

Abstract
In Escherichia coli cytochrome c maturation requires a set of eight proteins including the heme chaperone CcmE, which binds heme transiently, yet covalently. Several variants of CcmE were purified and analyzed by continuous-wave electron paramagnetic resonance, electron nuclear double resonance, and hyperfine sublevel correlation spectroscopy to investigate the heme axial coordination. Results reveal the presence of a number of coordination environments, two high-spin heme centers with different rhombicities, and at least one low-spin heme center. The low-spin species was shown to be an artifact induced by the presence of available histidines in the vicinity of the iron. Both of the high-spin forms are five-coordinated, and comparison of the spectra of the wild-type CcmE with those of the mutant CcmEY134H proves that the higher-rhombicity form is coordinated by Tyr134. The low-rhombicity (axial) form does not have a histidine residue or a water molecule as an axial ligand. However, we identified exchangeable protons coupled to the iron ion. We propose that the axial form can be coordinated by a carboxyl group of an acidic residue in the flexible domain of the protein. The two species would represent two different conformations of the flexible α-helix domain surrounding the heme. This conformational flexibility confers CcmE special dynamic properties that are certainly important for its function.
INTRODUCTION
During maturation of c-type cytochromes the heme cofactor is attached covalently to the Cys-Xaa-Xaa-Cys-His (CXXCH) signature motif of the apo-cytochrome. In E. coli eight cytoplasmic membrane proteins, encoded by the ccmABCDEFGH operon, are required for maturation (1,2). The heme chaperone CcmE binds heme covalently in the periplasm and delivers it to the apo-cytochrome (3). The nature of the binding of heme to CcmE has been determined by digestion of holo-CcmE with trypsin, isolation of the tryptic heme peptide, and subsequent analysis by NMR (3,4). Heme was found to be cross-linked at the β-carbon of one of the two vinyl groups to the Nδ1 of CcmE His130 imidazole. Various biochemical and biophysical studies on the heme-free apo-CcmE and the heme-bound holo-CcmE have been undertaken; however, the chemistry of heme transfer to CcmE and the heme delivery to the apo-cytochrome remain unclear. One attempt to tackle this chemical reaction is to determine the structure of holo-CcmE, which can be purified as a stable intermediate of the cytochrome c maturation pathway (3). Attempts to solve the structure of holo-CcmE have failed so far, but the structures of the soluble domain of the E. coli and Shewanella putrefaciens apo-CcmE have been solved by NMR (5,6). CcmE was found to consist of a rigid core followed by a poorly structured, flexible C-terminal domain. Heme was modeled to a hydrophobic platform at the surface of the core close to the heme-binding histidine (see Fig. 1). These structural features favor the model where the poorly structured domain of CcmE may enclose the bound heme in holo-CcmE. It has been proposed that heme binding by apo-CcmE might first involve the formation of a noncovalent complex in which the heme can be coordinated by His130 (5). In a second step, a covalent bond to His130 is formed, and the flexible C-terminal domain could bend to shield the heme group. Recently, resonance Raman spectroscopy has provided some clues on the coordination of the heme iron (7). Based on the position of the spin and coordination state marker bands, the ferric protein was suggested to contain a five-coordinate high-spin heme, and the ferrous protein to have heme in a six-coordinate low-spin state. In the same study both Tyr and His residues have been suggested to ligate the heme iron. By comparing the Raman spectra of the wild-type (WT) and the mutant CcmEY134F, the authors assigned a band at 600 cm−1 in the Raman spectrum to a Fe-Tyr stretching mode, and they proposed Tyr134 to be an axial ligand of the iron for both the ferrous and the ferric protein.
FIGURE 1.

Structural model of the heme binding site for holo-CcmE (5). CcmE consists of a rigid core formed by a β-barrel and a poorly structured C-terminal domain. His130, which covalently binds one of the heme vinyl groups, is near a hydrophobic platform at the surface of the barrel formed by the residues Phe37, Val110, and Leu127. The conserved residues linking the two subdomains, His130, Asp131, and Glu132, and the three amino acids Tyr134, Pro136, and Glu138, face the same side of the C-terminal subdomain.
In this study we analyzed by electron paramagnetic resonance (EPR) several preparations of WT soluble ferric holo-CcmE and CcmE derivatives mutated in putative native ligands. The potential of this technique derives from the fact that the electronic structure of the ferric iron in the protein is determined by the surroundings of the Fe ion. Consequently, the electron magnetic moment of the iron, which is characterized by g-values, is very sensitive to the axial coordination of the heme iron and can provide reliable information about spin state, electronic symmetry of the environment, and strength of the ligands (8–10). Continuous-wave EPR (cw EPR) probes the heme environment, conveying information about the chemical nature of the axial ligands and their geometric arrangement. In addition, hyperfine interactions of the unpaired electrons with the nearby magnetic nuclei can be used to identify ligands and provide information about the electronic structure of the heme environment. Such hyperfine interactions are not resolved in the cw EPR spectra of heme proteins and have to be studied by other EPR techniques such as ENDOR (electron nuclear double resonance) or ESEEM (electron spin echo envelope modulation) that directly detect the nuclear frequencies of the surrounding magnetic nuclei.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Strain EC06 (1), harboring either pEC86 (11) or pEC101 (12), was used for expression of CcmE derivatives. Final antibiotic concentrations were 200 μg/ml for ampicillin and 10 μg/ml for chloramphenicol. For expression of CcmE derivatives, Luria-Bertani broth was inoculated 100:1 with overnight cultures of the appropriate strain at 30°C. Cultures were grown to an OD600 of 0.8 and were induced with 0.1% arabinose. Seventeen hours after induction, cells were harvested by centrifugation at 3300 × g.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Preparation details |
|---|---|---|
| E. coli strains | ||
| DH5α | supE44 ΔlacU169 (Φ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | (13) |
| EC06 | ΔccmA-H derivative of MC1061; KmR | (1) |
| Plasmids | ||
| pEC86 | ccmABCDEFGH cloned into pACYC184, CmR | (11) |
| pEC101 | ccmABCD cloned into pACYC184, CmR | (12) |
| pEC301 | ompA’-‘ccmEsol, ApR | (19) |
| pEC304 |
ompA’-‘ , ApR , ApR
|
(19) |
| pEC600 | pEC412 expressing solu5ble CcmE30–140-strep | This study |
| pEC616 | pEC304 expressing soluble CcmE30–138 H120V | This study |
| pEC619 | pEC301 expressing soluble CcmEY134H | This study |
Site-directed mutagenesis
E. coli strain DH5α (13) was used for cloning procedures. A truncated version of CcmE was fused with a Strep-tag consisting of the amino acid sequence SAWSHPQFEK for affinity purification by ExSite PCR-based site-directed mutagenesis (Stratagene). For this, PCR was performed with primer pair solCcmE140strep (tttttcgaactgcgggtggctccaagcgctctcaacttctggcggcgtatatgttttcatcgtg) and mp19sss (tgagtcgactctagaggatccccggg) and pEC415 (3) as template resulting in plasmid pEC600 (Table 1). The pEC600 expresses C-terminally truncated soluble CcmE30–140-strep. pEC616 (Table 1) expressing C-terminally truncated soluble CcmE30–138 H120V that only contains one His residue (His130), was constructed by QuikChange site-directed mutagenesis (Stratagene) using pEC304 (Table 1) as template. The plasmid pEC619 expressing soluble CcmEY134H was also constructed by QuikChange site-directed mutagenesis using pEC301 as template. Sequences of resulting plasmids were verified by Microsynth (Balgach, Switzerland).
