

Summary
Arrestins serve as multi-functional regulators of G-protein coupled receptors, interacting with hundreds of different receptor subtypes and a variety of other signaling proteins. Here we identify calmodulin as a novel arrestin interaction partner using three independent methods in vitro and in cells. Arrestin preferentially binds calcium-loaded calmodulin with a Kd of ~7μM, which is within range of endogenous calmodulin concentrations. The calmodulin binding site is localized on the concave side of the C-domain and a loop in the center of the arrestin molecule, significantly overlapping with receptor- and microtubule-binding sites. Using purified proteins, we found that arrestins sequester calmodulin, preventing its binding to microtubules. Nanomolar affinity of arrestins for their cognate receptors makes calmodulin an ineffective competitor for arrestin binding at relatively high receptor concentrations. The arrestin-calmodulin interaction likely regulates the localization of both proteins and their availability for other interaction partners.
Keywords: arrestin, calmodulin, receptor, signaling, EPR
Introduction
Arrestins are key regulators of G-protein coupled receptor (GPCR) signaling and trafficking. They bind active phosphorylated receptors and block further G-protein activation 1. Arrestins also link GPCRs to alternative signaling pathways by virtue of their interaction with numerous non-receptor partners, such as Src family kinases, MAP kinases, the ubiquitin ligase Mdm2, etc (reviewed in 2; 3). Receptor binding induces a global conformational change in the arrestin molecule 4. Some of the arrestin partners preferentially interact with the receptor-bound (active) conformation of arrestin, whereas others also bind the free (basal) form 3. Thus, arrestins are versatile adaptors in a multitude of signaling processes. Vertebrates have four distinct arrestin subtypes. Arrestin1 (visual arrestin) and arrestin4 (cone arrestin) are predominantly expressed in rod and cone photoreceptors, respectively, where they regulate the signaling of rhodopsin and cone opsins5. The non-visual arrestins, arrestin2 (β-arrestin1) and arrestin3 (β-arrestin2), modulate the activity of hundreds of other GPCRs6.
Calmodulin (CaM) is an intracellular calcium sensor in eukaryotes. It undergoes a dramatic conformational change upon Ca2+ binding and serves as the most ubiquitous transducer of Ca2+ signaling 7; 8. CaM regulates the activity of various cellular proteins, including enzymes, ion channels, transcription factors and cytoskeletal proteins. CaM also participates in GPCR signaling via direct interactions with GRKs 9, type III adenylyl cyclase 10, CaM-dependent protein kinases 7, as well as certain receptors themselves including the 5-hydroxytrptamine 1A 11 and 2A 12 receptors, the V1 13 and V2 vasopressin receptors 14, parathyroid hormone receptor 1 15, metabotropic glutamate receptors 16; 17; 18, and the opioid receptor 19.
Here we describe the direct calcium-dependent interaction between the two ubiquitous signaling proteins, arrestin and CaM, in vitro and in living cells. The binding of all four members of the arrestin family to calmodulin suggests that the crosstalk between arrestin- and calmodulin-dependent pathways may be a common feature of cell signaling.
Results and Discussion
Arrestin2 binds directly to Ca2+-loaded calmodulin
Both arrestins and calmodulin interact with numerous GPCRs and regulate their signaling. Direct interactions of participating proteins often underlie the crosstalk between different signaling pathways in the cell. To determine whether these two proteins interact, we used purified wild type arrestin2 and a truncated (1-382) form of arrestin2 (which has been shown previously to interact with receptors in a phosphorylation-independent manner20; 21) in a pull-down assay on CaM-agarose. As shown in Fig.1a, both forms of arrestin2 bind calmodulin. Importantly, this interaction is strongly calcium-dependent: in the presence of 100μM Ca2+, CaM-agarose binds a significantly higher proportion of the arrestins compared to the calcium-free form. In contrast, low (less than 2% input) non-specific arrestin2 binding to “empty” agarose was Ca2+-independent (data not shown). Thus, arrestin2 directly interacts with Ca2+-loaded, but not with Ca2+-free calmodulin.
Figure 1. Calcium-dependent interaction of arrestin2 with calmodulin.
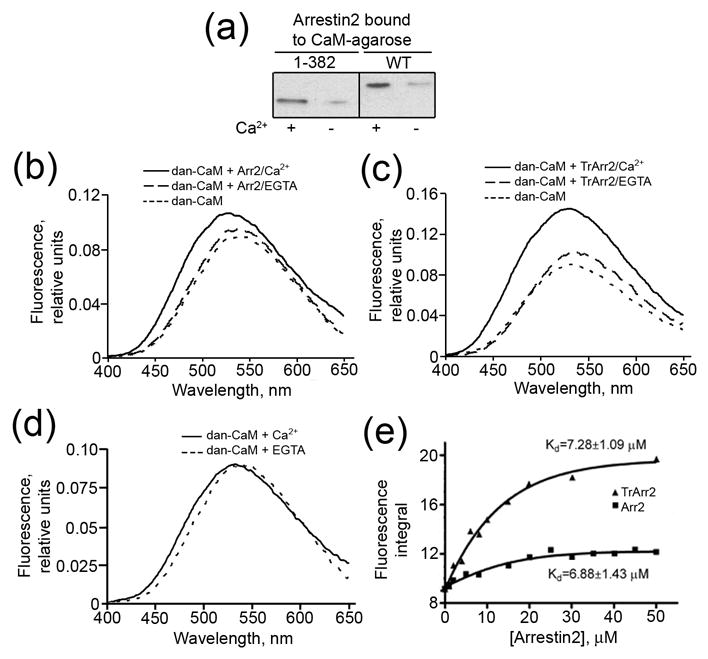
(a) Equal proportions of wild type (WT) and truncated (1-382) arrestin2 bound to CaM-agarose in the presence of 0.1mM CaCl2 or 5mM EGTA were run on SDS-PAGE and analyzed by Western blot with anti-arrestin antibody. “Empty” CNBr-activated Sepharose treated and blocked under the same conditions as CaM-agarose was used as a negative control. Non-specific arrestin binding was low (<2% of the input) and independent of Ca2+ (not shown). (b–d) Representative spectra of dansyl-CaM (5μM) in the presence of 20μM WT (b), truncated (Tr) (c) arrestin2 (Arr2), and in the absence of arrestin2 (d) in buffer containing 0.1mM CaCl2 or 5mM EGTA. The average spectra of three experiments are shown. (e) The integrated area under the emission spectrum plotted vs the concentration of arrestin2. The data were fit to the Boltzmann one-site binding equation and the dissociation constant was calculated according to Bertrand et al 51 as described in the Methods.
