

Abstract
Melittin is a cationic hemolytic peptide isolated from the European honey bee, Apis mellifera. The organization of membrane-bound melittin has earlier been shown to be dependent on the physical state and composition of membranes. In this study, we covalently labeled the N-terminal (Gly-1) and Lys-7 of melittin with an environment-sensitive fluorescent probe, the NBD group, to monitor the influence of negatively charged lipids and cholesterol on the organization and dynamics of membrane-bound melittin. Our results show that the NBD group of melittin labeled at its N-terminal end does not exhibit red edge excitation shift in DOPC and DOPC/DOPG membranes, whereas the NBD group of melittin labeled at Lys-7 exhibits REES of ∼8 nm. This could be attributed to difference in membrane microenvironment experienced by the NBD groups in these analogs. Interestingly, the membrane environment of the NBD groups is sensitive to the presence of cholesterol, which is supported by time-resolved fluorescence measurements. Importantly, the orientation of melittin is found to be parallel to the membrane surface as determined by membrane penetration depth analysis using the parallax method in all cases. Our results constitute the first report to our knowledge describing the orientation of melittin in cholesterol-containing membranes. These results assume significance in the overall context of the role of membrane lipids in the orientation and function of membrane proteins and peptides.
INTRODUCTION
Melittin, the principal toxic component in the venom of the European honey bee, Apis mellifera, is a cationic hemolytic peptide (1). It is a small linear peptide composed of 26 amino acids (NH2-GIGAVLKVLTTGLPALISWIKRKRQQ-CONH2) in which the amino-terminal region (residues 1–20) is predominantly hydrophobic whereas the carboxy-terminal region (residues 21–26) is hydrophilic due to the presence of a stretch of positively charged amino acids. This amphiphilic property of melittin makes it water soluble and yet it spontaneously associates with natural and artificial membranes (2,3). Such a sequence of amino acids, coupled with its amphiphilic nature, is characteristic of many membrane-bound peptides and putative transmembrane helices of membrane proteins (2,4). This has resulted in melittin being used as a convenient model for monitoring lipid-protein interactions in membranes. Apart from its powerful hemolytic activity, melittin causes bilayer micellization and membrane fusion and has also been reported to form voltage-dependent ion channels across planar lipid bilayers (2,5).
Melittin adopts predominantly random coil conformation as a monomer in aqueous solution (6). However, at high ionic strength, pH, or peptide concentration, it self-associates to form an α-helical tetrameric structure driven by the formation of a hydrophobic core (2,6,7). Interestingly, melittin adopts an α-helical conformation when bound to membranes of varying lipid composition (2,8–11) or membrane-mimetic systems (12,13). Despite the availability of a high-resolution crystal structure of tetrameric melittin in aqueous solution (14), the structure of the membrane-bound form is not yet resolved by x-ray crystallography. Yet, the importance of the membrane-bound form stems from the observation that the amphiphilic α-helical conformation of this hemolytic toxin in membranes resembles those of apolipoproteins and peptide hormones (15,16), signal peptides (17), and the envelope glycoprotein gp41 from the human immunodeficiency virus (HIV) (18). Furthermore, understanding melittin-membrane interaction assumes greater significance due to the observation that melittin mimics the N-terminal of HIV-1 virulence factor Nef1-25 (19).
Melittin is intrinsically fluorescent due to the presence of a single tryptophan residue, Trp-19, in the C-terminal region, which has been extensively used as a sensitive probe to obtain information on the interaction of melittin with membranes and membrane-mimetic systems (9–13,20–22). We have previously monitored the microenvironment experienced by the sole tryptophan in membrane-bound melittin and its modulation by anionic phospholipids, cholesterol, and unsaturation utilizing the wavelength-selective fluorescence approach (9–11,23). We showed that the tryptophan residue is located in a motionally restricted interfacial region of the membrane in all cases. However, a disadvantage of utilizing tryptophan fluorescence is that the information essentially comes only from the C-terminal region of melittin, and information about the N-terminal region is lacking. The lack of information about the N-terminal region of melittin makes it difficult to comment about the orientation of melittin in membranes.
In this article, we covalently labeled the amino terminal (Gly-1) and Lys-7 of melittin (see Fig. 1) with an environment-sensitive fluorescent group 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) to obtain information regarding the orientation and dynamics of N-terminal region of membrane-bound melittin using NBD fluorescence. The NBD group is widely used to fluorescently label proteins, peptides, and lipids (24), and has previously been shown by our group (25,26) and others (27) to be a very sensitive probe of the environment in which it resides. Because the sole tryptophan is located at the C-terminal region of melittin, the information obtained from NBD-melittin analogs would provide an additional handle to monitor the conformation and dynamics of melittin bound to membranes of various lipid composition. In addition, this approach would be useful in monitoring the dynamics of the microenvironment of the functionally important Lys-7 residue, which has previously been shown to be crucial for the hemolytic activity of melittin (28). Since the orientation (parallel or perpendicular to the plane of the membrane bilayer) and the lytic activity of melittin have been reported to be dependent on the physical condition and the composition of the membrane in which it is bound (9,10,29–31), we have monitored the effect of negatively charged lipids and the presence of cholesterol on the organization and dynamics of NBD-labeled melittin analogs. Importantly, we show here that NBD labeling does not affect the secondary structure and hemolytic activity of membrane-bound melittin. Overall, our results suggest that the orientation of melittin is parallel to the plane of the membrane surface in all the membrane systems studied, which assume significance in the context of the mechanism of action of melittin.
FIGURE 1.

Chemical structures of the NBD-labeled melittin analogs used.
MATERIALS AND METHODS
Materials
1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC), cholesterol, 3-(N-morpholino)propanesulfonic acid (MOPS), and cobalt chloride were obtained from Sigma Chemical (St. Louis, MO). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG), 1,2-dioleoyl-sn-glycero-3-phosphotempocholine (Tempo-PC), and 1-palmitoyl-2-(5-doxyl)stearoyl-sn-glycero-3-phosphocholine (5-PC) were purchased from Avanti Polar Lipids (Alabaster, AL). The synthetic NBD-labeled melittin analogs were custom synthesized by Mimotopes (Clayton, Australia). Anthroyloxy-labeled fatty acids such as 2-(9-anthroyloxy)stearic acid (2-AS) and 12-(9-anthroyloxy)stearic acid (12-AS) were from Molecular Probes (Eugene, OR). Lipids were checked for purity by thin layer chromatography on silica gel precoated plates (Sigma) in chloroform/methanol/water (65:35:5, v/v/v) and were found to give only one spot in all cases with a phosphate-sensitive spray and on subsequent charring (32). The concentration of DOPC was determined by phosphate assay subsequent to total digestion by perchloric acid (33). DMPC was used as an internal standard to assess lipid digestion. The concentration of melittin in aqueous solution was calculated from the molar extinction coefficient (ɛ) of the tryptophan residue, which is 5570 M−1cm−1 at 280 nm (9). All other chemicals used were of the highest purity available. The solvents used were of spectroscopic grade. Water was purified through a Millipore (Bedford, MA) Milli-Q system and used throughout.
Assay for hemolytic activity
The hemolytic activity of melittin was evaluated using Wistar rat erythrocytes as described previously (31). Erythrocytes were isolated from heparinized blood by centrifugation at 3000 rpm for 5 min and washed three times with HEPES buffer (5 mM HEPES, 150 mM NaCl, pH 7.4). Increasing concentrations of native melittin and NBD-melittin analogs (N-ter and Lys-7) were added to aliquots of 0.5 ml suspension that correspond to ∼107 cells in Eppendorf tubes and were incubated at 37°C in duplicates for 30 min with gentle mixing. The tubes were then centrifuged and the absorbance of the released hemoglobin in the supernatants was measured at 540 nm in a 96-well microtitre plate. The hemolysis obtained with water was taken as 100%. The cell suspension in buffer in the absence of peptide served as background control. All hemolytic assays were carried out on the same day of blood collection.
