

Abstract
A formalism for membrane protein structure determination was developed. This method is based on steady-state FRET data and information about the position of the fluorescence maxima on site-directed fluorescent labeled proteins in combination with global data analysis utilizing simulation-based fitting. The methodology was applied to determine the structural properties of the N-terminal domain of the major coat protein from bacteriophage M13 reconstituted into unilamellar DOPC/DOPG (4:1 mol/mol) vesicles. For our purpose, the cysteine mutants A7C, A9C, N12C, S13C, Q15C, A16C, S17C, and A18C in the N-terminal domain of this protein were produced and specifically labeled with the fluorescence probe AEDANS. The energy transfer data from the natural Trp-26 to AEDANS were analyzed assuming a two-helix protein model. Furthermore, the polarity Stokes shift of the AEDANS fluorescence maxima is taken into account. As a result the orientation and tilt of the N-terminal protein domain with respect to the bilayer interface were obtained, showing for the first time, to our knowledge, an overall α-helical protein conformation from amino acid residues 12–46, close to the protein conformation in the intact phage.
INTRODUCTION
M13 major coat protein is a small protein composed of 50 amino acid residues. The protein is involved in the membrane-bound assembly and disassembly of the phage M13 in the Escherichia coli host cytoplasmic membrane and has been the subject of several biophysical studies (for a recent review see (1)). Generally it is believed that approximately half of the protein is located in the membrane, whereas the remaining N-terminal residues are sticking out of the membrane. Despite intensive studies, the topology of the coat protein in lipid bilayers is still a matter of debate. This is mainly due to biophysical inabilities to study the structure and dynamics of the N-terminal domain of the protein in detail. Models for the overall topology of the protein varied 90° from an L-shape to an I-shape (1). This arises because the protein is a single membrane-spanning system that has no internal stability based on segment-segment interactions (1). This means that there is no tertiary structure to hold the protein together. Also, recently it was suggested that the protein is strongly affected by the environment into which it is inserted, i.e., micelles, vesicles, liposomes, or oriented membranes (2). These factors are most important for the N-terminal domain of the protein that emerges from the membrane.
To resolve this problem we have produced several cysteine mutants in the N-terminal domain of the protein and specifically labeled them with the fluorescence probe AEDANS. Analysis of the energy transfer data from the natural Trp-26 to AEDANS using a two-helix protein model and the application of the polarity Stokes shift of the AEDANS fluorescence maxima results in a low-resolution structure of the entire protein, including the tilt and orientation of the N-terminal domain with respect to the transmembrane domain.
EXPERIMENTAL METHODS
Sample preparation
Lipid bilayer systems were prepared from DOPC and DOPG lipids in a 4:1 molar ratio, denoted as DOPC/DOPG, as described before (3). Site-specific cysteine mutants of M13 major coat protein were prepared, purified and labeled with AEDANS as described previously (4). Wild-type protein and AEDANS-labeled M13 coat protein mutants were reconstituted into phospholipid bilayers as reported earlier (5).
Protein titration experiments were carried out using the same protocol as described previously (3). AEDANS-labeled cysteine mutants of M13 coat protein were used with the cysteine residue at positions 7 (A7C), 9 (A9C), 12 (N12C), 13 (S13C), 15 (Q15C), 16 (A16C), 17 (S17C), and 18 (A18C). Titration experiments were performed in which the wild-type protein concentration was increased whereas the mutant concentration was kept constant. The sample conditions for these titrations are given in Table 1. The labeling efficiencies were determined as reported previously (6) and are given in Table 1 as well. The labeling efficiency is explicitly taken into account in Table 1 in the ratio of the number of unlabeled to labeled proteins (rul), as it affects the acceptor concentration and therefore the energy transfer efficiency.
TABLE 1.
Sample composition of M13 major coat protein incorporated into DOPC/DOPG vesicles
| Mutant | A7C | A9C | N12C | S13C | Q15C | A16C | S17C | A18C |
|---|---|---|---|---|---|---|---|---|
| nA | 7 | 9 | 12 | 13 | 15 | 16 | 17 | 18 |
| Acceptor fluorescence maximum λmax, nm | 497.6 | 496.5 | 496.7 | 499.5 | 493.6 | 495.1 | 494.2 | 491.1 |
| Labeling efficiency | 0.44 | 0.78 | 0.79 | 0.55 | 0.53 | 0.85 | 0.54 | 0.56 |
| rLP | 336.0 | 217.0 | 276.9 | 561.5 | 422.6 | 239.7 | 267.7 | 248.6 |
| rul | 1.27 | 0.28 | 0.27 | 0.82 | 0.89 | 0.18 | 0.85 | 0.79 |
| E | 0.172 | 0.463 | 0.505 | 0.338 | 0.443 | 0.880 | 0.448 | 0.488 |
| rLP | 213.2 | 158.2 | 184.8 | 286.2 | 245.1 | 169.9 | 180.7 | 171.8 |
| rul | 2.58 | 0.76 | 0.90 | 2.57 | 2.25 | 0.66 | 1.74 | 1.58 |
| E | 0.119 | 0.366 | 0.360 | 0.184 | 0.261 | 0.650 | 0.323 | 0.354 |
| rLP | 156.2 | 124.5 | 138.7 | 192.0 | 172.6 | 131.6 | 136.3 | 131.2 |
| rul | 3.89 | 1.24 | 1.53 | 4.32 | 3.62 | 1.14 | 2.64 | 2.38 |
| E | 0.095 | 0.307 | 0.279 | 0.129 | 0.200 | 0.513 | 0.261 | 0.286 |
| rLP | 123.2 | 102.6 | 111.0 | 144.5 | 133.2 | 107.4 | 109.5 | 106.2 |
| rul | 5.20 | 1.71 | 2.16 | 6.07 | 4.99 | 1.63 | 3.53 | 3.18 |
| E | 0.084 | 0.260 | 0.238 | 0.102 | 0.152 | 0.436 | 0.217 | 0.248 |
| rLP | 101.7 | 87.2 | 92.5 | 115.8 | 108.5 | 90.7 | 91.5 | 89.1 |
| rul | 6.51 | 2.19 | 2.79 | 7.82 | 6.35 | 2.11 | 4.42 | 3.98 |
| E | 0.076 | 0.231 | 0.199 | 0.086 | 0.129 | 0.383 | 0.191 | 0.225 |
| rLP | 86.6 | 75.9 | 79.3 | 96.6 | 91.5 | 78.5 | 78.5 | 76.8 |
| rul | 7.81 | 2.67 | 3.42 | 9.57 | 7.72 | 2.59 | 5.31 | 4.78 |
| E | 0.067 | 0.207 | 0.184 | 0.075 | 0.111 | 0.340 | 0.175 | 0.198 |
| rLP | 57.0 | 52.2 | 52.9 | 61.2 | 59.1 | 53.4 | 52.6 | 51.8 |
| rul | 12.4 | 4.33 | 5.63 | 15.69 | 12.50 | 4.28 | 8.43 | 7.57 |
| E | 0.055 | 0.160 | 0.137 | 0.055 | 0.079 | 0.254 | 0.138 | 0.149 |
Values are given in terms of rLP and rul, labeling efficiencies, and observed acceptor fluorescence maxima and energy transfer efficiencies E for mutants with acceptor positions nA at 7, 9, 12, 13, 15, 16, 17, and 18.