The affinity of the Ca2+/CaM interaction
To confirm this observation using an independent method, we used dansylated CaM and monitored the interaction via changes in the fluorescence emission spectra. This method is based on the ability of the interaction partner (in this case arrestin2) to shield the fluorophore on dansyl-CaM from the aqueous environment, thereby enhancing the fluorescence signal 11. We found that wild type and truncated arrestin2 dramatically enhance the fluorescence of dansyl-CaM in the presence of Ca2+ but not in the presence of EGTA (Fig.1b,c). Importantly, the increase in fluorescence of dansyl-CaM was not associated with a Ca2+–induced conformational change (Fig.1d). The magnitude of the arrestin-induced change allowed us to measure the affinity of this interaction by titrating dansyl-CaM with increasing concentrations of arrestin2. The affinity of wild type arrestin2 and its 1-382 truncated form for Ca2+/CaM was found to be 6.88 ± 1.43μM and 7.28 ± 1.09μM, respectively (Fig.1e). These values are comparable to the >10μM endogenous calmodulin concentrations found in cells 22. Although not all CaM becomes Ca2+-liganded in a stimulated cell, high concentrations can develop in the process of localized Ca2+ signaling 8. Calmodulin has the same affinity for full-length and truncated arrestin2, suggesting that the arrestin C-tail is not involved in the interaction. However, the magnitude of the increase in the fluorescence of dansyl-CaM was greater in the case of truncated arrestin (Fig 1e), indicating that this conformationally “loose” mutant, that demonstrates binding to both phosphorylated and unphosphorylated forms of its cognate receptors 20; 21, shields the dansyl moiety on CaM more efficiently.
All arrestin subtypes interact with Ca2+/CaM in cells
Next we tested whether arrestin2 interacts with calmodulin in intact cells. To this end we co-expressed Flag-tagged arrestin2 with HA-tagged CaM, force-loaded the cells with Ca2+ (by the addition of Ca2+ and a Ca2+–ionophore) or depleted intracellular Ca2+ (by the addition of EGTA and the same Ca2+-ionophore), and immunoprecipitated either arrestin2 or calmodulin from the cell lysate (Fig. 2). We found that arrestin2 and calmodulin from Ca2+-loaded cells co-immunoprecipitate when pulled down with either antibody, whereas no appreciable co-immunoprecipitation of these two proteins was observed in the absence of Ca2+ or upon omission of the primary antibody during the immunoprecipitation step (Fig. 2). Thus, in agreement with our in vitro data, arrestin2 binds calmodulin in cells in a Ca2+-dependent manner (Figs. 1,2).
Figure 2. All four mammalian arrestins bind Ca2+/CaM in cells.
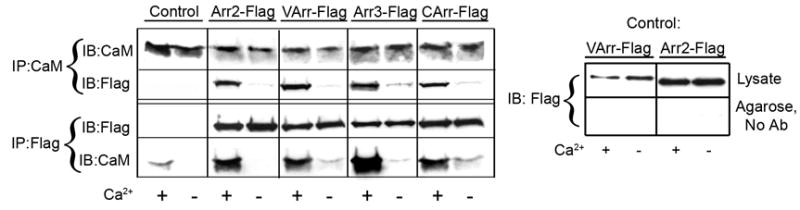
The lysates of cells expressing HA-CaM and the indicated flag-tagged arrestins (or empty vector; control) were immunoprecipitated (IP) with rabbit anti-Flag or rabbit anti-calmodulin (CaM) antibodies as described in the Methods. Aliquots of the total lysate and immunoprecipitated proteins were run on 12.5% (calmodulin) or 10% (arrestin) SDS-PAGE and blotted (IB) for HA-CaM or Flag-arrestin. Non-specific binding of arrestins to Protein G agarose (in the absence of antibody (No Ab)), was very low and calcium-independent (right panel). Visual (VArr), arrestin2 (Arr2), arrestin3 (Arr3), and cone arrestin (CArr) were expressed at 96+30, 74+26, 53+9, and 73+21 pmol/mg protein, respectively, yielding intracellular concentrations that are somewhat higher than those found in mature neurons for non-visual arrestins 39 and lower than those found in rod photoreceptors for visual arrestin 45; 54. HA-CaM was expressed at a level equal to that of endogenous CaM and was used to facilitate immunoblotting. Representative results from 2–4 experiments are shown.