Sample preparation
All experiments were done using large unilamellar vesicles (LUVs) of 100 nm diameter containing 1 mol % melittin except in vesicles containing 40 mol % cholesterol where 0.5 mol % of melittin was used to ensure complete binding (10). In general, 640 nmol (100 nmol for fluorescence quenching experiments and 2560 nmol for circular dichroism (CD) measurements) of total lipid (DOPC or DOPC/60% DOPG or DOPC/cholesterol) was dried under a stream of nitrogen while being warmed gently (∼35°C). After further drying under a high vacuum for >3 h, 1.5 ml of 10 mM MOPS, 150 mM NaCl, pH 7.2 buffer was added, and the sample was vortexed for 3 min to disperse the lipid and form homogeneous multilamellar vesicles. LUVs of 100 nm diameter were prepared by the extrusion technique using an Avestin Liposofast Extruder (Ottawa, Ontario, Canada) as previously described (34). Briefly, the multilamellar vesicles were freeze-thawed five times using liquid nitrogen to ensure solute equilibration between trapped and bulk solutions, and then extruded through polycarbonate filters (pore diameter of 100 nm) mounted in the extruder fitted with Hamilton syringes (Hamilton, Reno, NV). The samples were subjected to 11 passes through polycarbonate filters to give the final LUV suspension. To incorporate NBD-melittin analogs into membranes, a small aliquot containing 6.4 nmol (1 nmol and 25.6 nmol for quenching and CD measurements, respectively) of the melittin analog was added from a stock solution in water to the preformed vesicles and mixed well. The samples were incubated in dark for 12 h at room temperature (∼23°C) for equilibration before making measurements. Background samples were prepared the same way except that NBD-labeled melittin was not added to them. All experiments were done at room temperature (∼23°C).
Depth measurements using the parallax method
The actual spin (nitroxide) content of the spin-labeled phospholipids (Tempo- and 5-PC) was assayed using fluorescence quenching of anthroyloxy-labeled fatty acids (2- and 12-AS) as described earlier (35). For depth measurements, liposomes were made by the ethanol injection method (36). These samples were made by drying 160 nmol of total lipid (DOPC, or DOPC/60% DOPG, or DOPC/cholesterol) containing 10 mol % spin-labeled phospholipid (Tempo- or 5-PC) under a stream of nitrogen while being warmed gently (∼35°C) followed by further drying under a high vacuum for at least 3 h. The dried lipid film was dissolved in ethanol to give a final concentration of 40 mM. The ethanolic lipid solution was then injected into 10 mM MOPS, 150 mM NaCl, pH 7.2 buffer, while vortexing to give a final concentration of 0.11 mM total lipid in the buffer. NBD-melittin analogs (N-ter and Lys-7) were incorporated into membranes by adding a small aliquot containing 1.6 nmol of peptide from a stock solution in water to the preformed vesicles and mixed well to give membranes containing 1% melittin (0.5% in the case of 40 mol % cholesterol). The lipid composition of these samples were as follows: i), DOPC (90%) and 5- (or 12)-PC (10%); ii), DOPC (30%), DOPG (60%), and Tempo- (or 5)-PC (10%); iii), DOPC (70%), Tempo- (or 5)-PC (10%), and cholesterol (20%); and iv), DOPC (50%), Tempo- (or 5)-PC (10%), and cholesterol (40%). Duplicate samples were prepared in each case except for samples lacking the quencher (Tempo- or 5-PC) where triplicates were prepared. Background samples lacking the fluorophore (NBD-melittin analog) were prepared in all experiments, and their fluorescence intensity was subtracted from the respective sample fluorescence intensity. Samples were kept in the dark for 12 h before measuring fluorescence.
Steady-state fluorescence measurements
Steady-state fluorescence measurements were performed with a Hitachi F-4010 spectrofluorometer using 1 cm pathlength quartz cuvettes. Excitation and emission slits with a nominal bandpass of 5 nm were used for all measurements. Background intensities of samples in which melittin was omitted were subtracted from each sample spectrum to cancel out any contribution due to the solvent Raman peak and other scattering artifacts. The spectral shifts obtained with different sets of samples were identical in most cases. In other cases, the values were within ±1 nm of the ones reported. Fluorescence polarization measurements were performed at room temperature (∼23°C) using a Hitachi polarization accessory. Polarization values were calculated from the equation (37):
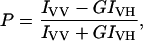 |
(1) |
where IVV and IVH are the measured fluorescence intensities (after appropriate background subtraction) with the excitation polarizer vertically oriented and emission polarizer vertically and horizontally oriented, respectively. G is the grating correction factor and is the ratio of the efficiencies of the detection system for vertically and horizontally polarized light, and is equal to IHV / IHH. All experiments were done with multiple sets of samples and average values of polarization are shown in Table 3 and Fig. 6.
TABLE 3.
Fluorescence polarization of NBD-melittin analogs in membranes of varying lipid composition
| Fluorescence polarization
|
Apparent rotational correlation time (τc)* (ns)
|
|||
|---|---|---|---|---|
| Membrane | N-ter | Lys-7 | N-ter | Lys-7 |
| DOPC | 0.278 ± 0.005 | 0.296 ± 0.003 | 13.0 | 10.9 |
| DOPC/60% DOPG | 0.213 ± 0.004 | 0.246 ± 0.001 | 6.1 | 6.4 |
| DOPC/20% chol | 0.302 ± 0.004 | 0.294 ± 0.002 | 13.4 | 9.5 |
The excitation wavelength was 465 nm; emission was monitored at 525 and 530 nm for N-ter and Lys-7 NBD-melittin analogs, respectively. The polarization values represent mean ± SE of at least three independent measurements. All other conditions are as in Table 1. See Materials and Methods for other details.
FIGURE 6.

Fluorescence polarization of (A) N-ter and (B) Lys-7 NBD-melittin analogs bound to vesicles containing DOPC (○), DOPC/60%DOPG (▵), and DOPC/20%cholesterol (▪) as a function of excitation (left panel) and emission (right panel) wavelengths. The polarization values were recorded at an emission wavelength of 530 nm (left panel), and the excitation wavelength used was 465 nm (right panel). The data points shown are the means of three independent measurements. The lines joining the data points are provided merely as viewing guides. All other conditions are as in Fig. 4. See Materials and Methods for other details.
For depth measurements, samples were excited at 465 nm and emission was collected at 530 nm. Excitation and emission slits with a nominal bandpass of 5 nm were used. Fluorescence was measured at room temperature and averaged over two 5-s readings. Intensities were found to be stable over time. In all cases, the intensity from background samples without fluorophore (NBD-melittin) was subtracted. Membrane penetration depths were calculated using Eq. 8 (see Results).
Time-resolved fluorescence measurements
Fluorescence lifetimes were calculated from time-resolved fluorescence intensity decays using a Photon Technology International (London, Western Ontario, Canada) LS-100 luminescence spectrophotometer in the time-correlated single-photon counting mode. This machine uses a thyratron-gated nanosecond flash lamp filled with nitrogen as the plasma gas (16 ± 1 inches of mercury vacuum) and is run at 18–20 kHz. Lamp profiles were measured at the excitation wavelength using Ludox (colloidal silica) as the scatterer. To optimize the signal/noise ratio, 10,000 photon counts were collected in the peak channel. All experiments were performed using excitation and emission slits with a bandpass of 6 nm or less. The sample and the scatterer were alternated after every 5% acquisition to ensure compensation for shape and timing drifts occurring during the period of data collection. This arrangement also prevents any prolonged exposure of the sample to the excitation beam thereby avoiding any possible photodamage of the fluorophore. The data stored in a multichannel analyzer were routinely transferred to an IBM PC for analysis. Fluorescence intensity decay curves so obtained were deconvoluted with the instrument response function and analyzed as a sum of exponential terms:
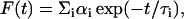 |
(2) |
where F(t) is the fluorescence intensity at time t and αi is a preexponential factor representing the fractional contribution to the time-resolved decay of the component with a lifetime τi such that Σi αi = 1. The decay parameters were recovered as described previously (12). Mean (average) lifetimes 〈τ〉 for biexponential decays of fluorescence were calculated from the decay times and preexponential factors using the following equation (37):
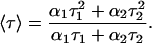 |
(3) |
Fluorescence quenching measurements
Cobalt quenching experiments of NBD fluorescence in membranes of varying lipid composition were carried out by measurement of fluorescence intensity of melittin analogs after serial addition of small aliquots of a freshly prepared stock solution of 0.4 M cobalt chloride in water to a stirred sample followed by incubation for 2 min in the sample compartment in the dark (shutters closed). The excitation wavelength used was 465 nm and emission was monitored at 530 nm. The fluorescence intensities were corrected for dilution. Corrections for inner filter effect were made using the following equation (37):
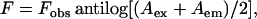 |
(4) |
where F is the corrected fluorescence intensity and Fobs is the background subtracted fluorescence intensity of the sample. Aex and Aem are the measured absorbance at the excitation and emission wavelengths. The absorbance of the samples was measured using a Hitachi U-2000 UV-visible absorption spectrophotometer. Quenching data were analyzed by fitting to the Stern-Volmer equation (37):
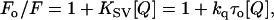 |
(5) |
where Fo and F are the fluorescence intensities in the absence and presence of the quencher, respectively, [Q] is the molar quencher concentration, and KSV is the Stern-Volmer quenching constant. The Stern-Volmer quenching constant KSV is equal to kqτo, where kq is the bimolecular quenching constant and τo is the lifetime of the fluorophore in the absence of quencher.