For the fluorescence experiments, stock solutions of protein mutants and wild-type protein solubilized in cholate buffer were mixed with solutions of lipids in the same buffer, as described previously (5). Repeated dialysis of the mixtures in cholate-free buffer was performed to remove the cholate in the sample. The lipid loss during dialysis can vary between 20–30% (3,5) and is accounted for in the analysis of the experimental data.
Fluorescence experiments
Fluorescence emission and fluorescence excitation measurements were performed at 20°C as described elsewhere (2,3). The position of the AEDANS emission maximum was different for different labeled mutants because the Stokes shift of AEDANS fluorescence significantly depends on the local polarity of the environment of the label and thus on the distance between the label and the center of the lipid bilayer (7,8). The position of the AEDANS emission maxima was determined using a polynomial approximation of the top part of the emission peak as in (7) and given in Table 1.
For fluorescence excitation measurements, the detection wavelength was set at the maximum of the acceptor (AEDANS) fluorescence of a particular mutant, and the excitation wavelength was scanned from 260 to 400 nm. The resulting AEDANS emission spectra for all mutants and examples of excitation spectra for mutant N12C are presented in Fig.1, A and B, respectively.
FIGURE 1.

(A) Emission spectra of M13 coat protein mutants A7C, A9C, N12C, S13C, Q15C, A16C, S17C, and A18C with AEDANS-labeled Cys in DOPC/DOPG vesicles after subtraction of the fluorescence of equimolar wild-type samples. The histogram shows the values of the acceptor emission maxima of the mutants. (B) Experimental excitation spectra obtained for mutant N12C at different titration points of wild-type proteins. The emission was detected at 496 nm. Labels 1–3 correspond to rul values of 0.27, 2.16, and 5.63, respectively. The lipid/protein ratios rLP are 277, 111, and 53, respectively (see Table 1). The sample showing the highest peak at 290 nm (spectrum 3) has the highest protein density (lowest rLP) and rul. Although the efficiency of energy transfer (Fig. 3) for this case is smallest, the overall energy absorbed by the donors in such a system, and therefore the transferred (intermolecular), is higher than for the other values of rLP and rul (3).
The energy transfer efficiency E (which is an average efficiency for all donors in the system and includes both intra- and intermolecular energy transfer) was calculated from the fluorescence intensities by
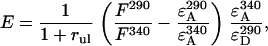 |
(1) |
where rul is the ratio of the number of unlabeled to labeled proteins. The derivation of this equation is described in detail elsewhere (3). For every sample the ratio of the fluorescence intensity at 290 nm, F290, (mainly donor excitation) to that at 340 nm, F340, (exclusively acceptor excitation) was calculated as a measure of the donor-to-acceptor energy transfer. The ratio F290/F340 was corrected for direct excitation of AEDANS at 290 nm by subtracting the ratio of the extinction coefficients 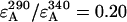 (calculated using mutant Y21A/Y24A/W26A/G23C). The ratio of the extinction coefficients of the acceptor at 340 nm (
(calculated using mutant Y21A/Y24A/W26A/G23C). The ratio of the extinction coefficients of the acceptor at 340 nm (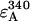 ) and donor at 290 nm (
) and donor at 290 nm (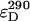 ) is 1.2.
) is 1.2.
METHODOLOGY
Model for M13 major coat protein incorporated into a lipid bilayer
In this study we will extend our previous single-helix model for the M13 major coat protein (3) to a two-helix model. This model consists of two flexibly linked helical domains connected via a kink (Fig. 2). One domain reflects the transmembrane protein part, and the other domain is the N-terminal protein part that is supposed to stick out of the membrane (1,7,9–11). The conformation of each domain is assumed to be a perfect α-helix. The main axis of the protein O is parallel to the transmembrane protein domain and defines the z axis of the axes system of the protein. The orientation of the x axis is defined by the location of the Cα of Trp-26 (donor), which is used as the reference amino acid residue. The complete set of structural parameters that determines the location and conformation of the protein is presented in Table 2. The protein parameters related to position, orientation, and tilt of the transmembrane domain are taken from a previous study (3). The parameter ranges given in Table 2 indicate the range of values considered in the simulations. It should be noted that the two-helix model could be easily generalized to other membrane proteins.