Overall sequence homology among the four vertebrate arrestins is fairly high (60–70%) 23; 24, suggesting that other members of the arrestin family may also interact with calmodulin. We performed similar co-immunoprecipitation experiments with the other three arrestins: arrestin3, and the visual rod and cone arrestins. We found that all four arrestins bind calmodulin (Fig.2). Importantly, the interaction was detected in Ca2+-loaded, but not in Ca2+-depleted cells in all cases, indicating its strong Ca2+-dependence (Fig.2). Thus, the binding of Ca2+-loaded calmodulin is a common functional characteristic of the four subtypes of mammalian arrestins.
The binding site of Ca2+/CaM on arrestin2
This finding provides a new link between GPCR-dependent and Ca2+/CaM-dependent signaling pathways. To elucidate the molecular mechanism of the arrestin2-Ca2+/CaM interaction we used site-directed spin labeling (SDSL) electron paramagnetic resonance (EPR) spectroscopy. Application of SDSL requires the elimination of reactive native cysteines from the protein, the introduction of a cysteine residue into the position of interest, and chemical modification of this unique cysteine with a spin labeling reagent (reviewed in 25; 26). For this purpose, we designed a cysteine-less base mutant of arrestin2 by substituting native cysteines with serines, valines, or leucines (see Methods) to maximally preserve intra-molecular interactions, based on its high-resolution crystal structure 27. We ascertained that cysteine-less arrestin2 interacts with known arrestin binding partners like wild type protein 5; 28; 29: purified phosphorylated carbachol-activated m2 muscarinic cholinergic receptor (P-m2 mAChR*), light-activated phosphorhodopsin (P-Rhodopsin*), and microtubules (Fig.3a). Next we introduced 17 single cysteine substitutions spanning the entire molecule of arrestin2 on the cysteine-less background, expressed the mutants in E. coli, purified, and spin-labeled them with a methanethiosulfonate nitroxide reagent to produce the side chain designated as R1 (Fig.3b). The EPR spectra of spin-labeled proteins encode information on the dynamics of R1, and thus provide a means of mapping protein-protein interaction surfaces through changes in R1 motion 29; 30; 31. A highly mobile spin label on the surface of a protein has a characteristic narrow spectrum with sharp peaks, whereas the reduction in its mobility due to intramolecular constraints or occlusion in a protein-protein interaction interface is reflected in the broadening of the spectrum and/or the appearance of an additional peak in the low-field region 25; 26. We have successfully used this method to identify important residues in the binding of both dark and light phosphorylated rhodopsin and microtubules on visual rod arrestin 29; 31.
Figure 3. The localization of the calmodulin binding site on arrestin2 using site-directed spin labeling EPR spectroscopy.
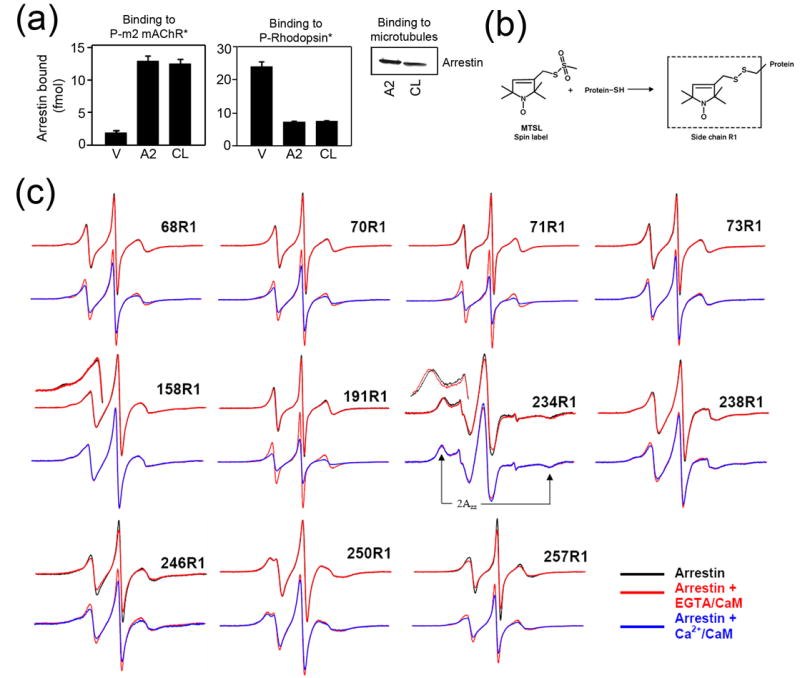
(a) Binding of visual (V), arrestin2 (A2), and cysless arrestin2 (CL) to the phosphorylated carbachol-activated m2 muscarinic receptor (P-m2 mAChR*) (left panel), light-activated phosphorylated rhodopsin (P-Rhodopsin*) (center panel), and microtubules (right panel) was performed as described in the Methods. Functionally cysless arrestin2 was identical to wild type arrestin2 in all cases. (b) The R1 side chain generated by reacting the arrestin cysteine mutants with the methanethiosulfonate (MTSL) nitroxide spin label reagent. (c) For each spin-labeled arrestin, normalized spectra in the absence (black) or presence of CaM + 1mM EGTA (red) are compared in the top row, and spectra in the presence of CaM + 1mM EGTA (red) and CaM + 0.1mM Ca2+ (blue) are compared in the bottom row. Portions of the overlaid spectra for 158R1 and 234R1 are magnified to better illustrate the spectral changes and the location of the hyperfine splitting (2Azz'). The spectrum of each arrestin mutant alone in solution or with 1mM EGTA or 0.1mM Ca2+ in the absence of calmodulin (black) were identical (spectra not shown). Only the spectra showing significant changes in the presence of calmodulin are presented.