Protein extraction, purification, and initial characterization
Periplasmic proteins were isolated from 0.9 liter cultures by extraction of cell pellets in 5 ml of 1 mg/ml polymyxin B sulfate/500 mM sucrose/100 mM Tris HCl, pH 8/5 mM EDTA. The suspension was stirred for 60 min at 4°C and was centrifuged at 15,000 × g for 20 min. For purification of soluble wild-type CcmE and CcmEY134H, the supernatant containing the periplasmic proteins was applied to a 2-ml bed volume DEAE Sepharose column and washed with 1 column volume each of 25 mM Tris, pH 8, containing no salt, 125 mM NaCl, and 175 mM NaCl. The fraction containing both holo- and apo-CcmE was eluted with two column volumes 275 mM NaCl in 25 mM Tris, pH 8. CcmE30–140-strep was purified from periplasmic extracts via strep-tactin affinity chromatography as recommended by the manufacturer (IBA GmbH, Göttingen, Germany), resulting in a fraction containing both holo- and apo-CcmE30–140-strep. Pure holo-CcmE30–140-strep was obtained by separation from apo-CcmE30–140-strep over a hydrophobic interaction column (HiTrap Phenyl HP, Amersham Biosciences) with an Aekta purifier using a gradient of 0.8 to 0 M ammonium sulfate in 10% EtOH and 50 mM TrisHCl, pH 8. Proteins were concentrated and buffers exchanged over 5 K NMWL Amicon Ultra-4 centrifugal filter devices (Millipore). The ratio of CcmE to total protein was estimated to be in the range of 50% by separation over SDS-15% polyacrylamide gels and subsequent staining with Coomassie brilliant blue R. Concentrations of holo-CcmE were determined by the pyridine hemochrome assay (14). All proteins were isolated to similar purity, and for all proteins both monomeric and dimeric forms were visible on Coomassie blue–stained gels and gels stained for covalently bound heme. Note that although the samples of CcmE are not completely pure, the signals described and analyzed below are clearly attributable to heme centers. These signals are identical for samples prepared using plasmid pEC86 and pEC101. For the latter preparations the only heme protein that could possibly be in the periplasm is CcmE, which ensures the assignation of the signals to the heme chaperone. Optical spectra were recorded for air-oxidized samples and for samples reduced with 5 mM Na-dithionite on a Hitachi model U-3300 spectrophotometer.
Sample preparation for EPR
Samples used for EPR spectroscopy were obtained from 10 liters of culture and contained 0.2 to 1 mM holo-CcmE in 100 μl 25 mM Tris HCl, pH 8. Then, they were mixed with glycerol to reach, unless stated otherwise, a protein solution:glycerol volume ratio of 7:3 and transferred to EPR quartz tubes (see the Supplementary Information for details about the effect of the glycerol amount on the EPR spectra). To prepare the deuterated sample, an exchange of the aqueous solvent by a buffer prepared with deuterated water (Cambridge Isotope Laboratories, Cambridge, UK) was performed three times by centrifugation over 5KMWL Amicon Ultra4 centrifugal filter devices followed by the addition of 30% of deuterated glycerol (Cambridge Isotope Laboratories,). The same procedure was followed to prepare the sample in 17O-water (Isotech, Basel, Switzerland), which achieved 70% labeling of the solvent molecules.
EPR spectroscopy
The cw EPR spectra were measured on a Bruker E500 X-band spectrometer (microwave (mw) frequency 9.45 GHz). The spectrometer was equipped with an Oxford ESR CF910 continuous-flow cryostat and a Bruker ER 4122 SHQ resonator. Measurements were carried out at a temperature of 15 K using 0.2 mW of mw power, 1 mT of modulation amplitude, and 100 kHz modulation frequency.
The ENDOR and hyperfine sublevel correlation (HYSCORE) spectra, as well as the measurements of the spin-lattice relaxation time T1 were recorded at X-band using a Bruker Elexsys E580 spectrometer (mw frequency 9.73 GHz) equipped with a liquid helium cryostat from Oxford. The pulse EPR spectra were taken at a temperature of 3.8 K. The following sequences of mw pulses were used:
HYSCORE experiments (15,16) were performed using the pulse sequence, π/2–τ–π/2– t1–π–t2–π/2–τ –echo, with pulse lengths tπ /2 = tπ = 16 ns. The time intervals t1 and t2 were varied from 96 to 4192 ns in steps of 24 ns. An eight-step phase cycle was used to eliminate unwanted echoes. Spectra were measured using several τ values (100, 124, and 192 ns) to avoid blind spots. Davies-ENDOR spectra (16,17) were measured with the sequence, π–T–π /2–τ–π–τ –echo, with, unless stated otherwise, pulse lengths of tπ /2 = 40 ns and tπ = 80 ns, and time intervals τ = 160 ns and T = 4.5 μs. A selective radio frequency (rf) π pulse of length 4 μs and variable frequency νrf was applied during time T.
Saturation recovery
The electron spin-lattice relaxation times were measured with the pulse sequence 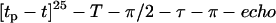 . A burst of 25 pulses with length tp = 32 ns separated by a time interval t = 320 ns was used at the maximal available mw power (∼1 kW). The recovery curve was monitored with primary echo sequence using mw pulses
. A burst of 25 pulses with length tp = 32 ns separated by a time interval t = 320 ns was used at the maximal available mw power (∼1 kW). The recovery curve was monitored with primary echo sequence using mw pulses  and
and 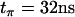 . The time interval τ was chosen to have maxima in echo amplitude, which is strongly modulated as a result of ESEEM effects. A long repetition time (5 ms) was used in this experiment to avoid heating effects.
. The time interval τ was chosen to have maxima in echo amplitude, which is strongly modulated as a result of ESEEM effects. A long repetition time (5 ms) was used in this experiment to avoid heating effects.
Data manipulation and simulations
The HYSCORE time traces were baseline corrected using a second-order polynomial, windowed with an asymmetric Gaussian or a Hamming function, and zero filled. Then the data were Fourier transformed in the dimensions t1 and t2, and the absolute-value spectra were calculated. The simulations of the cw EPR spectra were performed with the program EasySpin (18).
RESULTS
To characterize the axial heme ligands of ferric CcmE by EPR spectroscopy, the different preparations of the protein were expressed with a cleavable leader peptide, as described previously (19), resulting in a periplasmic soluble form. The soluble periplasmic form of CcmE (henceforth, simply, CcmE) was enriched in two steps, by periplasmic extraction and subsequently by DEAE-Sepharose anion exchange, and contained both holo- and apo-CcmE.
Optical characteristics
Optical spectra of wild-type CcmE and CcmEY134H were recorded in their ferric and ferrous forms, and reduced minus oxidized difference spectra were calculated. Difference spectra of the wild-type and the two truncated forms of CcmE corresponded well with the one previously published for soluble holo-CcmE (12). The wild-type form of CcmE had peak maxima for the γ band at 421 nm, for the β band at 526 nm, and for the α band at 555 nm (Fig. 2 A). For the CcmEY134H mutant a significant shift of 2 nm in the β band and of 3 nm in the α band was visible, whereas no shift was found for the γ band (Fig. 2 A). Under denaturizing conditions in the presence of excess pyridine as heme ligand, the peak maxima of both wild-type and CcmEY134H shifted to 415, 520, and 551 nm for the γ, β, and α bands, respectively (Fig. 2 B).
FIGURE 2.

Optical difference spectra of WT CcmE and soluble CcmEY134H. The difference spectra of WT CcmE (wt) and CcmEY134H (Y134H) are shown under native conditions in 25 mM Tris, pH 8 (A) and under denaturing conditions in the presence of excess pyridine as ligand (B) according to the protocol used for the pyridine-hemochrome assay (14). The concentrations for holo-CcmE are ∼2 μM.