FIGURE 2.

Schematic drawing of the two-helix protein model with a donor (Trp-26, solid circle, located at a distance lD from the protein helix axis) and acceptor (AEDANS, shaded circle, located at a distance lA from the protein helix axis) attached at positions 26 and 9, respectively, in its own protein axis system (x, y, z). The orientation of the x axis is defined by the location of Trp-26, which is used as the reference amino acid residue. The complete set of structural parameters that describes the protein-lipid system is presented in Table 2.
TABLE 2.
Definition of the parameters used in the two-helix model of proteins embedded in lipid bilayers
| Parameter | Range/value | Unit | Description |
|---|---|---|---|
| N0 | 26 | – | The position of a reference amino acid residue. The projection of its Cα to the helix axis of the protein O gives the origin of the coordinate system of the protein. Position n0 = 26 was selected for the transmembrane domain of M13 major coat protein. |
| h | 1.5 | Å | Translation per amino acid residue along the helix; 1.5 Å for a perfect α-helix. |
| nr | 3.6 | – | Number of amino acid residues per one turn; 3.6 for a perfect α-helix. |
| nD | 26 | – | Donor position; position of amino acid residue given by the donor. For M13 coat protein the donor is Trp-26, which is located in the transmembrane domain. |
| nA | 1–50 | – | Acceptor position; position of amino acid residue labeled by the acceptor. For the transmembrane domain of M13 coat protein, the acceptor positions are 24, 38, and 46. |
| lD | 6.5 | Å | Donor arm, the average distance from the donor moiety to the helix axis. A value lD = 6.5 Å was taken (7). |
| lA | 9.5 | Å | Acceptor arm, the average distance from the acceptor moiety to the helix axis. A value lA = 9.5 Å was taken (7). |
| nk | 1–25 | – | Position of helix kink; position of amino acid residue from which the N-terminal helix starts. |
| θ | 18 | ° | Protein tilt angle; the angle between the helix axis and the normal to the membrane. The value of 18° is found in the previous study (3). |
| d | 8.5 | Å | Distance from the origin of the coordinate system of the protein to the centre of the bilayer is 8.5 Å (3). |
| ψ | 60 | ° | Protein tilt direction; the direction of the protein transmembrane domain tilting is ∼60° as found earlier (3,7). |
| NP | 500 | – | Number of proteins in the system. All simulations were performed for models containing 500 proteins. |
| SL | 72 | Å2 | Area occupied by a lipid in one leaflet of a bilayer; the average area for the DOPC/DOPG system is 72 Å2 (17). |
| L | 0.0–1.0 | – | Lipid loss; ratio of lipids lost during dialysis to their initial quantity. |
| DP | 10 | Å | Protein exclusion distance; minimal protein-protein distance. For M13 coat protein a value DP = 10 Å was taken. |
| rLP | ≥0 | – | Lipid to protein molar ratio. |
| rul | ≥0 | – | Ratio between the number of unlabeled and labeled proteins. |
| k | 0 | – | Protein-protein association probability, defined as the percentage of clustered proteins with respect to the total number of proteins, for considered case ∼0 (3,30). |
| R0 | 24 | Å | Förster distance. A value of 24 Å is calculated using the data about the photophysical properties of the donor and acceptor. |
| ϕ | 0–90 | ° | N-terminal helix tilt angle; the angle between the main axes of the two protein domains. |
| ξ | −180–180 | ° | N-terminal helix tilt direction; the direction of the N-terminal helix with respect to the x axis of the protein axis system. |
| ω | −180–180 | ° | N-terminal helix coaxial rotation; the turning angle of N-terminal helix around its main axis defining the direction of amino acid residues (toward water or lipid phase). The case ω = 0° corresponds to an ideal α-helix, bent at position nk by angle ϕ. |
In the simulations the parameters nk, ϕ, ω, ξ, and L are varied. Parameters nA, rLP and rul are determined by the experiment; the other parameters are taken from previous work (3) and are fixed as shown in the table.
Furthermore, a one-helix protein is a special case of the two-helix model with ϕ = ω = 0°.
The two-helix protein model is incorporated in a membrane as described before (3,12). A square region of a bilayer containing a certain number of randomly incorporated proteins (NP) is considered. By using a three-dimensional mathematical description, protein molecules as shown in Fig. 2 are inserted randomly (both in location as well as in orientation) into the lipid bilayer so that the angle θ between the normal membrane and their main axis O of the transmembrane domain is between 0° and 90°. The direction of the protein tilt is given by ψ. A value ψ = 0 means that the protein is tilted toward the Cα of the reference (n0) amino acid residue. The depth of protein insertion is given by parameter d. It is assumed that when inserted into the membrane, the proteins occupy a cylindrical region in both bilayer leaflets with a protein exclusion distance DP. Within this region, no lipids or other proteins can be located. For this study the value of the protein-protein association probability k (3) is ∼0 and can be neglected. Therefore, the distribution of proteins in the bilayer is considered as uniformly random.
FRET model and Förster distance
To analyze the experimental steady-state fluorescence data for our system, a steady-state FRET simulation is employed as described previously (3). The simulation starts with the generation of the spatial model for the protein-lipid system. This model provides the coordinates of each donor and acceptor. Based on this information, the energy transfer efficiency E is calculated. Because of the stochastic nature of the spatial model, the resulting energy transfer efficiency contains stochastic deviations. Therefore, the simulations are executed several times (in our case, 50) to make the results statistically relevant. In the simulations the value of Förster distance R0 was 24 Å (3).