Figure 3c shows the EPR spectra for R1 at sites in arrestin2 where significant changes in R1 mobility were observed upon calmodulin binding. Spectra for the sites where no changes were observed (positions 33, 47, 81, 167, 269, 306) are not shown but their locations are highlighted in Figure 4. The spectrum of each arrestin alone in solution (Fig.3c, black traces) provides a baseline to detect increases or decreases in mobility upon interaction with calmodulin. It is important to note that the presence of EGTA or Ca2+ did not affect label mobility in arrestin (data not shown). As a control we first tested mobility changes that occur in the presence of Ca2+-free calmodulin (with 1mM EGTA) (Fig.3c, red traces). Three sites show specific changes under these conditions: R1 in position 158 in the N-domain of arrestin becomes slightly more mobile (shown magnified in Fig.3c), whereas C-domain residues 234 and 257 become less mobile. In the case of 234, the outermost hyperfine splitting increases by ~1 Gauss (2Azz', Fig.3c), which is indicative of even further immobilization of an already immobile site. These minor changes likely reflect very low affinity interactions of Ca2+-free calmodulin with arrestin2 that are hard to detect by less sensitive methods, similar to the low-affinity binding of arrestin proteins to the non-preferred forms of their cognate receptors described earlier 31; 32; 33.
Figure 4. Summary of the changes in spin label mobility induced by arrestin interaction with Ca2+/CaM.
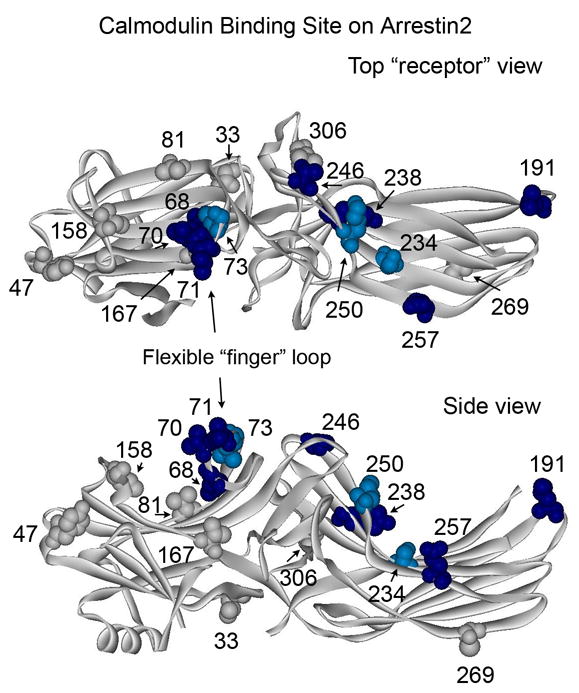
The magnitude of Ca2+/CaM-induced changes in spin label mobility (Fig.3) is color-coded on the arrestin2 crystal structure (PDB ID: 1G4R) 27 as follows: light blue/dark blue, small and large changes in mobility, respectively; gray, no change (spectral data not shown for sites 33, 47, 81, 167, 269, and 306). Top panel: view from the “receptor binding” side of arrestin. Bottom panel: side view.
The binding of Ca2+-loaded calmodulin to arrestin produced numerous dramatic spectral changes (Fig.3c, blue traces). Both 234 and 257 changed further in the presence of Ca2+/CaM. Site 257 becomes even more immobile whereas 234 reverts to an intermediate state between the free and EGTA/CaM forms. Residues 191, 238, 246, and 250 in the β-strands on the concave surface of the C-domain all become less mobile to varying degrees, whereas N-domain residue 158, which changed slightly in the presence of EGTA/CaM, does not change further. Interestingly, Ca2+/CaM binding also greatly reduces the mobility of every site tested in the flexible loop between β-strands V and VI (the “finger loop”, residues 64–74) (Fig.4).
Ca2+/CaM-induced mobility changes in arrestin2 mapped on the arrestin2 crystal structure identify a distinct binding site (Fig.4). This site encompasses a large part of the concave surface of the C-domain and the finger loop in the middle of the molecule. Sites on the rest of the arrestin2 N-domain and elsewhere are not affected by Ca2+/CaM binding. The binding site covers a fairly extensive surface (~60x25Å). However, the finger loop is highly flexible (has a high B factor) 27 and exists in two distinct conformations in the visual arrestin crystal (either folded down as in Figure 4 or in an upright strand-turn-strand configuration) 23. In an upright position this loop would be much closer to the C-domain, making the dimensions of the site ~50x25Å, which is more likely the case in the Ca2+/CaM-bound form based on the extent of mobility changes in the finger loop residues. Sites 68, 70, and 71 all show very large changes upon binding, whereas in the “folded over” position (Fig.4), residues 68 and 70 actually point down toward the N-domain cavity where the mobility of nearby residues 167 and 81 does not change. Therefore the “folded over” position of this loop seems less likely, given the motional data presented in Fig.3. However, both “extended” and “compact” conformations of Ca2+/CaM 34; 35 are large enough to fit this binding site on arrestin2.