Continuous-wave EPR
The cw EPR spectrum of the protein WT CcmE30–140-strep is shown in Fig. 3 A. As is usual practice (4,5,20), a tag was added to the C terminus in this sample to facilitate the protein purification. In this spectrum a number of features are present that reveal the existence of different iron centers. The signals below 200 mT, together with the feature at g = 2.0, belong to high-spin (HS) iron centers. In addition, the lines at g values of 3.2, 2.96, 2.27, and 1.54 can be assigned to two different heme iron centers in a low-spin (LS) state. The appearance of the LS spectrum with g = (2.96, 2.27, 1.54) is a typical signature of a heme center coordinated by two imidazole rings of histidine residues (21). When the holo-CcmE30–140-strep was purified by hydrophobic interaction chromatography from the above sample, the relative proportion of LS centers decreased substantially (Fig. 3 B), but no changes in the position or line shape of the HS signals were observed. Because the strep tag at the end of the flexible tail contains a histidine residue, it might be that most of the LS form in the previous spectrum is related to the presence of this tag. The two histidine ligands of the heme are thus most probably histidine residues located in the flexible domain of the same and/or in another protein unit. These results indicate that the heme group, which is thought to bind to the surface of the protein, can have a strong affinity for available histidines. To confirm this, an excess of imidazole (final concentration of imidazole 250 mM) was added to WT CcmE. As shown in Fig. 3 C, all the heme centers are now in the LS state (the 4.3 signal is an impurity, see below), with g values 2.96, 2.27, and 1.54, again values typical of bis-histidine coordination. To determine whether the effect of the imidazole can be reversed and thus whether purification procedures with imidazole, as used for His6-tag purification, may irreversibly change the heme coordination, the samples were dialyzed with 25 mM Tris, pH 8. The spectrum of the sample after dialysis shows only HS signals, but the amount of heme iron has decreased considerably (not shown).
FIGURE 3.

X-band cw EPR spectra of different preparations of WT CcmE taken at 15 K: (A), CcmE30–140-strep, mixture of holo and apoprotein; (B), Holo-CcmE30–140-strep; (C), WT CcmE + 250 mM imidazole; (D), CcmE30–138 H120V. The arrows indicate the g values of the most intense lines of the spectrum. The spectra have been rescaled to display approximately the same intensity. Spectrometer settings are described in the Materials and Methods section.
The histidine in the tag cannot be a natural ligand of the heme, neither His147, as it is not conserved in other sequences (5), or His120, because it is on the opposite side of the protein (5). Therefore, we constructed the C-terminally truncated mutant protein CcmE30–138 H120V resulting from expression of plasmid pEC616 (Table 1), which has only one histidine, His130, covalently bound to one of the vinyl groups of the heme. The EPR spectrum (Fig. 3 D) has no LS species; however, all the HS features are retained. The same spectrum was found in the control CcmE30–138 peptide (not shown). These experiments provide strong evidence indicating that the LS species are formed by interaction with available histidines around the heme and probably result in proteins with nonnative structures. Because the LS forms of CcmE are thought to be artifacts, we next focus on the HS forms of the protein.
To enable heme coordination to be as natural as possible, the preparation chosen for further study was CcmE30–159, whose EPR spectrum is shown in Fig. 4 A. The major contributions in the spectrum are from HS iron species. The most intense signal corresponds to an HS heme iron in an axially symmetric environment that shows a g⊥ feature at g ≈ 6.1 and a g‖ feature at g ≈ 2.0. The low-field feature of this center has two satellite lines, a shoulder at g ≈ 6.7, and a derivative shaped signal at g ≈ 4.9. These originate from different HS heme species with less symmetric environments, whose gz values are close to g ≈ 2.0 and cannot be resolved. The derivative-shaped sharp line at g ≈ 4.3 is caused by an impurity of nonheme iron in a rhombic ligand field, which can have considerable intensity even for small amounts of impurity in the sample (22).
FIGURE 4.

X-band cw EPR spectra of CcmE taken at 15 K. (A) WT CcmE. (B) CcmEY134H. Note that the relative increase of the signal at g = 4.3 is mainly a result of the decrease of the HS signal. The insets show the comparison of the experimental spectra with simulations performed as described in the text with the following parameters: axial HS, Δ1/λ = 7.2, Δ2/λ = 19.5, Δ3/λ = 20.8, and a Gaussian distribution of Δ1/λ and Δ2/λ of width 1.7; rhombic HS, Δ1/λ = 3.7, Δ2/λ = 5.73, Δ3/λ = 5.96, and a Gaussian distribution of Δ1/λ and Δ2/λ of width 0.02. (C) Simulation of the two main signals in spectrum B with the above parameters for the axial HS and gx = 1.52, gy = 2.27, gz = 2.96 for the LS. Ratio of the two species HS:LS is 1:3.
According to published data (8,23–26), rhombic HS signals can be observed in heme centers coordinated to tyrosine. To substantiate if tyrosine at position 134 is actually coordinating the heme center, a variant of soluble CcmE was produced, where the putative heme ligand was mutated. Expression from plasmid pEC619 (Table 1) resulted in mutant CcmEY134H where the Tyr134 is replaced by a His residue. The EPR spectrum of CcmEY134H (Fig. 4 B) shows no signal at g ≈ 4.9, and the shoulder at g ≈ 6.7 has decreased considerably. This result confirms that Tyr134 coordinates some of the heme iron, giving rise to the rhombic HS EPR spectrum. On the other hand, the axial HS signal is still observed in the spectrum and retains approximately the same width, suggesting that Tyr134 is not involved in the coordination of the axial CcmE species. All the other signals in the spectrum of the wild-type chaperone remain in the spectrum of the mutant. Noteworthy is the relative increase in intensity of the features associated with LS heme centers, especially the lines of the bis-histidine heme iron (see below for a definite proof of this coordination).
The behavior of the different features in the spectrum on increasing mw power and temperature is the one expected for low/high-spin heme centers (27). The LS signal was saturated at T = 15 K and an mw power of 12 mW, with the exception of the signal at g ≈ 3.1 which was more difficult to saturate. On the other hand, the HS signals (both axial and rhombic behave the same way) could not be saturated with the highest mw power available (200 mW), even at 5.5 K. By increasing the temperature, both signals lost intensity. The HS signals did this to a greater extent than the LS signals, but both were still visible at 70 K.
Experimental determination of the zero-field splitting and cw EPR simulations
Taking the peaks at g = 6.7 and g = 4.9 to represent the gx and gy components of one rhombic center, one could determine the zero-field splitting parameters according to a standard spin Hamiltonian with S = 5/2 (22). However, the deviation between the experimental g-values and the ones predicted by the calculation is nearly 3%. Both experimental g-values are lower than the ones predicted. Consequently, the center of gravity of these two levels falls below the calculated value that, for small rhombicities, is ∼6. Such unusual splitting of gx and gy has already been observed in some catalase-peroxidases and has been attributed to a small amount of mixing of the S = 5/2 ground state with the slightly higher lying S = 3/2 excited state (28,29). The degree of mixing and the peculiarities of the ground state could be a signature of a certain heme environment. The behavior of this kind of mixed spin states has been studied theoretically by Maltempo (30) for systems in an axial environment. Opposed to conventional HS centers, he considered that the exited state is close enough so that the mixing of the ground state (6A1) and the exited state (4T1) through spin-orbit coupling cannot be described using perturbation theory. We performed an analysis of our system following this approach, but, unlike the cases studied by Maltempo, at least one of the heme centers in CcmE has rhombic symmetry. This means that the mixing with all states coming from the 4T1 exited state needs to be considered. The energy diagram is shown in Fig. 5. The 18 × 18 Hamiltonian, which considers the spin-orbit interaction, was constructed following Weissbluth (31) and solved to find the eigenfunctions of the system, which are admixtures of the states 6A1 and 4T1. The actual amount of admixture depends on Δ1/λ, Δ2/λ, and Δ3/λ. In particular, the ground state 6A1 is split into three doublets, of which the lowest in energy is  and the next one,
and the next one,  , is ΔE higher in energy.
, is ΔE higher in energy.
FIGURE 5.

Crystal field splitting diagrams for high-spin Fe3+. The energy levels of the system, whose Hamiltonian is increasingly containing the terms above, are represented. The wave functions corresponding to some of the levels are also depicted.
The ΔE in hemeproteins can be determined experimentally by measuring the temperature dependence of the spin-lattice relaxation rate (32). This method is based on the characteristic temperature dependence of the Orbach processes of electron spin relaxation. In this process, phonons with energies equal to the splitting ΔE are absorbed and emitted by the spin system, resulting in a relaxation rate (1/T1) given by (33)
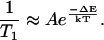 |
(1) |
T1 was measured as a function of temperature in the range from 3.6 K to 5.3 K, above which the electron spin echo disappears. 1/T1 plotted against 1/T has a linear dependence (see Fig. S1 in the Supplementary Material) and from the slope ΔE can be obtained for both centers (ΔEaxial = 12 ± 1.4 cm−1 and ΔErhombic = 11.4 ± 1.4 cm−1). In the cases where the state of the iron can be described as a pure S = 5/2, the splitting between the two lowest Kramers doublets is 2D (D is the zero-field splitting parameter). The values of ΔE obtained here are within the range of 2D reported in literature for other high-spin heme proteins (22,32).