To calculate the Förster distance, a value for the orientation factor κ2 = 2/3 was used, assuming a random orientation and mobility of the transition dipoles of the donor and acceptor relative to each other. The orientation factor depends both on the orientation and reorientation speed of the acceptor and donor. This approximation is believed to be generally valid for proteins (13) and in particular for the Trp-AEDANS donor-acceptor pair (14). In our case, this assumption is further supported by the fact that the Trp donor (15,16) and AEDANS acceptor (17) have a high degree of motion. In addition, all our measurements were carried out at room temperature in a mobile lipid environment, well above the gel-to-liquid crystalline phase transition temperature of the lipids used (TM, DOPC = −20°C and TM, DOPG = −18°C) (2). In fact, even in the case when one of the labels would be significantly limited in motion but another would be very mobile, the approximation of κ2 = 2/3 is valid (2,14,18–20). Another point in favor of our approximation is the varying direction of the absorption dipole of dansyl compounds. If this is the case for both the donor and acceptor, it causes an intrinsic averaging over κ2 for each donor-acceptor pair, even without rotation (21). In our case AEDANS is fairly mobile, as judged from the experimentally observed steady state anisotropy, which ranges from 0.05 to 0.1 depending on its location in the bilayer (data not shown). Considering also the varying polarization of its absorption that overlaps with the tryptophan fluorescence, we believe the approximation of κ2 = 2/3 is valid.
Simulation-based fitting approach to experimental data analysis
As a measure of the goodness of the fit the following criterion was introduced:
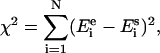 |
(2) |
where N is the number of data points, 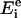 the experimentally obtained energy transfer efficiency, and
the experimentally obtained energy transfer efficiency, and 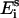 the simulated energy transfer efficiency. To fit the modeled energy transfer efficiencies to the experimental ones, the Nelder-Mead “simplex” method (22) was used. To increase the robustness of the method and the precision of the solution a global analysis approach was chosen, and therefore all experimental data were fitted simultaneously (23). To deal with this stochastic nature of the error function χ2 and to avoid possible local minima, the fitting procedure was performed a number of times with different initial estimations of the fitting parameters (3).
the simulated energy transfer efficiency. To fit the modeled energy transfer efficiencies to the experimental ones, the Nelder-Mead “simplex” method (22) was used. To increase the robustness of the method and the precision of the solution a global analysis approach was chosen, and therefore all experimental data were fitted simultaneously (23). To deal with this stochastic nature of the error function χ2 and to avoid possible local minima, the fitting procedure was performed a number of times with different initial estimations of the fitting parameters (3).
Handling of Stokes shift information
The fluorescence emission of molecules in different solvents is significantly affected by the solvent polarity. The dependency of the fluorescence emission maximum and related Stoke shift with respect to the polarity of the local environment of AEDANS-labeled cysteine mutants of M13 major coat protein incorporated in lipid bilayers was discussed recently (7,8). Therefore, we decided to use the Stokes shift information as an additional filtering for the structures obtained after fitting of the FRET data.
Unfortunately, analytical expressions describing the behavior of the Stokes shift exist only for the internal hydrophobic part of lipid bilayers (7). However, a monotonic behavior of the polarity with respect to the absolute value of the z coordinate in a bilayer system is demonstrated (24,25). This result is probably related to the presence of motional averaging in the liquid crystalline phase (we are working with bilayer systems above the gel-to-liquid crystalline phase transition temperature). Possible effects of different polarity of neighboring amino acid residues can be neglected in our case because of the long link between AEDANS moiety center and the protein backbone. This monotonic behavior enables us to build qualitative rules characterizing the relative z coordinates for a polarity probe that can be applied for sites on the protein in the headgroup region of the membrane or in the water phase. For example, consider two mutants with AEDANS emission maxima at wavelengths λ1 and λ2, and λ1 < λ2. Consequently the relation for the z coordinates of the fluorescent labels |z1| < |z2|, is also true. This relation can be considered as a qualitative rule: “the AEDANS position in the first mutant is closer to the membrane center then of the second mutant.”
Three types of qualitative relations were selected to describe the positions of AEDANS in various mutants, each associated with a characterizing number ∈[–1, 0, 1]. These numbers can be combined into a matrix M, presenting the polarity rules for all mutants that are taken into account. The matrix elements Mij describe the relation between the depth of ith and jth mutant. Assuming constant data precision for all mutants and denoting the maximal spread in the determined λ values as Δλ, the value of element Mij is set according to the following scheme:
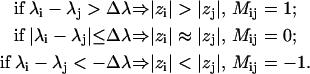 |
(3) |
The resulting matrix is symmetric with zero diagonal elements.
To quantify the deviation between experimental relations and modeled ones, the following parameter is introduced:
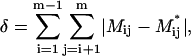 |
(4) |
where Mij* is the matrix element describing the relations obtained from the model of the protein for the ith and jth mutant.
In our approach, the value of δ is used for an additional filtering of the results coming out from the simulation-based fittings.
All models were realized as C++ classes. The Borland C++ Builder 6.0 environment was used to combine the developed models, OpenGL visualization, and simulation-based fitting algorithms into a software tool called FRETsim. The C++ classes and software are available from the authors upon request.
RESULTS
Analysis of FRET data
We started the study of the protein structure with a simultaneous analysis of all eight data series, measured for AEDANS at positions 7, 9, 12, 13, 15, 16, 17, and 18. The best-achieved fit, characterized by χ2 = 0.074 is presented in Fig. 3 by dotted lines. It can be seen that the simulation results for the acceptor at positions 7 and 9 clearly show a significant deviation between simulated and experimental data points. This deviation cannot be explained by small concentration inaccuracies in our sample preparation. Moreover, the high contribution of positions 7 and 9 to the χ2 value results in imperfections of the fit for positions 12–18, because the global optimization algorithm tries to decrease the large deviations for positions 7 and 9, rather than to precisely fit all data.