Circular dichroism measurements
CD measurements were carried out at room temperature (∼23°C) on a JASCO J-715 spectropolarimeter that was calibrated with (+)−10-camphorsulfonic acid. The spectra were scanned in a quartz optical cell with a pathlength of 0.1 cm. All spectra were recorded in 0.2 nm wavelength increments with a 4-s response and a bandwidth of 1 nm. For monitoring changes in secondary structure, spectra were scanned in the far-UV range from 205 to 250 nm at a scan rate of 50 nm/min. Each spectrum is the average of 15 scans with a full-scale sensitivity of 50 mdeg. All spectra were corrected for background by subtraction of appropriate blanks and were smoothed making sure that the overall shape of the spectrum remains unaltered. Data are represented as molar ellipticities and were calculated using the equation:
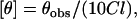 |
(6) |
where θobs is the observed ellipticity in mdeg, l is the pathlength in cm, and C is the concentration of melittin analog in mol/L.
RESULTS
Characterization of NBD-melittin analogs
To confirm that labeling of melittin at the amino terminal (Gly-1) and at Lys-7 with the NBD group did not affect the lytic activity of melittin, we carried out hemolytic assay of these analogs using rat erythrocytes. Fig. 2 shows the extent of hemolysis in rat erythrocytes as a function of increasing concentration of melittin (control) and its NBD analogs. As shown in the figure, increasing concentration of peptides drastically increases the release of hemoglobin, which is indicative of cell lysis. This clearly shows that NBD-melittin analogs are functionally similar to native melittin. This result confirms that NBD labeling of melittin does not affect the lytic activity of melittin significantly.
FIGURE 2.

Hemolysis of rat erythrocytes as a function of increasing concentration of peptides corresponding to native melittin (○), N-ter (•), and Lys-7 (□) NBD-melittin analogs. The hemolytic assay was carried out at room temperature (23°C). The data points shown are the mean ± SE of at least three independent measurements. Data for native melittin are taken from Raghuraman and Chattopadhyay (31). See Materials and Methods for other details.
It is well established that monomeric melittin in aqueous solution shows essentially random coil conformation as reported earlier (9,10). On the other hand, membrane-bound melittin shows a CD spectrum that is characteristic of an α-helical conformation (8–11,29,38). To examine the secondary structural characteristics of NBD-melittin analogs, we carried out far-UV CD spectroscopy of these analogs in membranes of varying lipid composition. The CD spectra of NBD-melittin analogs in water and when bound to vesicles containing DOPC and DOPC/20%cholesterol are shown in Fig. 3. These results show that the secondary structure of membrane-bound melittin (α-helical) does not appear to be sensitive to NBD labeling. Taken together, these results suggest that NBD-melittin analogs are structurally and functionally similar to native melittin and therefore could be used to monitor the organization and dynamics of the N-terminal region of melittin in membranes.
FIGURE 3.

Representative far-UV CD spectra of (A) N-ter and (B) Lys-7 NBD-melittin analogs in water (short-dashed line) and bound to vesicles containing DOPC (long-dashed line) and DOPC/20% cholesterol (solid line). The ratio of melittin/lipid was 1:100 (mol/mol) and the concentration of melittin was 17 μM in all cases. See Materials and Methods for other details.
Red edge excitation shift of membrane-bound NBD-melittin analogs
Red edge excitation shift (REES) represents a powerful approach that can be used to directly monitor the environment and dynamics around a fluorophore in a complex biological system (39–41). A shift in the wavelength of maximum fluorescence emission toward higher wavelengths, caused by a shift in the excitation wavelength toward the red edge of the absorption band, is termed REES. REES arises from slow rates of solvent relaxation (reorientation) around an excited state fluorophore that depends on the motional restriction imposed on the solvent molecules in the immediate vicinity of the fluorophore. Utilizing this approach, it becomes possible to probe the mobility parameters of the environment itself (which is represented by the relaxing solvent molecules) using the fluorophore merely as a reporter group. The unique feature of REES is that whereas all other fluorescence techniques (such as fluorescence quenching, energy transfer, and polarization measurements) yield information about the fluorophore (either intrinsic or extrinsic) itself, REES provides information about the relative rates of solvent (water in biological systems) relaxation dynamics that is not possible to obtain by other techniques. This makes REES extremely useful because hydration plays a crucial modulatory role in a large number of important cellular events including protein folding, lipid-protein interactions, and ion transport (42). We have previously shown that REES serves as a powerful tool to monitor the organization and dynamics of fluorescent probes and peptides/proteins bound to membranes and membrane mimetic media such as micelles and reverse micelles (39,40,43–47).
The fluorescence emission maximum of N-ter and Lys-7 NBD-melittin analogs bound to membranes of varying lipid composition is ∼525 and 530–533 nm, respectively, when excited at 465 nm (see Fig. 4). The shifts in the maxima of fluorescence emission of the NBD group of NBD-melittin analogs when bound to vesicles containing DOPC, DOPC/60% DOPG, and DOPC/20% cholesterol as a function of excitation wavelength are shown in Fig. 4. The center of mass of emission has been reported as the fluorescence maximum. As the excitation wavelength is changed from 465 to 510 nm, the emission maximum of membrane-bound melittin (N-ter) is invariant in zwitterionic and anionic membranes. However, a slight red shift in emission maximum is observed in cholesterol-containing membranes when excitation is carried out at 510 nm (see Fig. 4 A). In contrast, the emission maxima of Lys-7 NBD-melittin are shifted from 531 to 538 nm (in DOPC), 530–538 nm (DOPC/60%DOPG), and 533–543 nm (DOPC/20% cholesterol) when the excitation wavelength is changed from 465 to 520 nm (see Fig. 4 B). The resultant REES in these cases are shown in Table 1. Such dependence of the emission maximum on excitation wavelength is characteristic of REES. This implies that the NBD group of Lys-7 NBD melittin is localized in a motionally restricted region of the membrane in all these cases. This is consistent with the interfacial localization of the melittin when bound to membranes (9–11,23,48). The membrane interface is characterized by unique motional and dielectric characteristics distinct from both the bulk aqueous phase and the more isotropic hydrocarbon-like deeper regions of the membrane (39,49). This specific region of the membrane exhibits slow rates of solvent relaxation and is also known to participate in intermolecular charge interactions and hydrogen bonding through the polar headgroup. These structural features that slow down the rate of solvent reorientation have previously been recognized as typical features of microenvironments giving rise to significant REES effects. It is therefore the membrane interface that is most likely to display red edge effects (39).
FIGURE 4.

Effect of changing excitation wavelength on the wavelength of maximum emission of (A) N-ter and (B) Lys-7 NBD-melittin analogs bound to vesicles containing DOPC (○), DOPC/60%DOPG (▵), and DOPC/20%cholesterol (▪). The lines joining the data points are provided merely as viewing guides. The ratio of melittin/lipid was 1:100 (mol/mol) and the concentration of melittin was 4.27 μM in all cases. See Materials and Methods for other details.
TABLE 1.