On the introduction of a magnetic field, all doublets are split. If ΔE ≫ hν, the Zeeman interaction does not produce further mixing, and the EPR transition occurs within the doublet  whose main contribution is
whose main contribution is 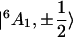 . Thus, the effect of the magnetic field can be considered solely on the ground state
. Thus, the effect of the magnetic field can be considered solely on the ground state 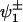 . In this way we can calculate the effective g values of our system, considered as an Seff = ½. Following this procedure, and taking a spin-orbit coupling constant for Fe3+ of 340 cm−1 (34), the values of Δ1, Δ2, and Δ3 that yield the observed values of gx, gy, gz, and ΔE have been obtained for each of the HS species of CcmE. To obtain a line shape describing the one observed experimentally, it is necessary to consider distributions of environments (22), which are expressed by distributions in Δ1, Δ2, and Δ3. In the simulations depicted in Fig. 4, we have considered Gaussian distributions of these parameters that result in asymmetric g distributions. These distributions give rise to distinctive features in the spectrum, such as the shoulder observed at the low-field region of the axial signal, its extreme broadening in the high-field side (see inset in Fig. 4 B), or the tail at the g = 2 feature. The simulations suggest that the axial and rhombic species have, respectively, a 5% and 10% mixture of S = 3/2. The ratio rhombic versus axial HS species is ∼1:3 in the WT (inset, Fig. 4 A).
. In this way we can calculate the effective g values of our system, considered as an Seff = ½. Following this procedure, and taking a spin-orbit coupling constant for Fe3+ of 340 cm−1 (34), the values of Δ1, Δ2, and Δ3 that yield the observed values of gx, gy, gz, and ΔE have been obtained for each of the HS species of CcmE. To obtain a line shape describing the one observed experimentally, it is necessary to consider distributions of environments (22), which are expressed by distributions in Δ1, Δ2, and Δ3. In the simulations depicted in Fig. 4, we have considered Gaussian distributions of these parameters that result in asymmetric g distributions. These distributions give rise to distinctive features in the spectrum, such as the shoulder observed at the low-field region of the axial signal, its extreme broadening in the high-field side (see inset in Fig. 4 B), or the tail at the g = 2 feature. The simulations suggest that the axial and rhombic species have, respectively, a 5% and 10% mixture of S = 3/2. The ratio rhombic versus axial HS species is ∼1:3 in the WT (inset, Fig. 4 A).
The LS species was simulated as an S = ½ center with a Gaussian g-strain. The simulation of the two main signals in Fig. 4 B gives an estimation of 3:1 for LS:axial HS in CcmEY134H (see Fig. 4 C). Note that species with different effective g values (as found in LS and HS centers) give EPR signals with different integral intensity per unit spin (35,36). In our case this factor is lower for the LS centers, which accounts for its larger amount despite similar intensities for both centers.
ENDOR and HYSCORE
To study the hyperfine interactions with the surrounding nitrogen nuclei, ENDOR measurements were performed on the preparations of WT CcmE and CcmEY134H, whose cw EPR spectra are presented in Fig. 4. Because of the very rapid decay of the echo intensity with temperature, the pulse experiments were recorded at 3.8 K. The magnetic field was set at g‖, where only the molecules with the porphyrin ring perpendicular to the magnetic field contribute to the ENDOR spectrum. This observer position (single-crystal-like) allows sharp and intense ENDOR lines of nuclei in the axial positions to be obtained (37,38). Therefore, ENDOR has been used to confirm the presence or absence of axial nitrogens in other HS heme complexes (39).
The ENDOR spectrum of CcmE is shown in Fig. 6 A. Two broad lines are observed at frequencies around 3.5 and 5.5 MHz. They are separated by approximately twice the Larmor frequency of 14N (νN), in agreement with the first-order equation for the resonance frequencies (40),
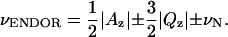 |
(2) |
FIGURE 6.

X-band Davies-ENDOR spectra taken at 348 mT and 3.8 K. (A) WT CcmE. (B) Metmyoglobin 5 mM in a 50 mM HEPES solution (pH 7). (C) Hemin 5 mM in a 1:1 DMSO:chloroform solution. (D) CcmEY134H. (E) WT CcmE in 17O-water. The lines point at the locations of the lines in the spectrum and identify their assignment. For all the spectra an rf pulse of 4 μs was used except for spectrum D, where it was 2 μs long.
The center of the spectrum is around 4.0 MHz, which results from a hyperfine interaction (Az) of ∼8 MHz. The nuclear quadrupole coupling is not resolved but, according to the width of the lines the nuclear quadrupolar coupling constant (Qz), is smaller than 0.4 MHz. The parameters for this nucleus are close to those reported for the heme nitrogens in other HS centers (40–43). In Fig. 6, B and C, the spectra of met-myoglobin and a DMSO:chloroform solution of hemin are shown for comparison. In the spectrum of myoglobin, the lines corresponding to the axial histidine nitrogen can be distinctly seen and lead to the determination of the coupling parameters Az ≈ 11.5 MHz and Qz ≈ 1.2 MHz. On the other hand, the spectrum of hemin, with no axial nitrogen, is much more like the one of CcmE, again consisting of two broad lines separated by 2νN. Note that the hyperfine constant of heme nitrogens in both myoglobin and hemin is ∼1 MHz less than in CcmE.
The ENDOR spectrum of CcmEY134H shown in Fig. 6 D is virtually the same as the one of the wild-type protein, and again only interactions with heme nitrogens are observed. This provides evidence that none of the CcmE HS species has a proximal nitrogen nucleus, and in particular a histidine residue, as an axial ligand. In the case of the mutant, the coordinating tyrosine has been replaced by a histidine, which has a strong affinity for coordinating the heme. However, there is no histidine coordinating the HS form. This means that the coordination of His134 has to be related to the considerable increase of the low-spin species in the CcmEY134H sample.
The hyperfine interactions have also been studied using ESEEM spectroscopy; in particular, the 2D HYSCORE experiment that correlates nuclear frequencies in the two mS manifolds of the electron spin (16) was found specially informative. Although both techniques, ENDOR and HYSCORE, detect nuclear frequencies, the signal intensities are different; thus, the two techniques are often complementary. Fig. 7 shows the HYSCORE spectra of several CcmE preparations again recorded at g‖ for the HS species. The spectrum of WT CcmE shows signals from strongly coupled nuclei with I = 1 in the (−, +) quadrant (Fig. 7 A). For the single-quantum (sq) nuclear transitions, the frequencies to first-order are the ones given in Eq. 2, and for the double-quantum (dq) transitions they can be expressed as
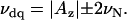 |
(3) |
FIGURE 7.

X-band HYSCORE spectra taken at 348 mT and 3.8 K. (A) WT CcmE. (B) CcmEY134H. (C) WT CcmE in deuterated water. The spectra shown are to the sum of three sets of HYSCORE data taken for τ values of 100, 124, and 192 ns.
The most intense correlations in the spectrum are the ridges centered at (−10.3, 6.0) MHz and (−6.0, 10.3) MHz (from now, we only mention one line of the pair). The difference between the two frequencies is ∼4νN, indicating that they correspond to the dq frequencies of a nitrogen nucleus in the two electron spin manifolds. Two less intense correlations were detected in the same quadrant at (−5.5, 2.6) MHz and (−4.8, 3.4) MHz. They were assigned to the sq frequencies of the same nucleus. With these experimental data we find Az = 8.1 MHz and Qz = 0.25 MHz, and, consequently, the signals are assigned to the four approximately equivalent nitrogen nuclei of the porphyrin ring (40). The sq frequencies of these nitrogens were also detected in the ENDOR experiments; however, the 2D HYSCORE spectra allow much better resolution. Again, no signals that could be attributed to axial nitrogens were detected even using matched pulses (16) to enhance the contribution of axial ligands.