FIGURE 3.

Experimental energy transfer efficiencies E (solid circles) and their approximation by the model (dotted and solid lines) after global analysis versus the ratio between unlabeled and labeled proteins rul. The mutant names are given in the right top corner of each plot. The dotted line corresponds to initial fit of data for acceptor label positions 7–18. The solid line presents efficiencies obtained after fitting data for acceptor label positions 12–18.
These high deviations lead to the conclusion that a rigid two-helix model cannot describe the protein structure around positions 7 and 9. Therefore, it was decided to exclude positions 7 and 9 from the final data analysis and concentrate our research on the data from acceptor positions 12-18. To deal with possible local minima and the stochastic nature of χ2, the fitting was performed with different initial estimations for 500 times. The best fit is shown in Fig. 3 by solid lines. The exclusion of positions 7 and 9 leads to a significant decrease of χ2; the minimal χ2 value obtained now is 0.008, which is a factor of 10 smaller than for the previous case.
As in our previous study (3), we took into account only the best 20% of all solutions found with χ2 ∈[0.008, 0.022] and discarded solutions with χ2 ∈[0.022, 0.203]. This results in 100 solutions with a good fit to the FRET data (Fig. 4 A). Despite the high quality of the fit, a significant uncertainty remains in the angular parameters that describe the tilt and orientation of the N-terminal helix: ϕ =15 ±13°, ξ = –161 ± 99°, and ω = 61 ± 62°. However, the resulting lipid loss parameter L = 0.20 ± 0.04 is quite well defined. To reduce the uncertainty in the angular parameters found, we decided to use additional information coming from the positions of the acceptor fluorescence maxima. Therefore, a filtering of the solutions was performed based on the Stokes shift information (7).
FIGURE 4.

(A) Resulting 100 structures obtained from global analysis of experimental FRET data of AEDANS-labeled M13 coat protein mutants in DOPC/DOPG vesicles. The structures are presented in terms of Cα positions that are projected on the plane formed by the OZ axis and the direction of tilt of the transmembrane domain. The protein domain from amino acid residue 1–9 cannot be described by a rigid α-helix and is schematically presented as a “cloud” containing several gray “unstructured” conformations. (B) Final set of 52 structures obtained after fitting of experimental data and filtering using Stokes shift information. The resulting tilt angle of the N-terminal domain ϕ = 5.0 ± 4.7°.
Solution filtering using Stokes shift information
Applying the methods described in the section Handling of Stokes shift information to the experimental acceptor fluorescence maxima in Table 1 and the resulting protein structures, we were able to filter the solutions by discarding those that do not satisfy the δ criterion (Eq. 4). For the resulting 100 solutions, the value of δ varied between 1 and 20. We decided to take into account only solutions with δ ≤ 2, which was true for ∼50% of the set of solutions (this corresponds to ∼10% of the entire set of solutions); the other solutions were discarded. The final set of resulting structures is presented in Fig. 4 B.
The final set of resulting structures indicates a tilted protein. A kink is determined at position nk = 20 ± 2. However, the tilt angle of the two protein domains between amino acid residue position 10 and nk is small: ϕ = 5.0 ± 4.7°. The filtering of the solutions also results in a strong decrease in the uncertainty of the other angular parameters: ξ = −140 ± 43° and ω = 42.1 ± 10°. This result indicates that there is a small tilt ϕ of the N-terminal helix with respect to the transmembrane domain. For such a small tilt, the N-terminal helix tilt direction ξ is not a sensitive parameter since it describes a small wobble of the N-terminal domain with respect to the transmembrane domain. For an ideal continuous α-helix from the transmembrane to the N-terminal domain, ω would be 0°. The resulting value of ω indicates a relatively small distortion of an overall helix at the kink position.
DISCUSSION
Despite intensive studies, the structure of the membrane-bound state of the M13 major coat protein is still unknown. This is largely due to the difficulty in determining the structure of the N-terminal protein domain. In the literature many types of structures are proposed: I-shape (2), L-shape (11), dynamic (26), and banana-shape (4). One of the possible causes of such diversity is the difference in lipid environments. For example, in the solid-state NMR study of Marassi et al. (11) the proteins were inserted into dehydrated lipid bilayers. This can lead to squeezing of the proteins and may result in L-shape structures (2). It could even be that this domain has no rigid structure and dynamically exchanges between several conformations (27). Therefore, it is not surprising that in the literature there is no consistent view about the orientation and tilt of the N-terminal protein domain with respect to the bilayer interface.
In our study, FRET was applied to a range of AEDANS-labeled cysteine mutants covering the N-terminal domain of the protein with the goal to employ the FRET distance constraints to resolve its structure. We aimed at minimizing possible artifacts coming from unnatural environments (dehydrated bilayers, micelles) by working at relatively low-protein concentrations (high lipid/protein ratios) in large unilamellar vesicles. Under such conditions, the application of FRET is ideal since the technique has a high sensitivity. To analyze the FRET data, we extended our previous single helix model describing the transmembrane domain of M13 coat protein (3) to a model of two helical domains that are connected by a helix kink, i.e., the position of the amino acid residue from which the N-terminal helix starts. Furthermore, we took into account the polarity-dependent Stokes shift of the AEDANS fluorescence maximum by the application of “fuzzy rules” (Eq. 3) in our data analysis.