Red edge excitation shift of NBD-melittin analogs in membranes of varying lipid composition
| REES (nm)
|
||
|---|---|---|
| Membrane* | N-ter | Lys-7 |
| DOPC | 0 | 7 |
| DOPC/60% DOPG | 0 | 8 |
| DOPC/20% chol | 2 | 10 |
| DOPC/40% chol | 2 | 10 |
The ratio of melittin/total lipid was 1:100 (mol/mol) and the concentration of melittin analogs was 4.27 μM in all cases except in vesicles containing 40 mol % cholesterol in which the ratio of melittin/total lipid was 1:200. See Materials and Methods for other details.
These results suggest that there is a considerable difference in the relaxation of solvent molecules (dynamics of hydration) in different positions of the N-terminal region of membrane-bound melittin. These results could be due to ground state heterogeneity, if any, of the N-ter melittin analog. However, this is ruled out because the N-ter melittin analog is predominantly membrane-bound, i.e., there is no ground state heterogeneity. This is supported by the emission maximum of 525 nm in membranes (compared to emission maximum of ∼542 nm in water), the secondary structural features (Fig. 3), and functional evidence (Fig. 2). We therefore attribute this result to flexible motion of amino terminal region of membrane-bound melittin. This is consistent with previous observations that the first few residues in the N-terminal region of membrane-bound melittin do not adopt helical conformation (50) leading to a higher tilt angle of N-terminal region (51), which in turn could influence the motional restriction of solvent molecules.
Time-resolved fluorescence measurements of membrane-bound melittin
Fluorescence lifetime serves as a faithful indicator of the local environment in which a given fluorophore is placed (52). In addition, it is well known that fluorescence lifetime of the NBD group in particular is sensitive to its local environment (53). A typical decay profile of N-ter NBD melittin bound to DOPC membranes with its biexponential fitting and the statistical parameters used to check the goodness of the fit is shown in Fig. 5. The fluorescence lifetimes of NBD-melittin analogs bound to various membrane systems are shown in Table 2. As seen from the table, all fluorescence decays could be fitted well with a biexponential function. The mean fluorescence lifetime obtained for the membrane-bound N-ter and Lys-7 NBD-melittin analogs in DOPC membranes is 9.49 and 6.93 ns, respectively. Interestingly, upon incorporation of negatively charged lipid and cholesterol, the mean fluorescence lifetimes of NBD-melittin analogs decrease significantly. This indicates a marked increase in polarity around the excited state NBD group of melittin analogs in these membrane systems. In general, it is known that the presence of negatively charged lipid and cholesterol influences the hydration profile of membranes (9,54,55). The fluorescence lifetime of NBD-melittin in pure water reduces to ∼2.0 ns (not shown). This is consistent with previous observations in which it was reported that the fluorescence lifetime of the NBD group is reduced in presence of water (25,26,53). The change in polarity around the NBD group of membrane-bound NBD-melittin analogs could therefore be attributed to increased water penetration in the interfacial region of membranes containing negatively charged lipid and cholesterol.
FIGURE 5.

Time-resolved fluorescence intensity decay of N-ter NBD melittin analog bound to DOPC vesicles. Excitation wavelength was at 465 nm, and emission was monitored at 530 nm. The sharp peak on the left is the lamp profile. The relatively broad peak on the right is the decay profile, fitted to a biexponential function. The two lower plots show the weighted residuals and the autocorrelation function of the weighted residuals. All other conditions are as in Fig. 4. See Materials and Methods for other details.
TABLE 2.
Fluorescence lifetimes of NBD-melittin analogs in membranes of varying lipid composition
| N-ter NBD-melittin*
| |||||
|---|---|---|---|---|---|
| Membrane | α1 | τ1 (ns) | α2 | τ2 (ns) | 〈τ〉 (ns) |
| DOPC | 0.26 | 4.69 | 0.74 | 10.26 | 9.49 (±0.03) |
| DOPC/60% DOPG | 0.68 | 5.54 | 0.32 | 10.73 | 8.01 (±0.11) |
| DOPC/20% chol | 0.67 | 5.82 | 0.33 | 10.15 | 7.82 (±0.24) |
| DOPC/40% chol | 0.69 | 5.70 | 0.31 | 10.06 | 7.63 (±0.21) |
| Lys-7 NBD-melittin
| |||||
| Membrane | α1 | τ1 (ns) | α2 | τ2 (ns) | 〈τ〉 (ns) |
| DOPC | 0.59 | 4.02 | 0.41 | 8.83 | 6.93 (±0.06) |
| DOPC/60% DOPG | 0.74 | 3.93 | 0.26 | 9.25 | 6.34 (±0.02) |
| DOPC/20% chol | 0.74 | 3.88 | 0.26 | 8.70 | 6.00 (±0.08) |
| DOPC/40% chol | 0.73 | 3.50 | 0.27 | 8.19 | 5.68 (±0.03) |
The excitation wavelength was 465 nm; emission was monitored at 525 and 530 nm for N-ter and Lys-7 NBD-melittin analogs, respectively. The values in parentheses represent the mean ± SE of at least three independent measurements. The number of photons collected at the peak channel was 10,000. See Table 1 and Materials and Methods for other details.
Fluorescence polarization of membrane-bound melittin
The steady-state fluorescence polarization of NBD-melittin analogs in various membranes is shown in Table 3. These values in general indicate that the rotational mobility of NBD group of NBD-melittin analogs is considerably restricted and are representative of motionally restricted NBD environments. The fluorescence polarization of NBD-melittin analogs in membranes containing negatively charged lipids is found to be significantly lower when compared to polarization obtained in zwitterionic and cholesterol-containing membranes. The change in fluorescence polarization of NBD-melittin analogs in various membranes could be attributed to possible change in headgroup packing in these membranes.
To ensure that the polarization values measured for membrane-bound melittin are not influenced by lifetime-induced artifacts, the apparent (average) rotational correlation times were calculated using Perrin's equation (37):
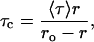 |
(7) |
where ro is the limiting anisotropy of tryptophan, r is the steady-state anisotropy (derived from the polarization values using r = 2P/(3-P)), and 〈τ〉 is the mean fluorescence lifetime taken from Table 2. Although Perrin's equation is not strictly applicable to this system, it is assumed that this equation will apply to a first-hand approximation, especially because we have used mean fluorescence lifetimes for the analysis of multiple component lifetimes. The values of the apparent rotational correlation times, calculated this way using a value of ro of 0.354 (56), are shown in Table 3. As is evident from the table, the apparent rotational correlation times of NBD-melittin analogs are considerably reduced in anionic membranes when compared to membranes containing zwitterionic lipid and cholesterol. This suggests increased rotational mobility experienced by NBD-melittin analogs in anionic membrane environment, in agreement with polarization values. This shows that the observed changes in polarization values are more or less free from lifetime-induced artifacts.
Fluorescence polarization is also known to be dependent on excitation and emission wavelengths in motionally restricted media (57). We measured polarization changes of NBD-melittin analogs bound to zwitterionic, anionic, and cholesterol-containing membranes as a function of excitation and emission wavelengths (see Fig. 6). Our results show that membrane-bound NBD-melittin analogs exhibit wavelength-dependent changes in polarization for both excitation and emission wavelengths. This reinforces our previous conclusion that melittin is localized in a motionally restricted interfacial region of the membrane in these cases (see results of depth analysis below).
Cobalt quenching of NBD fluorescence
The above results show that the presence of negatively charged lipid and cholesterol in DOPC membranes enhances the polarity of the interfacial region of membranes due to increased water penetration. We performed fluorescence quenching experiments using the aqueous quencher Co2+ to explore this issue further and to examine the accessibility and relative location of membrane-bound NBD-melittin analogs in the presence and absence of cholesterol. The paramagnetic cobaltous ion is soluble in water and is an efficient quencher of NBD fluorescence (43,58–60). We excluded anionic membranes from cobalt quenching measurements because the results could be complicated due to specific interaction of cobaltous ion with the negatively charged lipid headgroups. Representative results for quenching of the NBD group of NBD-melittin analogs bound to DOPC membranes by Co2+ are shown in Fig. 7 as Stern-Volmer plots. The slope of such a plot (KSV) is related to the degree of exposure (accessibility) of the NBD group to the aqueous phase. In general, the higher the slope, the greater the degree of exposure, assuming that the difference in fluorescence lifetime is not large. The quenching parameters (KSV) obtained by analyzing the Stern-Volmer plots are shown in Table 4. Interestingly, incorporation of cholesterol in DOPC membranes leads to an apparent increase in the accessibility of the NBD group to Co2+ as evident from a significant increase in KSV (see Table 4). This suggests that the NBD group of membrane-bound NBD melittin analogs is more exposed, which could be due to increased water penetration in the membrane interfacial region. Interestingly, KSV values show that the Lys-7 NBD melittin is more exposed to aqueous phase when compared to N-ter NBD melittin analog irrespective of the presence or absence of cholesterol. However, interpretation of KSV values is complicated this way due to its intrinsic dependence on fluorescence lifetime (see Eq. 5). The bimolecular quenching constant (kq) is a more accurate measure of the degree of exposure because it takes into account the differences in fluorescence lifetime. The kq values, calculated using mean fluorescence lifetimes from Table 2 and Eq. 5, are shown in Table 4. It is encouraging to note that the kq values are also increased in the presence of cholesterol, which is in overall agreement with KSV values. This reinforces the idea that there is an increased water penetration in the interfacial region of membranes where the NBD group of NBD-melittin analogs is localized in the presence of cholesterol (also see Table 2).