The low resolution of the signals in the ENDOR spectra and the ridge-shaped dq features in the HYSCORE spectra indicate that there are distributions of centers with slightly different environments which are translated to distributions of the coupling parameters, e.g., g-strain or A-strain. As a consequence, the g values and the hyperfine interactions are not the same for species with the same field orientation, which results in a broadening of the lines. This is especially remarkable in the ENDOR spectrum of CcmE, where the resolution is much poorer than that in myoglobin.
In the (+, +) quadrant of Fig. 7 A, a proton ridge perpendicular to the diagonal is observed at νH = 14.8 MHz and spans a frequency range of ∼5 MHz. This ridge indicates interactions with protons of the molecule or of the solvent. Fig. 7 B shows the corresponding HYSCORE spectrum of CcmEY134H. Apart from a poorer signal/noise ratio as a result of the very low concentration of HS centers, the spectrum is the same as that for WT CcmE, proving that these proton interactions are present in the axial HS species.
To gain a deeper insight into the nature of the interacting protons, the spectrum of CcmE WT prepared in deuterated water was measured (Fig. 7 C). The features in the (−, +) quadrant were again the same. However, there are changes in the (+, +) quadrant caused by the replacement of exchangeable protons by deuterons. Only the proton matrix line is still visible, indicating that the detected interactions with nonexchangeable protons of the protein are smaller than 2 MHz. On the other hand, the strong matrix line caused by small hyperfine interactions with deuterons of the solvent can also be seen in the (+, +) quadrant of Fig. 7 C (νD = 2.3 MHz). The interaction with the more strongly coupled deuterons is not resolved. If we assume that the stronger interaction with the protons is ∼5 MHz, the deuterium hyperfine coupling constant would be <0.8 MHz, which is too low to be resolved from the strong matrix line. Consequently, we can assign the stronger couplings of the ridge to exchangeable protons or protons of the solvent that are close to the heme.
If we assume that the dipole interaction is the main contribution to the hyperfine coupling, an estimation can be made using the point-dipole approximation (16):
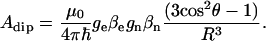 |
(4) |
To calculate Adip, the distance Fe-H and the angle θ between the Fe-H direction and the normal to the heme plane are needed. The closest position to the iron where an exchangeable proton can be found is bound to the axial ligand, which, as it does not seem to be nitrogen, must be an oxygen atom. Taking the structural data measured for the protons of the axial water in myoglobin (Fe-O distance of 2.22 Å and θ = 20° (44)), we find a value of Adip = 6.5 MHz. Interestingly, a hyperfine coupling of an interchangeable proton of ∼6 MHz has been reported in ENDOR studies of metmyoglobin (45) and was assigned to the heme-bound water protons. The difference between the experimental value and Adip was attributed to the isotropic contribution to the hyperfine coupling, Aiso.
It may be possible that slight variations in the Fe-O bond lengths and angles compared to metmyoglobin or a noticeable change in Aiso could lead to a reduction of the coupling to <5 MHz for an axial coordinating water. To check this possibility, and to explore the accessibility of the solvent to coordinating positions of the iron, we recorded the ENDOR spectrum for the WT CcmE prepared in  buffer (Fig. 6 E). 17O signals from the coordinating water molecule of myoglobin have been detected by ENDOR at g ≈ 2 previously (46). Nevertheless, in the case of CcmE, the ENDOR spectrum is the same to the one of the sample in nonlabeled buffer (Fig. 6 A) indicating that no water molecule coordinates the heme iron.
buffer (Fig. 6 E). 17O signals from the coordinating water molecule of myoglobin have been detected by ENDOR at g ≈ 2 previously (46). Nevertheless, in the case of CcmE, the ENDOR spectrum is the same to the one of the sample in nonlabeled buffer (Fig. 6 A) indicating that no water molecule coordinates the heme iron.
The next candidate for the proton signal would be a proton forming a hydrogen bond with the axial ligand. If we consider the structural parameters of such a proton (O-H distance of 1.6 Å, Fe-O-H angle of 107° and a Fe-O distance of 2 Å, which is what has been observed in cases where the heme iron coordinates an oxygen atom from an amino acid (47–49)), then Adip = 4.3 MHz. This value is in reasonable agreement with the value observed experimentally. Moreover, this proton would be covalently bound to a nitrogen or oxygen of another amino acid in the molecule or the solvent and in both cases would be exchangeable upon deuteration of the solvent. A hydrogen bond is also intrinsically more flexible in distance and bond angle, which could explain the ridge shape of the line in the HYSCORE spectrum. Nevertheless, the presence of more than one interacting exchangeable proton cannot be excluded.
Note that at B0 = 348 mT also some LS centers are in resonance and thus could contribute to the ENDOR and/or HYSCORE spectrum, especially in CcmEY134H. Despite this, no lines attributable to LS centers were detected with our experimental conditions (for example, the most intense LS correlation (dq-dq) would be expected to be around (−7, 3) MHz in the HYSCORE experiment (50)). Nevertheless, the magnetic interactions of the heme iron in the LS form can be probed choosing other magnetic field values. Fig. 8 shows the HYSCORE spectrum of CcmEY134H at B0 = 234 mT (g ≈ 2.96). This field position selects only the LS centers with the porphyrin plane perpendicular to the magnetic field, for this reason the signals in the spectrum are well resolved peaks. This spectrum is almost identical to the equivalent spectrum of the bis-imidazol model complex PPIX(Im)2 (50) and signals from both heme nitrogens and axial histidines are seen. This allows us to assign the signals to dq-dq and sq-sq transitions of the heme and histidine nitrogens (see Fig. 8) and confirms that the LS centers in CcmEY134H are bis-histidine coordinated.
FIGURE 8.

X-band HYSCORE spectra of LS centers in CcmEY134H taken at 234 mT and 9 K. The spectrum shown corresponds to a τ value of 96 ns for a better comparison with the spectrum obtained by Garcia-Rubio et al. (50). The arrows indicate the assignment of the features in the spectrum.
The position of the peaks in the different HYSCORE spectra, their assignment and the calculated coupling constants are summarized in Table 2.
TABLE 2.
Position, assignment, and calculated coupling parameters for the correlations found in the HYSCORE spectra
| Peak positions (MHz) | Nuclear transition type | Nucleus | Coupling parameters (MHz) |
|---|---|---|---|
| High-spin, B0 = 348 mT | |||
| Nitrogen | |||
| (−10.3, 6.0) | dq-dq | Az = 8.1 | |
| (−5.5, 2.6) | sq-sq | N-Heme | Qz = 0.25 |
| (−4.8, 3.4) | sq-sq | ||
| Proton | |||
| (18, 12) – (16, 14) | sq-sq | Exchangeable protons | 2 < A < 5 |
| Matrix line | |||
| Ridge to (16, 14) | sq-sq | Nonexchangeable H | A < 2 |
| Low-spin, B0 = 234 mT | |||
| Nitrogen | |||
| (−14.8, 9.2) | 2(dq-dq) | ||
| (−7.4, 4.6) | dq-dq | N-Heme | Az = 5.8 |
| (−4.3, 1.7) | sq-sq | Qz = 0.4 | |
| (−3.1, 2.9) | sq-sq | ||
| (−14.0, 8.4) | dq-dq + dq-dq | N-Heme & N-Imidazole | |
| (−6.6, 3.8) | dq-dq | ||
| (−4.5, 3.1) | sq-sq | Az = 5.1 | |
| (−3.1, 2.1) | sq-sq | N-Imidazole | Qz = 0.8 |
| (−3.1,0.7) | sq-sq | ||
| Proton | |||
| (11.7, 8.9) | sq-sq | H(δ) & H(ɛ) Imidazole | Az = 1.8 |
DISCUSSION
Maturation of c-type cytochromes requires the transfer of heme from holo-CcmE to apo-cytochrome c. During this process the covalent CcmE-heme bond has to be broken, and the two Cys residues of the CXXCH signature motif found in c-type cytochromes each have to stereospecifically form a covalent bond with one of the two heme vinyl groups, whereas the His residue of this motif ends up being an axial heme ligand (4). The chemical reaction of this transfer is unknown. However, we favor a model in which the His residue of the CXXCH motif binds heme as an axial ligand before the Cys residue binds, thereby enabling stereospecific heme attachment. For this to occur, the CcmE bound heme either has to have a free coordination site accessible for the CXXCH His residue or one of the CcmE heme ligands has to be exchanged by the CXXCH His residue. As we lack the structure of the holo-protein, the information about how the heme is bound in CcmE must be sought by spectroscopy. A first Raman study suggested Tyr and His as axial ligands and proposed a change on the coordination number with the oxidation state (7). The relative significance of the ferric and ferrous states is not known yet, but a change in the oxidation and coordination states could be involved in the maturation process. Here we analyzed the oxidized form of holo-CcmE by EPR spectroscopy and were able to add more information on the axial coordination of iron in ferric CcmE.