Effects of Ca2+/CaM binding on arrestin interactions with other partners
Using a wide variety of methods we have shown that the finger loop and the concave surfaces of both domains are important for arrestin binding to the phosphorylated receptor and microtubules 5; 29; 31; 32. Thus, calmodulin, receptors, and microtubules all compete for the same binding surface on arrestin. The relatively low affinity of the arrestin/calmodulin interaction (Fig.1) and nanomolar affinity of arrestin/receptor binding 28; 36 suggest that GPCRs should out-compete Ca2+/CaM. Indeed, we found that Ca2+/CaM, even at 10μM concentration, does not appreciably reduce the binding of visual and both non-visual arrestins to light-activated phosphorhodopsin (Fig.5a). Interestingly, Ca2+ alone reduces arrestin binding to the receptor; this effect is most pronounced for visual arrestin. It should be noted that relatively high concentrations of active phosphoreceptor (~150nM rhodopsin in Fig.5a) are used in our direct binding assay to obtain a measurable signal 28; 37. Rod photoreceptors function at very low light and demonstrate single-photon sensitivity 38. The endogenous expression levels of most GPCRs does not exceed 100fmol/mg protein, which translates into a concentration of ~ 10nM. Moreover, maximum physiological response often requires the activation of <1% of the receptor present. Under these conditions the effective concentration of arrestin binding-competent active phosphoreceptor may be low enough to make Ca2+/CaM competitive despite its much lower affinity for arrestin. On the other hand, intracellular concentrations of endogenous non-visual arrestins usually do not exceed 0.2μM 39, so that in the case of massive sustained receptor stimulation activated GPCRs can exhaust the arrestin pool 40. This would lead to the release of arrestin-bound calmodulin.
Figure 5. The effects of Ca2+/CaM on arrestin binding to the receptor and microtubules.
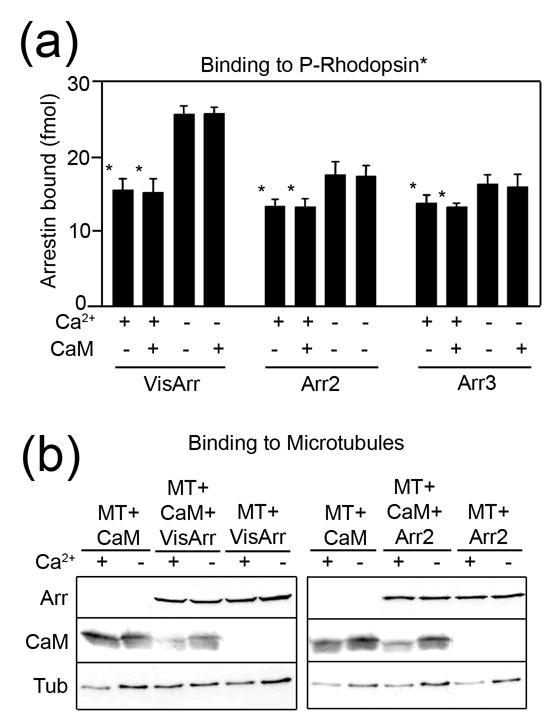
(a) Binding of radiolabeled arrestins to light-activated phosphorylated rhodopsin (P-Rhodopsin*) was performed in the presence (+) or absence (−) of 1.3μM free Ca2+ and in the presence (+) or absence (−) of 10μM purified calmodulin (CaM) as described in the Methods. VisArr, visual arrestin; Arr2, arrestin2; Arr3, arrestin3. (b) Binding of purified arrestins (0.44μM), and/or purified calmodulin (0.44μM), to pre-polymerized taxol-stabilized microtubules (MT) (1.8μM of tubulin αβ-dimer) was performed in the presence (+) or absence (−) of 1.3μM free Ca2+ as described in the Methods. Representative Western blots of the microtubule pellet fraction are shown.
Both arrestin and calmodulin bind microtubules (MTs) with relatively low affinity 29; 41; 42; 43 (Fig.5) that is comparable to the affinity of the arrestin-CaM interaction (Fig.1e), suggesting that arrestin can affect calmodulin binding to MTs and vice versa. We tested this possibility by incubating 20μg of taxol-stabilized microtubules (180pmol of αβ-dimer) with 44pmol of arrestin and 44pmol of calmodulin in the presence and absence of the physiological free Ca2+ concentration (1.3μM). We found that calmodulin did not affect arrestin binding to MTs (Fig.5b). Interestingly, arrestin reduced the amount of MT-bound calmodulin only in the presence of Ca2+ (Fig.5b), indicating that the sequestration of calmodulin by arrestin reduces its availability for microtubules. Ca2+/CaM has been shown to destabilize microtubules 44. Therefore arrestin may prevent MT destabilization upon elevation of cytoplasmic calcium. Arrestin binding to Ca2+/CaM may also dampen its signaling to other effectors, including those downstream of GPCRs.
In contrast to other cell types, rod photoreceptors contain more arrestin (>200μM) than calmodulin 45. In these cells the dramatic light-induced decrease in free calcium plays an important role in phototransduction and recovery 38. MTs in rods are important for the “storage” of arrestin in the dark 46, therefore Ca2+/CaM binding to arrestin in dark-adapted rods may shift the equilibrium toward free arrestin, which is critical for timely photoresponse shutoff 38. Additionally, the sequestration of Ca2+/CaM by arrestin in dark-adapted photoreceptors may participate in the CaM-dependent regulation of the sensitivity of cGMP-gated channels to cGMP, which is believed to play a particularly important role in the light adaptation of cone photoreceptors 47.
Although arrestins shuttle between the nucleus and the cytoplasm, they are mostly cytoplasmic proteins, with only arrestin2 demonstrating significant nuclear localization in certain types of neurons 48. Calmodulin is present in both compartments in fairly high concentrations 49, so arrestin-calmodulin interactions can occur both in the cytoplasm and in the nucleus. Interestingly, at low intracellular Ca2+ a large proportion of CaM diffuses freely, whereas an increase in cytoplasmic Ca2+ dramatically reduces the rate of CaM diffusion 49. This phenomenon is usually interpreted as a manifestation of Ca2+-dependent CaM binding to CaM-regulated protein kinases 49. Our data indicate that Ca2+ -dependent CaM binding to arrestin may also contribute.