FIGURE 7.

Representative data for Stern-Volmer analysis of Co2+ quenching of the N-ter (○) and Lys-7 (•) NBD-melittin analogs bound to DOPC vesicles. Fo is the fluorescence in the absence of quencher, F is the corrected fluorescence in the presence of quencher. The excitation wavelength was 465 nm and emission was monitored at 530 nm. See Materials and Methods and Table 4 for other details.
TABLE 4.
Cobalt quenching of NBD fluorescence of membrane-bound NBD-melittin analogs
|
KSV† (M−1)
|
kq (× 10−9)‡ (M−1s−1)
|
|||
|---|---|---|---|---|
| Membrane* | N-ter | Lys-7 | N-ter | Lys-7 |
| DOPC | 45.3 ± 3.4 | 73.9 ± 2.8 | 4.8 | 11.0 |
| DOPC/20% chol | 52.4 ± 1.4 | 115.1 ± 4.6 | 6.7 | 19.2 |
| DOPC/40% chol | 68.5 ± 5.8 | 161.4 ± 2.3 | 9.0 | 28.3 |
The ratio of melittin/total lipid was 1:100 (mol/mol) and the concentration of melittin analogs was 0.67 μM in all cases except in 40 mol % cholesterol sample in which the ratio of melittin/total lipid was 1:200. See Materials and Methods for other details.
Calculated using Eq. 5. The quenching parameter represents mean ± SE of at least three independent measurements. See Materials and Methods for other details.
Membrane penetration depth of membrane-bound NBD-melittin
Membrane penetration depth represents an important parameter in the study of membrane structure and organization (61,62). Knowledge of the precise depth of a membrane embedded group or molecule often helps define the conformation and topology of membrane probes and proteins. In addition, properties such as polarity, fluidity, segmental motion, ability to form hydrogen bonds, and the extent of solvent penetration are known to vary in a depth-dependent manner in the membrane. To gain a better understanding of the organization and conformation of the N-terminal region of membrane-bound melittin, penetration depths of the NBD group of NBD-melittin analogs in membranes of varying composition were determined. Depth of the NBD group was calculated by the parallax method (63) using the equation:
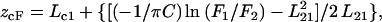 |
(8) |
where zcF = the depth of the fluorophore from the center of the bilayer, Lc1 = the distance of the center of the bilayer from the shallow quencher (5-PC in this case), L21 = the difference in depth between the two quenchers (i.e., the transverse distance between the shallow and the deep quencher), and C = the two-dimensional quencher concentration in the plane of the membrane (molecules/Å2). Here F1/F2 is the ratio of F1/Fo and F2/Fo in which F1 and F2 are fluorescence intensities in the presence of the shallow (5-PC) and deep quencher (Tempo-PC), respectively, both at the same quencher concentration C; Fo is the fluorescence intensity in the absence of any quencher. All the bilayer parameters used were the same as described previously (63). The depths of penetration of the NBD group of NBD-melittin analogs bound to zwitterionic, anionic, and cholesterol-containing membranes are shown in Table 5. The depth of penetration of the tryptophan residue of native melittin in these membranes is provided in the table for comparison. Our results show that the depth of penetration of the NBD group of both N-ter and Lys-7 NBD-melittin analogs is found to be, on an average, ∼18–21 Å from the center of the bilayer in all membranes studied. This clearly suggests that Gly-1 and Lys-7 residues of melittin are located at the relatively shallow interfacial region of the membrane (near headgroups) in all cases. We have previously shown that the tryptophan residue (Trp-19) of native melittin is located at the deeper interfacial region (near the carbonyl group, 10.6 Å from the center of the bilayer) in DOPC membranes (9,10). In addition, we showed that the depth of penetration of Trp-19 of native melittin is not sensitive to the presence of negatively charged lipid in membranes (see Table 5 and Ghosh et al. (9)), whereas the penetration depth of Trp-19 of melittin has been found to be extremely sensitive to the presence of membrane cholesterol (see Table 5 and Raghuraman and Chattopadhyay (10)). Taken together, these results suggest that the depth of penetration of the C-terminal region of membrane-bound melittin (sensed by Trp-19) is sensitive to the presence of membrane cholesterol when compared to the N-terminal region of melittin (sensed by the NBD group). In a control experiment, we determined the depth of penetration of Trp-19 of NBD-melittin analogs bound to DOPC vesicles to be ∼11 Å from the center of the bilayer (not shown), which is similar to that found for the tryptophan residue of native melittin bound to DOPC membranes (see Table 5). This shows that NBD labeling does not affect the membrane penetration ability of the C-terminal region of melittin, and validates our NBD labeling approach to monitor the organization and dynamics of membrane-bound melittin.
TABLE 5.
Penetration depth of the NBD group in membrane-bound NBD-melittin analogs by the parallax method
| Distance from the center of the bilayer, zcF (Å)*
|
|||
|---|---|---|---|
| Membrane | N-ter | Lys-7 | Trp-19 |
| DOPC | 17.9 ± 0.3 | 19.4 ± 0.6 | 10.6‡ |
| DOPC/60%DOPG | 19.2 ± 0.5 | 20.9 ± 0.7 | 10.3‡ |
| DOPC/20% chol† | 20.1 ± 0.6 | 18.8 ± 0.7 | 17.4§ |
Depths were calculated from fluorescence quenchings obtained with samples containing 10 mol % of Tempo-PC and 5-PC and using Eq. 8. Samples were excited at 465 nm, and emission was collected at 530 nm. The depth values represent mean ± SE of at least three independent measurements. The ratio of melittin/total lipid was 1:100 (mol/mol) and the concentration of melittin was 1.07 μM in all cases. See Materials and Methods for other details.
Corrections were made for the altered concentrations of spin-labeled lipids (for lateral distribution) and the depths of the quenchers used in membranes containing cholesterol (64).
From Ghosh et al. (9).
From Raghuraman and Chattopadhyay (10).
In membranes containing cholesterol, the (lateral) concentration of spin-labeled phospholipids and their depths in the membrane are altered. These values were calculated as described previously (64). It should be mentioned here that the phase properties of Tempo- and 5-PC are such that no phase separation is expected to be induced when these spin-labeled phospholipids are used in such small concentrations (10 mol %) with DOPC at room temperature (65,66). Another concern arises from the possibility of partitioning of the spin-labeled phospholipids into cholesterol-rich or -poor regions and its effect on penetration depth. However, this appears to be not a serious concern because it has been previously shown that in the case of a spin-labeled phospholipid (12-PC) incorporated in a mixture of DOPC and cholesterol, the membrane behaves as a single-phase mixture that rules out any specific partitioning of spin-labeled phospholipids into cholesterol-rich or cholesterol-poor domains (66).