Fe3+ has five d electrons. In an approximate octahedral symmetry Fe3+ can attain two spin states: S = 5/2 (HS), when the effect of the ligands is weaker than the electronic repulsion and S = 1/2 (LS), when the ligands are strong enough to force all five electrons into the t2g orbitals. The four nitrogen ligands of the porphyrin ring alone set the ligand field close to the transition between these two spin states, and therefore the spin state of the iron depends on the coordination strength of the axial ligands (9,51). Coordination by two strong ligands such as histidine, methionine, cysteine or lysine yields low spin, whereas in general five-coordinated centers or six-coordinated species involving a weak ligand (water, tyrosine, glutamate) attain high spin. The cw EPR experiments show the presence of a number of iron centers in CcmE preparations, both HS and LS, but in view of the results presented above, it can be concluded that the native axial coordination of CcmE yields a heme in the high spin state. However, two different HS forms were detected in ferric holo-CcmE; both have to be five-coordinated or have a weak sixth ligand.
In addition, evidence from several experiments suggests that both centers are five-coordinated. On one hand, the position of the coordination state marker bands of the Raman spectrum measured at room temperature (ν2 at 1579 nm and ν3 at 1492 nm) indicates a five-coordinated high-spin heme (7). On the other hand, the hyperfine coupling constant, Az, is ∼1 MHz larger than the one found in myoglobin (40). This implies that the change of coordination is large enough to generate a noticeable change in the electron spin density in the heme plane. Changes in the hyperfine coupling parameters of nuclei in the porphyrin plane have been observed by NMR and related to the coordination number in ferric HS hemoproteins (52). Moreover, the variation of heme nitrogen couplings on changes in heme axial coordination has been studied in a series of ENDOR experiments, and the authors have concluded that the larger couplings could be correlated to a five-coordinated heme, whereas smaller couplings would be associated with the six-coordinated hemin (42,43). All of this experimental evidence indicates, then, that CcmE is five-coordinated, in agreement with the structural model (5) that proposes the heme docked on the hydrophobic platform formed by the residues Phe37, Val110, and Leu127. None of those residues is an evident axial ligand for heme, and consequently, the heme is not expected to be coordinated on that side.
The two HS species differ in the rhombicity of their signals: the one that we called “rhombic” shows two resolved low-field features at g ≈ 6.7 and 4.9. These signals disappear on replacement of Tyr134 by a His, which definitely proves that they are related to a Tyr coordinated center. A rhombicity (R = (gx − gy)/16) of 11% can be calculated for this center, which is very close to the one reported for the form 2 of bovine liver catalase (8) and some mutants of myoglobin (25,26), in which the heme iron is known to be five-coordinated with a Tyr as the fifth ligand. As pointed out above, the two g values are not symmetrically placed around g = 6, but successful simulations of the spectra have been accomplished taking into account a 5–10% mixing of the S = 5/2 ground state with the slightly higher lying S = 3/2 state.
Additionally, an axial signal is present in the spectra of both WT and CcmEY134H. The position and shape of this line, its behavior on the addition/removal of glycerin (see Supplementary Material), and the ENDOR and HYSCORE spectra indicate that the axial species is the same in the WT and in the mutant protein, but its ligand is not obviously assigned. As described in the experimental section, no axial nitrogens were detected by ENDOR or HYSCORE either for WT CcmE or CcmEY134H; therefore, most likely the axial ligand is an oxygen atom. According to our simulations the width of the axial signal hides a small rhombicity of ∼4% or 5%. An axial coordination of tyrosine to the heme iron has been reported to give rise to HS signals with such small rhombicity, for example in the naturally occurring mutants of hemoglobin (9,23,53). However, Tyr134 can be ruled out because it is not present in CcmEY134H and the signal persists in this spectrum. The other Tyr residues in the globular part of the protein can also be discarded because they are far away from the heme binding site in the structure. Tyr156 is also not the axial ligand because the axial signal is still present in truncated CcmE constructs that lack this part of the C-terminal domain (CcmE30–138). The detection of an axial water molecule or OH− in the experiments performed in 17O-water was negative; this leads us to the consideration of the oxygen atoms in the carboxyl side chains of acidic residues. This kind of coordination would match our HYSCORE results because, at the pH of the experiments, the oxygen of the carboxyl group would be deprotonated but able to form a hydrogen bond with another residue of the protein or a water molecule. There is published work characterizing a mutant of cytochrome c peroxidase in which the axial nitrogen was replaced by a glutamic acid (47,54). It is not clear if the cw EPR spectrum reported corresponds to a six- or five-coordinated species, but it displays an axial signal similar to the one we find in CcmE. Unlike what was described for other iron coordinations, the authors did not find any specific bands characteristic of glutamate coordination in the Raman spectra. This fact could have caused the axial species to be overlooked in the Raman study of CcmE (7).
Glu138 in the flexible domain is conserved as an acidic residue and could be the fifth ligand of the iron. However, several indications favor Asp131 or Glu132. Both are also highly conserved (5). Additionally, their position in the sequence is apparently optimal to bind the heme. In the CXXCH pattern that typically binds heme in cytochromes c, the His axial ligand is next to one of the residues binding the heme covalently. In this case, Asp131 would be in an analogous position, next to His130, which is the residue binding covalently one of the vinyl groups of heme (4).
The effect of point mutations of several residues around the heme binding site has been reported (12). These results show that the heme binding to CcmE was strongly reduced in the mutant CcmEY134A. On the other hand, the single mutants CcmEE131A and CcmED132A were not so influenced by the mutation, but the double mutant CcmEE131A/D132A was. The biological significance of the axial signal and of the presence of two distinct heme environments needs to be further investigated, but there is experimental evidence that the two different species are interconvertible. The relative percentage of the two species can be altered by the addition of a large amount of glycerol to the solution (see Supplementary Material for details) or by mutation of the Tyr134 by His. Additionally, the results shown above indicate that the two species have different axial coordination. Because the structural model proposed for this protein (see Fig. 1) places the heme between a hydrophobic rigid platform and the flexible α-helical domain of the protein, where Tyr134, Glu138, Asp131, and Glu132 are located (5,7), this would mean the presence of two conformations of CcmE differing in the structure of the flexible domain. Even within each of the two well-defined conformations there are indications of a certain degree of flexibility given by the broad distribution of slightly different environments appearing in the spectra (distribution of Δ2 and Δ3 that was necessary to introduce to simulate the cw spectra, the intrinsic poor resolution in ENDOR, and the ridges in the HYSCORE spectra). This conformational flexibility fits perfectly to the highly dynamic properties that the function of this protein implies because it has to receive, transiently accommodate, and deliver heme and also allows specific interactions with the heme donor and acceptor proteins.