In conclusion, direct interaction between Ca2+/CaM and all four mammalian arrestins described here is a novel functional link between these ubiquitous signaling proteins. This binding likely regulates the availability of arrestin and calmodulin for their intracellular partners in a Ca2+-dependent fashion. The full scope of the cell-specific biological implications of the interaction between Ca2+-liganded calmodulin and arrestins remains to be elucidated.
Methods
Site-directed mutagenesis and arrestin purification
Site-directed mutagenesis and arrestin expression and purification were performed as described 50. All cysteine mutations for the SDSL EPR studies were generated on the background of cysteine-less arrestin2: (C59V, C125S, C140L, C150V, C242V, C251V, C269S). Similar to two cysteine-less based visual arrestins 31, cysteine-less arrestin2 is fully functional with regard to receptor binding (Fig.3a,b,c).
CaM-agarose binding assay
50μl CaM-agarose (Sigma) was washed in binding buffer (20mM Hepes, pH 7.5, 100mM NaCl) (BB) and incubated with 10μg of purified arrestin2 in BB in the presence of 0.1mM CaCl2 or 5mM EGTA at 4°C for 1 hour in a volume of 400μl. The resin was washed three times with the corresponding buffer and bound arrestin was eluted with SDS sample buffer (Sigma). Samples were analyzed by Western blot using an anti-arrestin2 antibody. CNBr-activated Sepharose treated and blocked under the same conditions as CaM-agarose with the omission of CaM in the coupling step was used as a negative control. Non-specific arrestin binding was found to be low (<2% of the input) and independent of Ca2+ (not shown).
Arrestin binding to dansylated calmodulin
500μg CaM (Calbiochem) was treated with 5μl 10mg/ml dansyl chloride (Molecular Probes) at 4°C for 5 hours in 20mM Hepes, pH 7.5, 100mM NaCl. Free dansyl chloride was removed by dialysis. The extent of labeling was 50%, as determined by absorbance at 340nm. Dansyl-CaM (5μM) was incubated with increasing concentrations of arrestin at 4°C overnight in the presence of 0.1mM CaCl2 or 5mM EGTA. The fluorescence emission spectrum was recorded following excitation at 340nm on a SLM 8000C spectrofluorimeter. Measurements were repeated three times for each solution. The integral under the fluorescence curve was plotted against the concentration of arrestin2 and the data were fit to the Boltzmann one-site binding function (Graphpad Prism4). The dissociation constant was calculated according to Bertrand et al 51. Briefly, the fractional degree of saturation of dansyl-CaM was determined by a= (F−F0)/(F∞−F0), where F0 is the fluorescence signal of dansyl-CaM in the absence of arrestin and F∞ is the fluorescence signal at saturation level. Next, 1/ (1−a) was plotted against the concentration of arrestin2 divided by a. The data were fit to a straight line by linear regression. Kd was determined as the reciprocal of the slope.
Arrestin-calmodulin co-immunoprecipitation
HEK-293A cells were transfected with HA-CaM and arrestins Flag-tagged at the C-terminus using Lipofectamine 2000. Cells were trypsinized 48 hours post-transfection, divided into two aliquots and washed in 50mM Hepes 7.3, 100mM NaCl. Cells were then incubated with 5μM Ca2+ ionophore ETH-129 (Sigma), and 0.1mM CaCl2 (+Ca2+) or 2mM EGTA (−Ca2+) for 10min at RT. The cross-linker DSP (2mM) (Pierce) was added for 30min at RT and quenched by the addition of 50mM Tris 7.4 for 15min. Cells were lysed by the addition of NP-40 (to 0.2%), 2mM benzamidine, and 2mM PMSF and centrifuged for 10min at 16,000xg. Cell lysate (100μg total protein) was pre-cleared with 10μl Protein G Sepharose (Sigma) for 30min at 4°C and then incubated with either 2μg rabbit anti-Flag (Sigma) or 3μg rabbit anti-calmodulin (Zymed) antibodies for 2 hours at 4°C, followed by the addition of 10μl Protein G Sepharose for an additional 2 hours. The Sepharose was collected and washed three times. The beads were resuspended in Laemmli’s sample buffer (Sigma), boiled for 5min, and aliquots of the supernatant were analyzed by SDS-PAGE and Western blot using anti-HA and anti-Flag monoclonal antibodies (Sigma). Non-specific binding of arrestin to protein G-agarose was determined by omitting the antibody was very low and calcium-independent (Fig.2). Western blotting protocol developed by Hulen et al 52 was used to increase the sensitivity for calmodulin. Briefly, before transfer the gels and PVDF membranes were soaked in 25mM KH2/K2HPO4, pH 7.0 (KP buffer) for 15min. Transfer was conducted at 20V overnight at 4°C in KP buffer. Membranes were then incubated for 45min in fresh 0.2% glutaraldehyde, rinsed several times in KP buffer, and blocked with 2%BSA/ 0.1%Tween/TBS for 30–60min at RT. Standard protocol for primary and secondary antibody incubation and detection by ECL were then followed.
Direct binding assays
In vitro transcription and translation to generate radiolabeled arrestins were performed, as described 5. Briefly, to determine rhodopsin binding tritiated arrestins (50fmol) were incubated in 50mM Tris-HCl, pH 7.5, 0.5mM MgCl2, 1.5mM dithiothreitol, 50mM potassium acetate with 7.5pmol (0.3μg) of light-activated phosphorylated rhodopsin in a final volume of 50μl for 5min at 37°C in room light. To determine the binding to m2 muscarinic cholinergic receptor, tritiated arrestins (50fmol) were incubated with 50fmol of phosphorylated purified receptor in the presence of 100μM carbachol for 35min at 30°C. After incubation with either receptor, the samples were immediately cooled on ice and loaded onto 2ml Sepharose 2B columns equilibrated with 10mM Tris-HCl, pH 7.5, 100mM NaCl. Bound arrestin eluted with receptor-containing membranes in the void volume (between 0.5 – 1.1ml). Nonspecific binding determined in the presence of 0.3μg liposomes (less than 10 % of the total binding and less than 0.5% of the arrestin present in the assay) was subtracted.