The N-terminal protein domain is dominated by the presence of negatively charged amino acid residues (Glu-2, Asp-4, and Asp-5), which will always try to extend into the aqueous phase and therefore act as a hydrophilic anchor (1). Furthermore, there is a Pro at position 6 (a helix breaker). Therefore, we limited our study to a range of site-directed AEDANS labels attached to the protein from positions 7–18. In this range, we decided to leave out positions 11 and 14, since in previous work it was found that these AEDANS-labeled mutants showed an anomalous behavior in the analysis of the fluorescence maximum (4). Taking into account the yield, quality and availability of mutants, this resulted in eight labeled positions: 7, 9, 12, 13, and 15–18. To discriminate between intramolecular energy transfer of acceptor-labeled proteins and intermolecular energy transfer, a titration with wild type proteins was performed (3). Intramolecular energy transfer efficiency is mainly sensitive to the distance between Trp-26 and the AEDANS label in one protein molecule whereas intermolecular efficiency is related to distances between planes, in which donors and acceptors are distributed in the membrane-protein system. Thus, by using FRET results from the titration experiments, we are able to get the structure and bilayer embedment of the protein. However, from intramolecular FRET only, the result would not be a single structure, but an infinite number of structures with equal intramolecular distances. This arises because in the case of our protein-lipid system setup, we have just a single donor position (Trp-26).
The structure of the protein is studied using a simulation-based fitting approach, which means adjusting all variable parameters of the model to fit the simulated data to experimental ones. To make our fitting analysis manageable, the parameters that describe the transmembrane helix are taken from our previous FRET study (3), whereas only the parameters describing the kink position, tilt, and orientation of the N-terminal helix domain are varied in our simulations (i.e., nk, ϕ, ω, ξ, and L). From the spatial model of the membrane-protein system the coordinates of donors and acceptors are obtained and used to calculate energy transfer efficiencies. To make the analysis more stable we used a global analysis approach and fit all the data points using the same model (changing only experimental conditions, such as acceptor position nA, and concentration-dependent ratios rLP and rul). A validation of our approach is given in Appendix A, where several numerical tests are described and analyzed to determine the precision of the parameters determined. The results indicate that the method can easily distinguish between I and L-shape protein structures and allows a precise determination of L, nk, and ϕ.
Interestingly, in the global analysis of the complete experimental data set, it is found that in our DOPC/DOPG vesicles positions 7 and 9 show a large deviation, indicating that these positions do not fit to the two-helix model. Because all other points for mutants 12–18 are fitting, this strongly suggests that these positions are located in a part of the protein for which the proposed structural model of two helices is not correct. This is consistent with a recent site-directed spin labeling study of M13 coat protein in phospholipid bilayers with increasing acyl chain length (28). In this study,the N-terminal domain contains 7 unstructured amino acid residues in 22:1PC and 14 residues in 14:1PC. Therefore, it is reasonable to assume that position 7 and 9 are in a flexible or unstructured part of the N-terminal protein domain, for which the rigid helix model does not apply. Consequently, our final analysis was based on a global analysis, excluding positions 7 and 9 since fitting data with a wrong model will only worsen the overall result. Clearly, the exclusion of these positions results in a dramatic reduction of the value of χ2, suggesting that for the remaining amino acid residues M13 coat protein is well described by a rigid two-helix model.
To support the idea that positions 7 and 9 arise from an unstructured protein domain, some additional calculations were performed. The model of the protein was enhanced by implementation of an unstructured domain for positions 1–10. The program randomly simulates the angles between the Cα connections, keeping the Cα-Cα distances constant and throwing away clashing conformations. As a result, instead of a fixed location of the acceptor attached to positions 7 or 9, a “cloud” of possible acceptor locations is obtained. To demonstrate this effect, the simulated data were recalculated for acceptor positions 7 and 9 using the new structural model and the previously determined orientation of two helices. The new results for mutants A7C and A9C are presented in Fig. 5. Clearly, a strong improvement of the fit is observed in favor of the unstructured protein model.
FIGURE 5.

Experimental energy transfer efficiencies E (solid circles) and their approximation by the model (solid lines) after introducing an unstructured domain to the protein model between positions 1 and 10. The dotted lines show the previous fits by a model without unstructured domain (see Fig. 3).
To enhance the quality of the resulting angular parameters ϕ, ω, and ξ, we used an additional filtering criterion δ, based on the polarity shift of the AEDANS fluorescence maxima. By applying the “fuzzy rules” polarity criterion given in Eq. 3, we assume that from two AEDANS labels the furthest to the bilayer center is the one that has a more red-shifted fluorescence (larger Stokes shift). The application of this criterion allows us to discard roughly half of the solutions and to more precisely determine the average tilt angle of the N-terminus. This can be seen by comparing Fig. 4, A and B, where the two α-helical domains of the protein are indicated with solid lines, and the proposed “unstructured” region between amino acid residues 1–9 is drawn as a gray cloud. The best fitting (in terms of both FRET and Stokes shift) structures for M13 coat protein embedded in DOPC/DOPG vesicles are collected in Fig. 4 B.
Overall, the protein is in a tilted α-helical state (by 18° with respect to the normal to the membrane) from positions 12 to 46 (i.e., the labeled mutants that we investigated here and in (7)), supporting a previous FRET analysis that was based on validating existing protein structures (2). The small kink around position 20 could indicate that the protein has a slightly weakened region in the helix here. This region is related to the previously called “hinge region” around amino acid residue 20 found in micellar systems (26). This position is close to the interface between the acyl chains of the phospholipids and the headgroup region (see Fig. 4). It should be reminded that in our two-helix protein model the simplest way of connection of the two helical domains is via a common point that is called a “kink” (Fig. 2). It is likely, however, that if the actual protein structure will show a smooth protein bend, the kink will denote the point of maximal curvature of the structure. The N-terminal α-helix starts at a position around amino acid residue 10. It is interesting to note that this position marks the interface between the headgroups of the lipids and the water phase, and reflects the end of the unstructured N-terminal hydrophilic anchor (28) that is emerging from the headgroup region into the water phase (see Fig. 4).