The additional LS signals at g = 2.96, 2.27, and 1.52 are caused by bis-histidine coordinated centers. Although the presence of LS centers was shown to be an artifact regarding the native state of CcmE, the interaction of the heme with histidines could potentially be interesting. A residual amount of LS is already present in WT CcmE, where the only histidines in the vicinity of the heme are His130, which binds the heme covalently, and His147 in the flexible tail. This latter residue must be involved in the coordination of the LS heme because it doesn't appear in the CcmE30–138 truncated version of the WT protein. In any case, the LS species most probably represents a misfolding of the protein. When a histidine is placed in the position 134, the protein folds in such a way that there is another histidine coordinating as a sixth ligand, most probably His147, because the coordination of His130 would be detected in the UV-VIS spectrum as a considerable amount of noncovalent complex in the sample. In any case, the point is that when one histidine is in a coordinating position, the flexibility of the tail is sufficient to stabilize the complex with a second histidine. Also, the stability of this LS form is clear by the change of proportions of the two species of CcmE. In the WT the proportion of species ligated to residue 134 to the one ligated to the carboxyl group is 1:3, versus 3:1 in CcmEY134H. Indeed, bis-histidine coordination is so stable that a stequiometric excess of imidazole displaces the other ligands. By removing the excess of imidazole, the HS are recovered, but the amount of heme is less, which might indicate that bis-histidine coordination could be important in the uptake and release of heme.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors thank C. Aldag and Prof. K. Pervushin for their help in the completion of this work.
This research has been supported by the Swiss National Science Foundation.
Arthur Schweiger was deceased January 2006.
Martin Braun's present address is BIOTECH, El Playón, Tecoluca, San Vicente, El Salvador.
References
- 1.Thöny-Meyer, L., F. Fischer, P. Künzler, D. Ritz, and H. Hennecke. 1995. Escherichia coli genes required for cytochrome c maturation. J. Bacteriol. 177:4321–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grove, J., S. Tanapongpipat, G. Thomas, L. Griffiths, H. Crooke, and J. Cole. 1996. Escherichia coli K-12 genes essential for the synthesis of c-type cytochromes and a third nitrate reductase located in the periplasm. Mol. Microbiol. 19:467–481. [DOI] [PubMed] [Google Scholar]
- 3.Schulz, H., H. Hennecke, and L. Thöny-Meyer. 1998. Prototype of a heme chaperone essential for cytochrome c maturation. Science. 281:1197–1200. [DOI] [PubMed] [Google Scholar]
- 4.Lee, D., K. Pervushin, D. Bischof, M. Braun, and L. Thöny-Meyer. 2005. Unusual heme-histidine bond in the active site of a chaperone. J. Am. Chem. Soc. 127:3716–3717. [DOI] [PubMed] [Google Scholar]
- 5.Enggist, E., L. Thöny-Meyer, P. Guntert, and K. Pervushin. 2002. NMR structure of the heme chaperone CcmE reveals a novel functional motif. Structure. 10:1551–1557. [DOI] [PubMed] [Google Scholar]
- 6.Arnesano, F., L. Banci, P. D. Barker, I. Bertini, A. Rosato, X. C. Su, and M. S. Viezzoli. 2002. Solution structure and characterization of the heme chaperone CcmE. Biochemistry. 41:13587–13594. [DOI] [PubMed] [Google Scholar]
- 7.Uchida, T., J. M. Stevens, O. Daltrop, E. M. Harvat, L. Hong, S. J. Ferguson, and T. Kitagawa. 2004. The interaction of covalently bound heme with the cytochrome c maturation protein CcmE. J. Biol. Chem. 279:51981–51988. [DOI] [PubMed] [Google Scholar]
- 8.Peisach, J., W. E. Blumberg, S. Ogawa, E. A. Rachmilewitz, and R. Oltzik. 1971. The effects of protein conformation on the heme symmetry in high spin ferric heme proteins as studied by electron paramagnetic resonance. J. Biol. Chem. 246:3342–3355. [PubMed] [Google Scholar]
- 9.Palmer, G. 1983. Electron paramagnetic resonance of hemeproteins. In Iron Porphyrins, Part II. A. B. P. Lever and H. B. Gray, editors. Addison Wesley, London. 43–88.
- 10.Walker, F. 1999. Magnetic spectroscopic (EPR, ESEEM, Mössbauer, MCD and NMR) studies of low-spin ferriheme centers and their corresponding heme proteins. Coord. Chem. Rev. 185–186:471–534. [Google Scholar]
- 11.Arslan, E., H. Schulz, R. Zufferey, P. Künzler, and L. Thöny-Meyer. 1998. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 251:744–747. [DOI] [PubMed] [Google Scholar]
- 12.Enggist, E., M. J. Schneider, H. Schulz, and L. Thöny-Meyer. 2003. Biochemical and mutational characterization of the heme chaperone CcmE reveals a heme binding site. J. Bacteriol. 185:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. [DOI] [PubMed] [Google Scholar]
- 14.Fuhrhop, J. H., and K. M. Smith. 1975. Laboratory methods. In Porphyrins and Metalloporphyrins. K. M. Smith, editor. Elsevier Science, Amsterdam. 757–869.
- 15.Höfer, P., A. Grupp, H. Nebenführ, and M. Mehring. 1986. Hyperfine Sublevel Correlation (HYSCORE) spectroscopy—a 2D electron-spin-resonance investigation of the squaric acid radical. Chem. Phys. Lett. 132:279–282. [Google Scholar]
- 16.Schweiger, A., and G. Jeschke. 2001. Principles of Pulse Electron Paramagnetic Resonance. Oxford University Press, Oxford.
- 17.Davies, E. R. 1974. New pulse endor technique. Phys. Lett. A. 47:1–2. [Google Scholar]
- 18.Stoll, S., and A. Schweiger. 2006. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178:42–55. [DOI] [PubMed] [Google Scholar]
- 19.Enggist, E., and L. Thöny-Meyer. 2003. The C-terminal flexible domain of the heme chaperone CcmE is important but not essential for its function. J. Bacteriol. 185:3821–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daltrop, O., J. M. Stevens, C. W. Higham, and S. J. Ferguson. 2002. The CcmE protein of the c-type cytochrome biogenesis system: unusual in vitro heme incorporation into apo-CcmE and transfer from holo-CcmE to apocytochrome. Proc. Natl. Acad. Sci. USA. 99:9703–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peisach, J., W. E. Blumberg, and A. Adler. 1973. Electron-paramagnetic resonance studies of iron porphin and chlorin systems. Ann. N. Y. Acad. Sci. 206:310–327. [DOI] [PubMed] [Google Scholar]
- 22.Gaffney, B. J., and H. J. Silverstone. 1993. Simulation of the EMR spectra of high-spin iron in proteins. In Biological Magnetic Resonance, Volume 13. L. J. Berliner and J. Reuben, editors. Plenum Press, New York. 1–57.
- 23.Nagai, M., K. Mawatari, Y. Nagai, S. Horita, Y. Yoneyama, and H. Hori. 1995. Studies of the oxidation states of hemoglobin M Boston and hemoglobin M Saskatoon in blood by EPR spectroscopy. Biochem. Biophys. Res. Commun. 210:483–490. [DOI] [PubMed] [Google Scholar]
- 24.Williams-Smith, D. L., and K. Patel. 1975. Induced changes in the electron paramagnetic resonance spectra of mammalian catalases. Biochim. Biophys. Acta. 405:243–252. [DOI] [PubMed] [Google Scholar]
- 25.Egeberg, K. D., B. A. Springer, S. A. Martinis, S. G. Sligar, D. Morikis, and P. M. Champion. 1990. Alteration of sperm whale myoglobin heme axial ligation by site-directed mutagenesis. Biochemistry. 29:9783–9791. [DOI] [PubMed] [Google Scholar]
- 26.Adachi, S., S. Nagano, K. Ishimori, Y. Watanabe, I. Morishima, T. Egawa, T. Kitagawa, and R. Makino. 1993. Roles of proximal ligand in heme proteins: replacement of proximal histidine of human myoglobin with cysteine and tyrosine by site-directed mutagenesis as models for P-450, chloroperoxidase, and catalase. Biochemistry. 32:241–252. [DOI] [PubMed] [Google Scholar]
- 27.Migita, C., and M. Iwaizumi. 1981. Low-temperature electron-paramagnetic-resonance studies of highly anisotropic low-spin (protoporphyrinato)iron(III) complexes. J. Am. Chem. Soc. 103:4378–4381. [Google Scholar]
- 28.Baker, R. D., C. O. Cook, and D. C. Goodwin. 2004. Properties of catalase-peroxidase lacking its C-terminal domain. Biochem. Biophys. Res. Commun. 320:833–839. [DOI] [PubMed] [Google Scholar]
- 29.Chouchane, S., S. Girotto, S. Kapetanaki, J. P. Schelvis, S. Yu, and R. S. Magliozzo. 2003. Analysis of heme structural heterogeneity in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 278:8154–8162. [DOI] [PubMed] [Google Scholar]
- 30.Maltempo, M. 1974. Magnetic state of an unusual bacterial heme protein. J. Chem. Phys. 61:2540–2547. [Google Scholar]
- 31.Weissbluth, M. 1967. The physics of hemoglobin. In Structure and Bonding, Volume 2. C. K. Jorgensen, J. B. Neilands, R. S. Nyholm, D. Reinen, and R. J. P. Williams, editors. Springer Verlag, Berlin. 1–125.