Binding of purified arrestins to microtubules was measured as described 29. Briefly, 2 μg arrestin was incubated for 20 min at 25°C in 50mM Tris-HCl, pH 7.4, 100mM NaCl, 2mM EDTA, and 1mM EGTA with 20μg of purified tubulin (Cytoskeleton, Inc.) pre-polymerized with taxol according to the manufacturer’s instructions. Microtubules along with bound arrestin were pelleted by centrifugation for 10 min at 25°C at 90,000 rpm in a TLA 120.1 rotor in a Beckman TL100 ultracentrifuge. Parallel samples with the same amount of each arrestin without microtubules served as controls. The pellet was dissolved in Laemmli’s sample buffer (Sigma) and the amount of arrestin was quantified by Western blot. The effects of calmodulin and calcium on arrestin binding to receptors and microtubules were determined using EGTA-Ca2+ buffers at 0 and 1.3μM free calcium as described 53.
Electron paramagnetic spectroscopy
Arrestin mutants with unique cysteines were dialyzed into 50mM MOPS, 100mM NaCl, pH7.2 buffer and labeled with a ten-fold molar excess of 2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (MTSL; Toronto Research Chemicals) overnight at 4°C as described 31. EPR samples contained 25 μM spin labeled arrestin and 50μM bovine calmodulin (Sigma) in the presence of either 1mM EGTA or 0.1mM Ca2+ in a final volume of 10 μL. The spectra of the spin labeled arrestins alone in solution (in the absence of calmodulin) did not change in the presence of EGTA or Ca2+. X-band EPR spectra were recorded for samples in glass capillaries (10μL) at room temperature over a 100 G range with an incident microwave power of 10mW on a Bruker ELEXSYS 500 fitted with a super high Q cavity. Spectra were typically the average of 25–36 scans, baseline corrected, and normalized to the same area for the overlays shown in Fig.3.
Acknowledgments
The authors are grateful to Drs. Mark E. Anderson and Xiufeng Song for expression constructs for HA-calmodulin and Flag-tagged cone arrestin, respectively. We thank Dr. Alexander Dizhoor for calcium buffers and expert advice. This work was supported by NIH grants HL39757 (MS and MT), EY11500 (VVG), and AI58024 and GM70642 (CSK).
References
- 1.Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 2.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 3.Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure (Camb) 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 4.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. TIPS. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 6.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klee CB, Vanaman TC. Calmodulin. Adv Protein Chem. 1982;35:213–321. doi: 10.1016/s0065-3233(08)60470-2. [DOI] [PubMed] [Google Scholar]
- 8.Persechini A, Stemmer PM. Calmodulin is a limiting factor in the cell. Trends Cardiovasc Med. 2002;12:32–37. doi: 10.1016/s1050-1738(01)00144-x. [DOI] [PubMed] [Google Scholar]
- 9.Chuang TT, Paolucci L, De Blasi A. Inhibition of G protein-coupled receptor kinase subtypes by Ca2+/calmodulin. J Biol Chem. 1996;27:28691–28696. doi: 10.1074/jbc.271.45.28691. [DOI] [PubMed] [Google Scholar]
- 10.Choi EJ, Xia Z, Storm DR. Stimulation of the type III olfactory adenylyl cyclase by calcium and calmodulin. Biochemistry. 1992;31:6492–6498. doi: 10.1021/bi00143a019. [DOI] [PubMed] [Google Scholar]
- 11.Turner JH, Gelasco AK, Raymond JR. Calmodulin interacts with the third intracellular loop of the serotonin 5-hydroxytryptamine1A receptor at two distinct sites: putative role in receptor phosphorylation by protein kinase C. J Biol Chem. 2004;279:17027–17037. doi: 10.1074/jbc.M313919200. [DOI] [PubMed] [Google Scholar]
- 12.Turner JH, Raymond JR. Interaction of calmodulin with the serotonin 5-hydroxytryptamine2A receptor. A putative regulator of G protein coupling and receptor phosphorylation by protein kinase C. J Biol Chem. 2005;280:30741–30750. doi: 10.1074/jbc.M501696200. [DOI] [PubMed] [Google Scholar]
- 13.Wu N, Dazard RM, Nithianantham S, Xu Z, Hanson SM, Vishnivetskiy SA, Gurevich VV, Thibonnier M, Shoham M. Soluble mimics of the cytoplasmic face of the human V1-vascular vasopressin receptor bind arrestin2 and calmodulin. Mol Pharmacol. 2006;70:249–258. doi: 10.1124/mol.105.018804. [DOI] [PubMed] [Google Scholar]
- 14.Nickols HH, Shah VN, Chazin WJ, Limbird LE. Calmodulin interacts with the V2 vasopressin receptor: elimination of binding to the C terminus also eliminates arginine vasopressin-stimulated elevation of intracellular calcium. J Biol Chem. 2004;279:46969–46980. doi: 10.1074/jbc.M407351200. [DOI] [PubMed] [Google Scholar]
- 15.Owen DJ, Vallis Y, Pearse BM, McMahon HT, Evans PR. The structure and function of the beta 2-adaptin appendage domain. EMBO J. 2000;19:4216–4227. doi: 10.1093/emboj/19.16.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minakami R, Jinnai N, Sugiyama H. Phosphorylation and calmodulin binding of the metabotropic glutamate receptor subtype 5 (mGLUR5) are antagonistic in vitro. J Biol Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima Y, Yamamoto T, Nakayama T, Nakanishi S. A relationship between protein kinase C phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J Biol Chem. 1999;274:27573–27577. doi: 10.1074/jbc.274.39.27573. [DOI] [PubMed] [Google Scholar]