DISCUSSION
Melittin is one of the most extensively studied amphipathic, membrane-lytic peptide to understand lipid-protein interactions in membranes. Despite a number of studies addressed to determine the orientation (parallel or perpendicular) of membrane-bound melittin with respect to the plane of the membrane bilayer (2,29,48,50,51,67–75), there is no consensus regarding the orientation of melittin in membranes (see Fig. 8). This is because the orientation of melittin in membranes is critically dependent on the peptide concentration, membrane lipid composition, and membrane properties, which include hydration and the phase state of the lipid (29). For example, it has been shown that the orientation of membrane-bound melittin depends on temperature, and melittin adopts perpendicular orientation in fluid phase DMPC (29,67), DLPC, and DTPC membranes (29). In contrast, melittin orients parallel to the membrane surface in DOPC (48) and DPhPC (29) membranes, in which there is very little dependence on temperature. Interestingly, the orientation of melittin in POPC membranes is complicated since it assumes parallel or perpendicular orientation depending upon the peptide concentration, though these membranes are in the fluid phase (29). In addition, the orientation of membrane-bound melittin is further complicated by the observation that melittin does not adopt a fully transmembrane orientation in membranes, but a pseudo-transmembrane orientation (50,51,73). Overall, these studies clearly point out the role of lipid composition and physical factors that affect the membrane properties, which in turn could influence the orientation of membrane-bound melittin. Interestingly, information about the orientation of membrane-bound melittin in mixed bilayer systems is lacking. We therefore monitored the influence of negatively charged lipids and cholesterol on the orientation of membrane-bound melittin using the site-specific fluorescence of NBD and the parallax method.
FIGURE 8.

A schematic representation of the membrane bilayer showing the various orientations and locations of membrane-bound melittin: (A) surface (2,29,48,68,70,72,74), (B) transmembrane (2,29,67,69,71,75), and (C) pseudo-transmembrane (50,51,73) orientations. The V-shaped bent rod represents the amphipathic, α-helical membrane-bound melittin. Our results using NBD fluorescence of NBD-melittin analogs support the surface (parallel) orientation (A) of membrane-bound melittin. Such a surface (parallel) orientation of melittin should be applicable to zwitterionic, anionic, and cholesterol-containing membranes under the experimental conditions used. See text for other details.
We have previously characterized the interaction of melittin with membranes containing anionic lipids (9) and cholesterol (10) utilizing the fluorescence of the sole tryptophan residue of melittin. We showed that anionic lipids and cholesterol not only affect the organization and dynamics of melittin bound to model membranes, but also inhibit the lytic activity of melittin (9,10). In these studies, the parallax method (63) was employed to monitor the location of the tryptophan residue of membrane-bound melittin. It was found that the presence of anionic lipids in membranes did not affect the membrane penetration ability of melittin (9). However, the presence of cholesterol in membranes was found to prevent the penetration of melittin in membranes (10) since the tryptophan residue was found to be localized at a relatively shallow interfacial region in membranes containing cholesterol (see Table 5).
In this article, we have monitored the organization and dynamics of NBD-melittin analogs bound to zwitterionic, anionic, and cholesterol-containing membranes to obtain information about the location and dynamics of the N-terminal region of membrane-bound melittin. This was achieved by covalently labeling the N-terminal end and Lys-7 of melittin with the environment-sensitive fluorescent NBD group, and monitoring the influence of negatively charged lipids and cholesterol on the organization and dynamics of these NBD-melittin analogs. Importantly, our results show that NBD labeling does not affect the structure and hemolytic activity of melittin (Figs. 2 and 3). Because NBD-melittin analogs are structurally and functionally similar to native melittin, these could be potentially useful in microscopic studies.
Fluorescence characteristics such as emission maximum, REES, polarization, lifetime, and quenching exhibit considerable differences between the NBD-melittin analogs in all the membrane systems studied. Interestingly, the NBD group of melittin labeled at its N-terminal end does not exhibit REES in zwitterionic and anionic membranes, whereas the NBD group of melittin labeled at Lys-7 exhibits considerable REES of ∼8 nm. This could be attributed to difference in membrane microenvironment experienced by the NBD groups in these analogs. Further, the polarity of NBD-melittin analogs is sensitive to the presence of negatively charged lipid and cholesterol, which is supported by time-resolved fluorescence measurements. In addition, cobalt quenching measurements show an increased accessibility of Lys-7 from the aqueous phase compared to the N-ter melittin analog. Based on the fluorescence properties of these two analogs, the NBD group in the N-ter NBD melittin analog appears to be located deeper in the membrane with respect to the Lys-7 analog. Importantly, analysis of membrane penetration depth shows that the NBD group of NBD-melittin analogs is located at a relatively shallow interfacial region in membranes of varying lipid composition.
The natural target membrane for the action of melittin is the erythrocyte membrane, which contains high amounts (∼45 mol %) of cholesterol (76). Recently, we have shown that cholesterol inhibits the lytic activity of melittin in its natural target membrane, i.e., the erythrocyte membrane (31). This study suggested that, though pore formation could be the mode of action of melittin, membrane destabilization induced by melittin cannot be ruled out as the mechanism of action of melittin. In this context, our membrane penetration depth results of NBD-melittin analogs in zwitterionic, anionic, and cholesterol-containing membranes assume significance. Comparison of the membrane penetration depths of Trp-19, Gly-1, and Lys-7 residues of melittin in membranes of varying lipid composition (see Table 5) clearly suggests that melittin may adopt parallel (or pseudo-parallel) orientation with respect to the plane of lipid bilayer even in membranes containing negatively charged lipids or cholesterol. Our results are in agreement with previous reports that show that membrane-bound melittin orients parallel to the membrane plane (2,29,48,68,70,72,74), and with the hypothesis that transmembrane orientation may not be an absolute requirement for the lytic activity of cationic, amphipathic peptides (77). Importantly, it appears that the C-terminal region of melittin (where Trp-19 is present) is more sensitive to the presence of high amounts of cholesterol in membranes (see Table 5). To the best of our knowledge, this is the first report that gives an insight about the orientation of melittin in cholesterol-containing membranes. In summary, our results are relevant in understanding the orientation of melittin in its natural target membrane (i.e., erythrocyte membrane), which in turn could significantly contribute to the understanding of the mechanism of action of melittin.
Acknowledgments
We thank Y. S. S. V. Prasad and G. G. Kingi for technical help and members of our laboratory for critically reading the manuscript.
This work was supported by the Council of Scientific and Industrial Research, Government of India. H.R. thanks the Council of Scientific and Industrial Research, Government of India, for the award of a Research Associateship. A.C. is an Honorary Professor of the Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore (India).
Abbreviations used: 2-AS, 2-(9-anthroyloxy)stearic acid; 12-AS, 12-(9-anthroyloxy)stearic acid; 5-PC, 1-palmitoyl-2-(5-doxyl)stearoyl-sn-glycero-3-phosphocholine; 12-PC, 1-palmitoyl-2-(12-doxyl)stearoyl-sn-glycero-3-phosphocholine; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; DOPG, 1,2-dioleoyl-sn-glycero-3-phosphoglycerol; DPhPC, 1,2-diphytanoyl-sn-glycero-3-phosphocholine; DLPC, 1,2-dilauroyl-sn-glycero-3-phosphocholine; DTPC, 1,2-ditetradecyl-sn-glycero-3-phosphocholine; HEPES, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonicacid; LUV, large unilamellar vesicle; MOPS, 3-(N-morpholino)propanesulfonic acid; NBD, 7-nitrobenz-2-oxa-1,3-diazol-4-yl; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; REES, red edge excitation shift; Tempo-PC, 1,2-dioleoyl-sn-glycero-3-phosphotempocholine.