In summary, our FRET work resolves the problem of the tilt and orientation of the N-terminal domain of M13 coat protein in the membrane-bound state and shows that, except for the N-terminal hydrophilic anchor, the protein conformation is almost a straight helix. It is for the first time that site-directed FRET emerges into such a detailed molecular model for a membrane-embedded protein. Overall the resulting membrane-embedded M13 coat protein structure does not differ much from the native α-helical structure of the protein in bacteriophage M13 (29). This finding may be important for the membrane-bound phage assembly since it could allow a fast and efficient incorporation of the protein into the bacteriophage with a low-energy cost. Probably the overall tilt of the protein of 18° is related to an efficient anchoring and integration of the protein in the membrane (1). Now that the structure of the coat protein in a membrane becomes evident, future questions about the membrane-bound phage assembly should address the dissociation of the coat protein from the membrane, i.e., studying the process of lifting the membrane anchors (1). FRET may be an excellent tool in monitoring the molecular details of such a process.
Acknowledgments
We would like to thank Ruud B. Spruijt for the preparation of the protein mutants and helpful comments on the work.
This work was supported by contract No. QLG-CT-2000-01801 of the European Commission (MIVase—New Therapeutic Approaches to Osteoporosis: Targeting the Osteoclast V-ATPase).
APPENDIX: SENSITIVITY OF THE MODEL PARAMETERS AND NOISE STABILITY
To determine the sensitivity to the model parameters and the noise stability, the following procedure was employed. For each of the two published structures of M13 major coat protein, I-shape (2) and L-shape (11), artificial FRET data were generated by our model and then used instead of experimental data in the simulation-based fitting algorithm. Because of the stochastic behavior of the χ2 function the fitting algorithm provides a distribution of solutions for the global minima. The spread of a parameter in this cluster of solutions allows characterizing its sensitivity. To study the experimental noise effects on the parameter distribution, the same operation was performed on data containing artificial noise, similar as is described in our previous work (3). The standard deviation of the noise varies for each data point (see error bars in Fig. 3).
The results of the numerical tests are given in Table 3. For an ideal α-helix the algorithm was able to determine the precise structure for the considered range of amino acid residues (12–26). For all solutions in the “elite” set (20% of solutions with smallest χ2) nk < 12, which means that an ideal helix was found for positions 12–26. The introduction of noise to the artificial data did not change this tendency. For an L-shape protein structure the parameters L, nk, and ϕ were determined quite well, although the noise in the artificial data increased the uncertainty for almost all parameters. The angular parameters ω and, especially ξ, showed a rather high spread. However, the mean values of the parameters found still were close to the initial values.
TABLE 3.
Original and calculated values of the model parameters after analysis of synthetic FRET data by means of a simulation-based fitting approach
| Parameter | Original value in synthetic data simulation | Value found after analysis with no noise added to synthetic data | Value found after analysis with additional noise in synthetic data |
|---|---|---|---|
| I-shape protein (ideal α-helix between positions 12 to 26) | |||
| L | 0.2 | 0.20 ± 0.01 | 0.19 ± 0.02 |
| nk | <12 | <12 | <12 |
| ϕ | 0° | 0° | 0° |
| ω | 0° | 0° | 0° |
| ξ | 0° | 0° | 0° |
| L-shape protein (11) | |||
| L | 0.2 | 0.21 ± 0.02 | 0.19 ± 0.04 |
| nk | 20 | 20 ± 1 | 20 ± 1 |
| ϕ | 110° | 107 ± 9° | 104 ± 14° |
| ω | 40° | 48 ± 12° | 53 ± 21° |
| ξ | –110° | –115 ± 17° | –120 ± 37° |
Abbreviations used: FRET, Förster (or fluorescence) resonance energy transfer; DOPC, dioleoylphosphatidylcholine; DOPG, dioleoylphosphatidylglycerol; AEDANS, N-(acetylaminoethyl)-5-naphthylamine-1-sulfonic acid; rLP, lipid/protein molar ratio; 22:1PC, 1,2-dierucoyl-sn-glycero-3-phosphocholine; 14:1PC, 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine.
