M. Barak, S. Eriksson, and N. Cohen. 2001. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 29:337-341. [DOI] [PubMed] [Google Scholar]
- 43.Melnick, L., and F. Sherman. 1993. The gene clusters ARC and COR on chromosomes 5 and 10, respectively, of Saccharomyces cerevisiae share a common ancestry. J. Mol. Biol. 233:372-388. [DOI] [PubMed] [Google Scholar]
- 44.Monteiro, G., G. A. Pereira, and L. E. Netto. 2002. Regulation of mitochondrial thioredoxin peroxidase I expression by two different pathways: one dependent on cAMP and the other on heme. Free Radic. Biol. Med. 32:278-288. [DOI] [PubMed] [Google Scholar]
- 45.Naviaux, R. K., W. L. Nyhan, B. A. Barshop, J. Poulton, D. Markusic, N. C. Karpinski, and R. H. Haas. 1999. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers' syndrome. Ann. Neurol. 45:54-58. [DOI] [PubMed] [Google Scholar]
- 46.Nebert, D. W., and D. W. Russell. 2002. Clinical importance of the cytochromes P450. Lancet 360:1155-1162. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen-nhu, N. T., and B. Knoops. 2002. Alkyl hydroperoxide reductase 1 protects Saccharomyces cerevisiae against metal ion toxicity and glutathione depletion. Toxicol. Lett. 135:219-228. [DOI] [PubMed] [Google Scholar]
- 48.Nieminen, A. L., A. M. Byrne, B. Herman, and J. J. Lemasters. 1997. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am. J. Physiol. 272:C1286-C1294. [DOI] [PubMed] [Google Scholar]
- 49.Nishino, I., A. Spinazzola, and M. Hirano. 1999. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283:689-692. [DOI] [PubMed] [Google Scholar]
- 50.Nisoli, E., E. Clementi, C. Paolucci, V. Cozzi, C. Tonello, C. Sciorati, R. Bracale, A. Valerio, M. Francolini, S. Moncada, and M. O. Carruba. 2003. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299:896-899. [DOI] [PubMed] [Google Scholar]
- 51.O'Rourke, T. W., N. A. Doudican, M. D. Mackereth, P. W. Doetsch, and G. S. Shadel. 2002. Mitochondrial dysfunction due to oxidative mitochondrial DNA damage is reduced through cooperative actions of diverse proteins. Mol. Cell. Biol. 22:4086-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Outten, C. E., and V. C. Culotta. 2003. A novel NADH kinase is the mitochondrial source of NADPH in Saccharomyces cerevisiae. EMBO J. 22:2015-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park, S. G., M. K. Cha, W. Jeong, and I. H. Kim. 2000. Distinct physiological functions of thiol peroxidase isoenzymes in Saccharomyces cerevisiae. J. Biol. Chem. 275:5723-5732. [DOI] [PubMed] [Google Scholar]
- 54.Pedrajas, J. R., A. Miranda-Vizuete, N. Javanmardy, J. A. Gustafsson, and G. Spyrou. 2000. Mitochondria of Saccharomyces cerevisiae contain one-conserved cysteine type peroxiredoxin with thioredoxin peroxidase activity. J. Biol. Chem. 275:16296-16301. [DOI] [PubMed] [Google Scholar]
- 55.Petrat, F., S. Pindiur, M. Kirsch, and H. De Groot. 2003. NAD(P)H, a primary target of 1O2 in mitochondria of intact cells. J. Biol. Chem. 278:3298-3307. [DOI] [PubMed] [Google Scholar]
- 56.Pinz, K. G., and D. F. Bogenhagen. 1998. Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol. Cell. Biol. 18:1257-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponamarev, M. V., M. J. Longley, D. Nguyen, T. A. Kunkel, and W. C. Copeland. 2002. Active site mutation in DNA polymerase gamma associated with progressive external ophthalmoplegia causes error-prone DNA synthesis. J. Biol. Chem. 277:15225-15228. [DOI] [PubMed] [Google Scholar]
- 58.Richter, C., J. W. Park, and B. N. Ames. 1988. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 85:6465-6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ropp, P. A., and W. C. Copeland. 1996. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics 36:449-458. [DOI] [PubMed] [Google Scholar]
- 60.Saada, A., A. Shaag, H. Mandel, Y. Nevo, S. Eriksson, and O. Elpeleg. 2001. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat. Genet. 29:342-344. [DOI] [PubMed] [Google Scholar]
- 61.Salazar, J. J., and B. Van Houten. 1997. Preferential mitochondrial DNA injury caused by glucose oxidase as a steady generator of hydrogen peroxide in human fibroblasts. Mutat. Res. 385:139-149. [DOI] [PubMed] [Google Scholar]
- 62.Santos, J. H., L. Hunakova, Y. Chen, C. Bortner, and B. Van Houten. 2003. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J. Biol. Chem. 278:1728-1734. [DOI] [PubMed] [Google Scholar]
- 63.Sheline, C. T., E. H. Choi, J. S. Kim-Han, L. L. Dugan, and D. W. Choi. 2002. Cofactors of mitochondrial enzymes attenuate copper-induced death in vitro and in vivo. Ann. Neurol. 52:195-204. [DOI] [PubMed] [Google Scholar]