References
- 1.Habermann, E. 1972. Bee and wasp venoms. Science. 177:314–322. [DOI] [PubMed] [Google Scholar]
- 2.Dempsey, C. E. 1990. The actions of melittin on membranes. Biochim. Biophys. Acta. 1031:143–161. [DOI] [PubMed] [Google Scholar]
- 3.Saberwal, G., and R. Nagaraj. 1994. Cell-lytic and antibacterial peptides that act by perturbing the barrier function of membranes: facets of their conformational features, structure-function correlation and membrane-perturbing abilities. Biochim. Biophys. Acta. 1197:109–131. [DOI] [PubMed] [Google Scholar]
- 4.Shai, Y. 1995. Molecular recognition between membrane-spanning polypeptides. Trends Biochem. Sci. 20:460–464. [DOI] [PubMed] [Google Scholar]
- 5.Bechinger, B. 1997. Structure and functions of channel-forming peptides: magainins, cecropins, melittin and alamethicin. J. Membr. Biol. 156:197–211. [DOI] [PubMed] [Google Scholar]
- 6.Bello, J., H. R. Bello, and E. Granados. 1982. Conformation and aggregation of melittin: dependence on pH and concentration. Biochemistry. 21:461–465. [DOI] [PubMed] [Google Scholar]
- 7.Raghuraman, H., and A. Chattopadhyay. 2006. Effect of ionic strength on folding and aggregation of the hemolytic peptide melittin in solution. Biopolymers. 83:111–121. [DOI] [PubMed] [Google Scholar]
- 8.De Jongh, H. H. J., E. Goormaghtigh, and J. A. Killian. 1994. Analysis of circular dichroism spectra of oriented protein-lipid complexes: toward a general application. Biochemistry. 33:14521–14528. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh, A. K., R. Rukmini, and A. Chattopadhyay. 1997. Modulation of tryptophan environment in membrane-bound melittin by negatively charged phospholipids: implications in membrane organization and function. Biochemistry. 36:14291–14305. [DOI] [PubMed] [Google Scholar]
- 10.Raghuraman, H., and A. Chattopadhyay. 2004. Interaction of melittin with membrane cholesterol: a fluorescence approach. Biophys. J. 87:2419–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghuraman, H., and A. Chattopadhyay. 2004. Influence of lipid chain unsaturation on membrane-bound melittin: a fluorescence approach. Biochim. Biophys. Acta. 1665:29–39. [DOI] [PubMed] [Google Scholar]
- 12.Raghuraman, H., and A. Chattopadhyay. 2003. Organization and dynamics of melittin in environments of graded hydration: a fluorescence approach. Langmuir. 19:10332–10341. [Google Scholar]
- 13.Raghuraman, H., and A. Chattopadhyay. 2004. Effect of micellar charge on the conformation and dynamics of melittin. Eur. Biophys. J. 33:611–622. [DOI] [PubMed] [Google Scholar]
- 14.Terwilliger, T. C., and D. Eisenberg. 1982. The structure of melittin. J. Biol. Chem. 237:6016–6022. [PubMed] [Google Scholar]
- 15.Kaiser, E. T., and F. J. Kezdy. 1983. Secondary structures of proteins and peptides in amphiphilic environment. Proc. Natl. Acad. Sci. USA. 80:1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morii, H. S., S. Honda, S. Ohashi, and H. Uedaira. 1994. Alpha-helical assembly of biologically active peptides and designed helix bundle protein. Biopolymers. 34:481–488. [DOI] [PubMed] [Google Scholar]
- 17.Golding, C., and P. O'Shea. 1995. The interactions of signal sequences with membranes. Biochem. Soc. Trans. 23:971–976. [DOI] [PubMed] [Google Scholar]
- 18.Rabenstein, M., and Y. K. Shin. 1995. A peptide from the heptad repeat of human immunodeficiency virus gp41 shows both membrane binding and coiled-coil formation. Biochemistry. 34:13390–13397. [DOI] [PubMed] [Google Scholar]
- 19.Barnham, K. J., S. A. Monks, M. G. Hinds, A. A. Azad, and R. S. Norton. 1997. Solution structure of polypeptide from the N terminus of the HIV protein Nef. Biochemistry. 36:5970–5980. [DOI] [PubMed] [Google Scholar]
- 20.Cajal, Y., and M. K. Jain. 1997. Synergism between melittin and phospholipase A2 from bee venom: apparent activation by intervesicle exchange of phospholipids. Biochemistry. 36:3882–3893. [DOI] [PubMed] [Google Scholar]
- 21.Bradrick, T. D., A. Philippetis, and S. Georghiou. 1995. Stopped-flow fluorometric study of the interaction of melittin with phospholipid bilayers: importance of the physical state of the bilayer and the acyl chain length. Biophys. J. 69:1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oren, Z., and Y. Shai. 1997. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: structure-function study. Biochemistry. 36:1826–1835. [DOI] [PubMed] [Google Scholar]
- 23.Chattopadhyay, A., and R. Rukmini. 1993. Restricted mobility of the sole tryptophan in membrane-bound melittin. FEBS Lett. 335:341–344. [DOI] [PubMed] [Google Scholar]
- 24.Chattopadhyay, A. 1990. Chemistry and biology of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: fluorescent probes of biological and model membranes. Chem. Phys. Lipids. 53:1–15. [DOI] [PubMed] [Google Scholar]
- 25.Chattopadhyay, A., S. Mukherjee, and H. Raghuraman. 2002. Reverse micellar organization and dynamics: a wavelength-selective fluorescence approach. J. Phys. Chem. B. 106:13002–13009. [Google Scholar]
- 26.Mukherjee, S., A. Chattopadhyay, A. Samanta, and T. Soujanya. 1994. Dipole moment change of NBD group upon excitation studied using solvatochromic and quantum chemical approaches: implications in membrane research. J. Phys. Chem. 98:2809–2812. [Google Scholar]
- 27.Fery-Forgues, S., J. P. Fayet, and A. Lopez. 1993. Drastic changes in the fluorescence properties of NBD probes with the polarity of the medium: involvement of a TICT state? J. Photochem. Photobiol. A. 70:229–243. [Google Scholar]
- 28.Blondelle, S. E., and R. A. Houghten. 1991. Hemolytic and antimicrobial activities of twenty-four individual omission analogues of melittin. Biochemistry. 30:4671–4678. [DOI] [PubMed] [Google Scholar]
- 29.Yang, L., T. A. Harroun, T. M. Weiss, L. Ding, and H. W. Huang. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subbarao, N. K., and R. C. MacDonald. 1994. Lipid unsaturation influences melittin-induced leakage of vesicles. Biochim. Biophys. Acta. 1189:101–107. [DOI] [PubMed] [Google Scholar]
- 31.Raghuraman, H., and A. Chattopadhyay. 2005. Cholesterol inhibits the lytic activity of melittin in erythrocytes. Chem. Phys. Lipids. 134:183–189. [DOI] [PubMed] [Google Scholar]
- 32.Dittmer, J. C., and R. L. Lester. 1964. Simple, specific spray for the detection of phospholipids on the thin-layer chromatograms. J. Lipid Res. 5:126–127. [PubMed] [Google Scholar]
- 33.McClare, C. W. F. 1971. An accurate and convenient organic phosphorus assay. Anal. Biochem. 39:527–530. [DOI] [PubMed] [Google Scholar]
- 34.MacDonald, R. C., R. I. MacDonald, B. P. Menco, K. Takeshita, N. K. Subbarao, and L. R. Hu. 1991. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta. 1061:297–303. [DOI] [PubMed] [Google Scholar]
- 35.Abrams, F. S., and E. London. 1993. Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipid polar headgroup. Biochemistry. 32:10826–10831. [DOI] [PubMed] [Google Scholar]
- 36.Kremer, J. M. H., M. W. van der Esker, C. Pathmamanoharan, and P. H. Wiersema. 1977. Vesicles of variable diameter prepared by a modified injection method. Biochemistry. 16:3932–3935. [DOI] [PubMed] [Google Scholar]
- 37.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- 38.Ladokhin, A. S., and S. H. White. 1999. Folding of amphipathic α-helices on membranes: energetics of helix formation by melittin. J. Mol. Biol. 285:1363–1369. [DOI] [PubMed] [Google Scholar]
- 39.Chattopadhyay, A. 2003. Exploring membrane organization and dynamics by the wavelength-selective fluorescence approach. Chem. Phys. Lipids. 122:3–17. [DOI] [PubMed] [Google Scholar]
- 40.Raghuraman, H., D. A. Kelkar, and A. Chattopadhyay. 2005. Novel insights into membrane protein structure and dynamics utilizing the red edge excitation shift. In Reviews in Fluorescence, Vol. 2. C. D. Geddes and J. R. Lakowicz, editors. Springer, New York. 199–222.