- 32.Scholes, C. P., R. A. Isaacson, and G. Feher. 1971. Determination of zero-field splitting of Fe3+ in heme proteins from temperature dependence of spin-lattice relaxation rate. Biochim. Biophys. Acta. 244:206–210. [DOI] [PubMed] [Google Scholar]
- 33.Abragam, A., and B. Bleaney. 1986. Electron Paramagnetic Resonance of Transition Ions. Dover Publications, New York.
- 34.Salmeen, I., and G. Palmer. 1968. Electron paramagnetic resonance of beef-heart ferricytochrome c. J. Chem. Phys. 48:2049–2052. [DOI] [PubMed] [Google Scholar]
- 35.Svistunenko, D., M. Sharpe, P. Nicholls, M. Wilson, and C. Cooper. 2000. A new method for quantitation of spin concentration by EPR spectroscopy: Application to methemoglobin and metmyoglobin. J. Magn. Reson. 142:266–275. [DOI] [PubMed] [Google Scholar]
- 36.Aasa, R., and T. Vanngard. 1975. EPR signal intensity and powder shapes re-examination. J. Magn. Reson. 19:308–315. [Google Scholar]
- 37.Tierney, D. L., P. Martasek, P. E. Doan, B. S. S. Masters, and B. M. Hoffman. 1998. Location of guanidino nitrogen of l-arginine substrate bound to neuronal nitric oxide synthase (nNOS): Determination by Q-band pulsed ENDOR spectroscopy. J. Am. Chem. Soc. 120:2983–2984. [Google Scholar]
- 38.Tierney, D. L., H. Huang, P. Martasek, B. S. S. Masters, R. B. Silverman, and B. M. Hoffman. 1999. ENDOR spectroscopic evidence for the position and structure of N-G-hydroxy-l-arginine bound to holo-neuronal nitric oxide synthase. Biochemistry. 38:3704–3710. [DOI] [PubMed] [Google Scholar]
- 39.Jiang, F., T. Zuberi, J. Cornelius, R. Clarkson, R. Gennis, and R. Belford. 1993. Nitrogen and proton endor of cytochrome-d, hemin, and metmyoglobin in frozen-solutions. J. Am. Chem. Soc. 115:10293–10299. [Google Scholar]
- 40.Scholes, C., A. Lapidot, R. Mascarenhas, T. Inubushi, R. Isaacson, and G. Feher. 1982. Electron nuclear double-resonance (ENDOR) from heme and histidine nitrogens in single-crystals of aquometmyoglobin. J. Am. Chem. Soc. 104:2724–2735. [Google Scholar]
- 41.Scholes, C. P., R. A. Isaacson, and G. Feher. 1972. Electron nuclear double resonance studies on heme proteins: determination of the interaction of Fe3+ with its ligand nitrogens in metmyoglobin. Biochim. Biophys. Acta. 263:448–452. [DOI] [PubMed] [Google Scholar]
- 42.Van Camp, H., C. Scholes, C. Mulks, and W. Caughey. 1977. Electron nuclear double-resonance of a series of axially liganded proto-hemins and deutero-hemins. J. Am. Chem. Soc. 99:8283–8290. [Google Scholar]
- 43.Van Camp, H., C. Scholes, and C. Mulks. 1976. Electron nuclear double resonance on heme compounds - endor from iron ligands in protohemin chloride and protohemin bromide. J. Am. Chem. Soc. 98:4094–4098. [DOI] [PubMed] [Google Scholar]
- 44.Oldfield, T. J., S. J. Smerdon, Z. Dauter, K. Petratos, K. S. Wilson, and A. J. Wilkinson. 1992. High-resolution x-ray structures of pig metmyoglobin and two CD3 mutants: Mb(Lys45→Arg) and Mb(Lys45→Ser). Biochemistry. 31:8732–8739. [DOI] [PubMed] [Google Scholar]
- 45.Mulks, C., C. Scholes, L. Dickinson, and A. Lapidot. 1979. Electron nuclear double-resonance from high-spin and low-spin ferric hemoglobins and myoglobins. J. Am. Chem. Soc. 101:1645–1654. [Google Scholar]
- 46.Veselov, A. V., J. P. Osborne, R. B. Gennis, and C. P. Scholes. 2000. Q-band ENDOR (electron nuclear double resonance) of the high-affinity ubisemiquinone center in cytochrome bo(3) from Escherichia coli. Biochemistry. 39:3169–3175. [DOI] [PubMed] [Google Scholar]
- 47.Choudhury, K., M. Sundaramoorthy, A. Hickman, T. Yonetani, E. Woehl, M. F. Dunn, and T. L. Poulos. 1994. Role of the proximal ligand in peroxidase catalysis - crystallographic, kinetic, and spectral studies of cytochrome-c peroxidase proximal ligand mutants. J. Biol. Chem. 269:20239–20249. [PubMed] [Google Scholar]
- 48.Fita, I., A. M. Silva, M. R. N. Murthy, and M. G. Rossmann. 1986. The refined structure of beef-liver catalase at 2.5 a resolution. Acta Crystallogr. Sect. B: Struct. Sci. 42:497–515. [Google Scholar]
- 49.Maurus, R., R. Bogumil, Y. Luo, H. L. Tang, M. Smith, A. G. Mauk, and G. D. Brayer. 1994. Structural characterization of heme ligation in the His64→Tyr variant of myoglobin. J. Biol. Chem. 269:12606–12610. [DOI] [PubMed] [Google Scholar]
- 50.García-Rubio, I., J. Martínez, R. Picorel, I. Yruela, and P. Alonso. 2003. HYSCORE spectroscopy in the cytochrome b(559) of the photosystem II reaction center. J. Am. Chem. Soc. 125:15846–15854. [DOI] [PubMed] [Google Scholar]
- 51.Moore, G. F., and G. W. Pettigrew. 1990. Cytochromes c. Evolutionary Structural and Physicochemical Aspects. Springer-Verlag, Berlin, Heidelberg.
- 52.Morishima, I., Y. Shiro, and T. Wakino. 1985. Meso deuterium NMR hyperfine shift as a probe for determining 5-coordination or 6-coordination at heme iron-binding site in ferric high-spin hemoproteins. J. Am. Chem. Soc. 107:1063–1065. [Google Scholar]
- 53.Hayashi, A., T. Suzuki, K. Imai, H. Morimoto, and H. Watari. 1969. Properties of hemoglobin M, Milwaukee-I variant and its unique characteristic. Biochim. Biophys. Acta. 194:6–15. [DOI] [PubMed] [Google Scholar]
- 54.Smulevich, G., F. Neri, O. Willemsen, K. Choudhury, M. P. Marzocchi, and T. L. Poulos. 1995. Effect of the His175 to Glu mutation on the heme pocket architecture of cytochrome-c peroxidase. Biochemistry. 34:13485–13490. [DOI] [PubMed] [Google Scholar]