- 64.Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3-21. [DOI] [PubMed] [Google Scholar]
- 65.Sia, E. A., C. A. Butler, M. Dominska, P. Greenwell, T. D. Fox, and T. D. Petes. 2000. Analysis of microsatellite mutations in the mitochondrial DNA of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 97:250-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sohal, R. S., S. Agarwal, M. Candas, M. J. Forster, and H. Lal. 1994. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech. Ageing Dev. 76:215-224. [DOI] [PubMed] [Google Scholar]
- 67.Spelbrink, J. N., F. Y. Li, V. Tiranti, K. Nikali, Q. P. Yuan, M. Tariq, S. Wanrooij, N. Garrido, G. Comi, L. Morandi, L. Santoro, A. Toscano, G. M. Fabrizi, H. Somer, R. Croxen, D. Beeson, J. Poulton, A. Suomalainen, H. T. Jacobs, M. Zeviani, and C. Larsson. 2001. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 28:223-231. [DOI] [PubMed] [Google Scholar]
- 68.Spencer, F., Y. Hugerat, G. Simchen, O. Hurko, C. Connelly, and P. Hieter. 1994. Yeast kar1 mutants provide an effective method for YAC transfer to new hosts. Genomics 22:118-126. [DOI] [PubMed] [Google Scholar]
- 69.Steele, D. F., C. A. Butler, and T. D. Fox. 1996. Expression of a recoded nuclear gene inserted into yeast mitochondrial DNA is limited by mRNA-specific translational activation. Proc. Natl. Acad. Sci. USA 93:5253-5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steinmetz, L. M., C. Scharfe, A. M. Deutschbauer, D. Mokranjac, Z. S. Herman, T. Jones, A. M. Chu, G. Giaever, H. Prokisch, P. J. Oefner, and R. W. Davis. 2002. Systematic screen for human disease genes in yeast. Nat. Genet. 31:400-404. [DOI] [PubMed] [Google Scholar]
- 71.Strain, J., and V. C. Culotta. 1996. Copper ions and the regulation of Saccharomyces cerevisiae metallothionein genes under aerobic and anaerobic conditions. Mol. Gen. Genet. 251:139-145. [DOI] [PubMed] [Google Scholar]
- 72.Strand, M. K., and W. C. Copeland. 2002. Measuring mtDNA mutation rates in Saccharomyces cerevisiae using the mtArg8 assay. Methods Mol. Biol. 197:151-157. [DOI] [PubMed] [Google Scholar]
- 73.Tamura, T., H. W. McMicken, C. V. Smith, and T. N. Hansen. 1997. Gene structure for mouse glutathione reductase, including a putative mitochondrial targeting signal. Biochem. Biophys. Res. Commun. 237:419-422. [DOI] [PubMed] [Google Scholar]
- 74.Thiele, D. J. 1988. ACE1 regulates expression of the Saccharomyces cerevisiae metallothionein gene. Mol. Cell. Biol. 8:2745-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tran, H. T., J. D. Keen, M. Kricker, M. A. Resnick, and D. A. Gordenin. 1997. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 17:2859-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turrens, J. F., and A. Boveris. 1980. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 191:421-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Goethem, G., B. Dermaut, A. Lofgren, J. J. Martin, and C. Van Broeckhoven. 2001. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 28:211-212. [DOI] [PubMed] [Google Scholar]
- 78.Van Goethem, G., A. Lofgren, J. J. Martin, and C. Van Broeckhoven. 2000. Further evidence for genetic heterogeneity of autosomal dominant disorders with accumulation of multiple deletions of mitochondrial DNA. J. Med. Genet. 37:547-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wallace, D. C. 1992. Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 61:1175-1212. [DOI] [PubMed] [Google Scholar]
- 80.Wallace, D. C. 1999. Mitochondrial diseases in man and mouse. Science 283:1482-1488. [DOI] [PubMed] [Google Scholar]
- 81.Wallace, D. C., G. Singh, M. T. Lott, J. A. Hodge, T. G. Schurr, A. M. Lezza, L. J. Elsas II, and E. K. Nikoskelainen. 1988. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242:1427-1430. [DOI] [PubMed] [Google Scholar]
- 82.Wei, Y., C. P. Scholes, and T. E. King. 1981. Ubisemiquinone radicals from the cytochrome b-c1 complex of the mitochondrial electron transport chain—demonstration of QP-S radical formation. Biochem. Biophys. Res. Commun. 99:1411-1419. [DOI] [PubMed] [Google Scholar]
- 83.Yakes, F. M., and B. Van Houten. 1997. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 94:514-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Young, P., J. M. Leeds, M. B. Slabaugh, and C. K. Mathews. 1994. Ribonucleotide reductase: evidence for specific association with HeLa cell mitochondria. Biochem. Biophys. Res. Commun. 203:46-52. [DOI] [PubMed] [Google Scholar]