- 18.O'Connor V, El Far O, Bofill-Cardona E, Nanoff C, Freissmuth M, Karschin A, Airas JM, Betz H, Boehm S. Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science. 1999;286:1180–1184. doi: 10.1126/science.286.5442.1180. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Sadee W, Quillan JM. Calmodulin binding to G protein-coupling domain of opioid receptors. J Biol Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- 20.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent β-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 21.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 22.Kakiuchi S, Yasuda S, Yamazaki R, Teshima Y, Kanda K, Kakiuchi R, Sobue K. Quantitative determinations of calmodulin in the supernatant and particulate fractions of mammalian tissues. J Biochem (Tokyo) 1982;92:1041–1048. doi: 10.1093/oxfordjournals.jbchem.a134019. [DOI] [PubMed] [Google Scholar]
- 23.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 Å crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 24.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3 Å: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 25.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 26.Klug CS, Feix JF. Biomedical EPR - Part B: Methodology and Instrumentation. In: Eaton SS, Eaton GR, Berliner LJ, editors. SDSL: A Survey of Biological Applications in Biological Magnetic Resonance. Vol. 24. Kluwer Academic/Plenum Publishers; New York, NY: 2005. pp. 269–308. [Google Scholar]
- 27.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 28.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 29.Hanson SM, Francis DF, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crane JM, Mao C, Lilly AA, Smith VF, Suo Y, Hubbell WL, Randall LL. Mapping of the docking of SecA onto the chaperone SecB by site-directed spin labeling: insight into the mechanism of ligand transfer during protein export. J Mol Biol. 2005;353:295–307. doi: 10.1016/j.jmb.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Hanson SM, Francis DF, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci USA. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 34.Fallon JL, Quiocho FA. A closed compact structure of native Ca(2+)-calmodulin. Structure (Camb) 2003;11:1303–1307. doi: 10.1016/j.str.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Babu YS, Bugg CE, Cook WJ. Structure of calmodulin refined at 2.2 A resolution. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 36.Osawa S, Raman D, Weiss ER. Heterologous expression and reconstitution of rhodopsin with rhodopsin kinase and arrestin. Methods Enzymol. 2000;315:411–422. doi: 10.1016/s0076-6879(00)15858-6. [DOI] [PubMed] [Google Scholar]
- 37.Gurevich VV, Benovic JL. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- 38.Burns ME, Baylor DA. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu Rev Neurosci. 2001;24:779–805. doi: 10.1146/annurev.neuro.24.1.779. [DOI] [PubMed] [Google Scholar]
- 39.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- 40.Schmidlin F, Dery O, Bunnett NW, Grady EF. Heterologous regulation of trafficking and signaling of G protein-coupled receptors: beta-arrestin-dependent interactions between neurokinin receptors. Proc Nat Acad Sci USA. 2002;99:3324–3329. doi: 10.1073/pnas.052161299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welsh MJ, Dedman JR, Brinkley BR, Means AR. Tubulin and calmodulin. Effects of microtubule and microfilament inhibitors on localization in the mitotic apparatus. J Cell Biol. 1979;81:624–634. doi: 10.1083/jcb.81.3.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eckmiller MS. Calmodulin immonolocalization in outer segments of Xenopus laevis photoreceptors. Cell Tissue Res. 2002;308:439–442. doi: 10.1007/s00441-002-0558-3. [DOI] [PubMed] [Google Scholar]
- 43.Fisher DD, Gilroy S, Cyr RJ. Evidence for opposing effects of calmodulin on cortical microtubules. Plant Physiol. 1996:1079–1087. doi: 10.1104/pp.112.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore RC, Durso NA, Cyr RJ. Elongation tactor-1α stabilizes microtubules in a calcium/calmodulin-dependent manner. Cell Motil Cytoskeleton. 1998;41:168–180. doi: 10.1002/(SICI)1097-0169(1998)41:2<168::AID-CM7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 45.Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rebrik TI, Korenbrot JI. In intact mammalian photoreceptors, Ca2+-dependent modulation of cGMP-gated ion channels is detectable in cones but not in rods. J Gen Physiol. 2004;123:63–75. doi: 10.1085/jgp.200308952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their "inactive" conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim SA, Heinze KG, Waxham MN, Schwille P. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc Nat Acad Sci USA. 2004;101:105–110. doi: 10.1073/pnas.2436461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurevich VV, Benovic JL. Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 2000;315:422–37. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- 51.Bertrand B, Wakabayashi S, Ikeda T, Pouyssegur J, Shigekawa M. The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins. Identification and characterization of calmodulin-binding sites. J Biol Chem. 1994;269:13703–9. [PubMed] [Google Scholar]
- 52.Hulen D, Baron A, Salisbury J, Clarke M. Production and specificity of monoclonal antibodies against calmodulin from Dictyostelium discoideum. Cell Motil Cytoskeleton. 1991;18:113–122. doi: 10.1002/cm.970180206. [DOI] [PubMed] [Google Scholar]
- 53.Tsien R, Pozzan T. Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 1989;172:230–262. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- 54.Hamm HE, Bownds MD. Protein complement of rod outer segments of frog retina. Biochemistry. 1986;25:4512–4523. doi: 10.1021/bi00364a010. [DOI] [PubMed] [Google Scholar]