- 41.Demchenko, A. P. 2002. The red-edge effects: 30 years of exploration. Luminescence. 17:19–42. [DOI] [PubMed] [Google Scholar]
- 42.Mentré, P. (Ed.). 2001. Water in the cell. Cell. Mol. Biol. 47:709–970. [PubMed] [Google Scholar]
- 43.Raghuraman, H., S. K. Pradhan, and A. Chattopadhyay. 2004. Effect of urea on the organization and dynamics of Triton X-100 micelles: a fluorescence approach. J. Phys. Chem. B. 108:2489–2496. [Google Scholar]
- 44.Mukherjee, S., and A. Chattopadhyay. 2005. Influence of ester and ether linkage in phospholipids on the organization and dynamics of the membrane interface: a wavelength-selective fluorescence approach. Langmuir. 21:287–293. [DOI] [PubMed] [Google Scholar]
- 45.Rawat, S. S., D. A. Kelkar, and A. Chattopadhyay. 2005. Effect of structural transition of the host assembly on dynamics of an ion channel peptide: a fluorescence approach. Biophys. J. 89:3049–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelkar, D. A., and A. Chattopadhyay. 2005. Effect of graded hydration on the dynamics of an ion channel peptide. Biophys. J. 88:1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raghuraman, H., S. Ganguly, and A. Chattopadhyay. 2006. Effect of ionic strength on the organization and dynamics of membrane-bound melittin. Biophys. Chem. 124:115–124. [DOI] [PubMed] [Google Scholar]
- 48.Hristova, K., C. E. Dempsey, and S. H. White. 2001. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys. J. 80:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White, S. H., and W. C. Wimley. 1994. Peptides in lipid bilayers: structural and thermodynamic basis of partitioning and folding. Curr. Opin. Struct. Biol. 4:79–86. [Google Scholar]
- 50.Bachar, M., and O. M. Becker. 2000. Protein-induced membrane disorder: a molecular dynamics study of melittin in a dipalmitoylphosphatidylcholine bilayer. Biophys. J. 78:1359–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toraya, S., N. Katsuyuki, and A. Naito. 2004. Dynamic structure of vesicle-bound melittin in a variety of lipid chain lengths by solid-state NMR. Biophys. J. 87:3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prendergast, F. G. 1991. Time-resolved fluorescence techniques: methods and applications in biology. Curr. Opin. Struct. Biol. 1:1054–1059. [Google Scholar]
- 53.Lin, S., and W. S. Struve. 1991. Time-resolved fluorescence of nitrobenzoxadiazole-aminohexanoic acid: effect of intermolecular hydrogen-bonding on non-radiative decay. Photochem. Photobiol. 54:361–365. [DOI] [PubMed] [Google Scholar]
- 54.Ho, C., S. J. Slater, and C. D. Stubbs. 1995. Hydration and order in lipid bilayers. Biochemistry. 34:6188–6195. [DOI] [PubMed] [Google Scholar]
- 55.Kurad, D., G. Jeschke, and D. Marsh. 2003. Lipid membrane polarity profiles by high-field EPR. Biophys. J. 85:1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukherjee, S., H. Raghuraman, and A. Chattopadhyay. 2004. Organization and dynamics of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: a fluorescence approach. Chem. Phys. Lipids. 127:91–101. [DOI] [PubMed] [Google Scholar]
- 57.Mukherjee, S., and A. Chattopadhyay. 1995. Wavelength-selective fluorescence as a novel tool to study organization and dynamics in complex biological systems. J. Fluoresc. 5:237–246. [DOI] [PubMed] [Google Scholar]
- 58.Chattopadhyay, A., and E. London. 1988. Spectroscopic and ionization properties of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids in model membranes. Biochim. Biophys. Acta. 938:24–34. [DOI] [PubMed] [Google Scholar]
- 59.Homan, R., and M. Eisenberg. 1985. A fluorescence quenching technique for the measurement of paramagnetic ion concentrations at the membrane/water interface. Intrinsic and X537A-mediated cobalt fluxes across lipid bilayer membranes. Biochim. Biophys. Acta. 812:485–492. [DOI] [PubMed] [Google Scholar]
- 60.Morris, S. J., D. Bradley, and R. Blumenthal. 1985. The use of cobalt ions as a collisional quencher to probe surface charge and stability of fluorescently labeled bilayer vesicles. Biochim. Biophys. Acta. 818:365–372. [DOI] [PubMed] [Google Scholar]
- 61.Chattopadhyay, A. 1992. Membrane penetration depth analysis using fluorescence quenching: a critical review. In Biomembrane Structure and Function: The State of the Art. B. P. Gaber and K. R. K. Easwaran, editors. Adenine Press, Schenectady, NY. 153–163.
- 62.London, E., and A. S. Ladokhin. 2002. Measuring the depth of amino acid residues in membrane-inserted peptides by fluorescence quenching. In Current Topics in Membranes. D. Benos and S. Simon, editors. Elsevier, San Diego, CA. 89–115.
- 63.Chattopadhyay, A., and E. London. 1987. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 26:39–45. [DOI] [PubMed] [Google Scholar]
- 64.Kaiser, R. D., and E. London. 1998. Location of diphenylhexatriene (DPH) and its derivatives within membranes: comparison of different fluorescence quenching analyses of membrane depth. Biochemistry. 37:8180–8190. [DOI] [PubMed] [Google Scholar]
- 65.Chen, S.-C., and B. J. Gaffney. 1978. Paramagnetic resonance evidence for phase transitions in bilayers of pure spin-labeled lipids. J. Mag. Res. 29:341–353. [Google Scholar]
- 66.Ahmed, S. N., D. A. Brown, and E. London. 1997. On the origin of sphingolipid/cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of detergent-insoluble, liquid-ordered lipid phase in model membranes. Biochemistry. 36:10944–10953. [DOI] [PubMed] [Google Scholar]
- 67.Vogel, H. 1987. Comparison of the conformation and orientation of alamethicin and melittin in lipid membranes. Biochemistry. 26:4562–4572. [DOI] [PubMed] [Google Scholar]
- 68.Altenbach, C., and W. L. Hubbell. 1988. The aggregation state of spin-labeled melittin in solution and bound to phospholipid membranes: evidence that membrane-bound melittin is monomeric. Proteins. 3:230–242. [DOI] [PubMed] [Google Scholar]
- 69.Frey, S., and L. K. Tamm. 1991. Orientation of melittin in phospholipid bilayers. A polarized attenuated total reflection infrared study. Biophys. J. 60:922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dempsey, C. E., and G. S. Butler. 1992. Helical structure and orientation of melittin in dispersed phospholipid membranes from amide exchange analysis in situ. Biochemistry. 31:11973–11977. [DOI] [PubMed] [Google Scholar]
- 71.Smith, R., F. Separovic, T. J. Milne, A. Whittaker, F. M. Bennett, B. A. Cornell, and A. Makriyannis. 1994. Structure and orientation of the pore-forming peptide, melittin, in lipid bilayers. J. Mol. Biol. 241:456–466. [DOI] [PubMed] [Google Scholar]
- 72.Bradshaw, J. P., C. E. Dempsey, and A. Watts. 1994. A combined X-ray and neutron diffraction study of selectively deuterated melittin in phospholipid bilayers: effect of pH. Mol. Membr. Biol. 11:79–86. [DOI] [PubMed] [Google Scholar]
- 73.Wall, J., C. A. Golding, M. Van Veen, and P. O'Shea. 1995. The use of fluoresceinphosphatidylethanolamine (FPE) as a real-time probe for peptide-membrane interactions. Mol. Membr. Biol. 12:183–192. [DOI] [PubMed] [Google Scholar]
- 74.Bernèche, S., M. Nina, and B. Roux. 1998. Molecular dynamics simulation of melittin in a dimyristoylphosphatidylcholine bilayer membrane. Biophys. J. 75:1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naito, A., T. Nagao, K. Norisada, T. Mizuno, S. Tuzi, and H. Saitô. 2000. Conformation and dynamics of melittin bound to magnetically oriented lipid bilayers by solid-state 31P and 13C NMR spectroscopy. Biophys. J. 78:2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yeagle, P. L. 1985. Cholesterol and the cell membrane. Biochim. Biophys. Acta. 822:267–287. [DOI] [PubMed] [Google Scholar]
- 77.Castano, S., I. Cornut, K. Buttner, J. L. Dasseux, and J. Dufourcq. 1999. The amphipathic helix concept: length effects on ideally amphipathic LiKj(i = 2j) peptides to acquire optimal hemolytic activity. Biochim. Biophys. Acta. 1416:161–175. [DOI] [PubMed] [Google Scholar]