References
- 1.Stopar, D., R. B. Spruijt, and M. A. Hemminga. 2006. Anchoring mechanisms of membrane-associated M13 major coat protein. Chem. Phys. Lipids. 141:83–93. [DOI] [PubMed] [Google Scholar]
- 2.Vos, W. L., R. B. M. Koehorst, R. B. Spruijt, and M. A. Hemminga. 2005. Membrane-bound conformation of M13 major coat protein: a structure validations through FRET-derived constrains. J. Biol. Chem. 280:38522–38527. [DOI] [PubMed] [Google Scholar]
- 3.Nazarov, P. V., R. B. M. Koehorst, W. L. Vos, V. V. Apanasovich, and M. A. Hemminga. 2006. FRET study of membrane proteins: simulation-based fitting for analysis of membrane protein embedment and association. Biophys. J. 91:454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spruijt, R. B., A. B. Meijer, C. J. A. M. Wolfs, and M. A. Hemminga. 2000. Localization and rearrangement modulation of the N-terminal arm of the membrane-bound major coat protein of bacteriophage M13. Biochim. Biophys. Acta. 1509:311–323. [DOI] [PubMed] [Google Scholar]
- 5.Spruijt, R. B., C. J. A. M. Wolfs, and M. A. Hemminga. 1989. Aggregation-related conformational change of the membrane-associated coat protein of bacteriophage M13. Biochemistry. 28:9158–9165. [DOI] [PubMed] [Google Scholar]
- 6.Spruijt, R. B., C. J. A. M. Wolfs, J. W. G. Verver, and M. A. Hemminga. 1996. Accessibility and environment probing using cysteine residues introduced along the putative transmembrane domain of the major coat protein of bacteriophage M13. Biochemistry. 35:10383–10391. [DOI] [PubMed] [Google Scholar]
- 7.Koehorst, R. B. M., R. B. Spruijt, F. J. Vergeldt, and M. A. Hemminga. 2004. Lipid bilayer topology of the transmembrane α-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys. J. 87:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spruijt, R. B., C. J. A. M. Wolfs, and M. A. Hemminga. 2004. Membrane assembly of M13 major coat protein: evidence for structural adaptation in the hinge region and a tilted transmembrane domain. Biochemistry. 43:13972–13980. [DOI] [PubMed] [Google Scholar]
- 9.Stopar, D., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 2003. Protein-lipid interactions of bacteriophage M13 major coat protein. Biochim. Biophys. Acta. 1611:5–15. [DOI] [PubMed] [Google Scholar]
- 10.Glaubitz, C., G. Grobner, and A. Watts. 2000. Structural and orientational information of the membrane embedded M13 coat protein by 13C-MAS NMR spectroscopy. Biochim. Biophys. Acta. 1463:151–161. [DOI] [PubMed] [Google Scholar]
- 11.Marassi, F. M., and S. J. Opella. 2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restrains. Protein Sci. 12:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nazarov, P. V., V. V. Apanasovich, V. M. Lutkovski, M. M. Yatskou, R. B. M. Koehorst, and M. A. Hemminga. 2004. Artificial neural network modification of simulation-based fitting: application to a protein-lipid system. J. Chem. Inf. Comput. Sci. 44:568–574. [DOI] [PubMed] [Google Scholar]
- 13.Lakowicz, J. R. 1999. Principles of fluorescence spectroscopy. Kluwer Academic/Plenum Publishers, New York.
- 14.Lakshmikanth, G. S., K. Sridevi, G. Krishnamoorthy, and J. B. Udgaonkar. 2001. Structure is lost incrementally during the unfolding of barstar. Nat. Struct. Biol. 8:799–804. [DOI] [PubMed] [Google Scholar]
- 15.Datema, K. P., A. J. W. G. Visser, A. Van Hoek, C. J. A. M. Wolfs, R. B. Spruijt, and M. A. Hemminga. 1987. Time-resolved tryptophan fluorescence anisotropy investigation of bacteriophage M13 coat protein in micelles and mixed bilayers. Biochemistry. 26:6145–6152. [DOI] [PubMed] [Google Scholar]
- 16.Johnson, I. D., and B. S. Hudson. 1989. Environmental modulation of M13 coat protein tryptophan fluorescence dynamics. Biochemistry. 28:6392–6400. [DOI] [PubMed] [Google Scholar]
- 17.Fernandes, F., L. M. S. Loura, M. Prieto, R. B. M. Koehorst, R. B. Spruijt, and M. A. Hemminga. 2003. Dependence of M13 major coat protein oligomerization and lateral segregation on bilayer composition. Biophys. J. 85:2430–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loura, L. M. S., A. Fedorov, and M. Prieto. 1996. Resonance energy transfer in a model system of membranes: application to gel and liquid crystalline phases. Biophys. J. 71:1823–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamal, J. K. A., and D. V. Behere. 2002. Spectroscopic studies on human serum albumin and methemalbumin: optical, steady-state, and picosecond time-resolved fluorescence studies, and kinetics of substrate oxidation by methemalbumin. J. Biol. Inorg. Chem. 7:273–283. [DOI] [PubMed] [Google Scholar]
- 20.Gustiananda, M., J. R. Liggins, P. L. Cummins, and J. E. Gready. 2004. Conformation of prion protein repeat peptides probed by FRET measurements and molecular dynamics simulations. Biophys. J. 86:2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haas, E., E. Katchalski-Katzir, and I. Z. Steinberg. 1978. Brownian motion of the ends of oligopeptide chains in solution as estimated by energy transfer between the chain ends. Biopolymers. 17:11–31. [Google Scholar]
- 22.Nelder, J. A., and R. Mead. 1965. A simplex method for function minimization. Comput. J. 7:308–313. [Google Scholar]
- 23.Beechem, J. M., and L. Brand. 1986. Global analysis of fluorescence decay: applications to some unusual experimental and theoretical studies. Photochem. Photobiol. 44:323–329. [DOI] [PubMed] [Google Scholar]
- 24.White, S. H., and W. C. Wimley. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1376:339–352. [DOI] [PubMed] [Google Scholar]
- 25.Marsh, D. 2001. Polarity and permeation profiles in lipid membranes. Proc. Natl. Acad. Sci. USA. 89:7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papavoine, C. H. M., M. L. Remerowski, L. M. Horstink, R. N. H. Konings, C. W. Hilbers, and F. J. M. van de Ven. 1997. Backbone dynamics of the major coat protein of bacteriophage M13 in detergent micelles by 15N nuclear magnetic resonance relaxation measurements using the model-free approach and reduced spectral density mapping. Biochemistry. 36:4015–4026. [DOI] [PubMed] [Google Scholar]
- 27.Meijer, A. B., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 2001. Configurations of the N-terminal amphipathic domain of the membrane-bound M13 major coat protein. Biochemistry. 40:5081–5086. [DOI] [PubMed] [Google Scholar]
- 28.Stopar, D., J. Štrancar, R. B. Spruijt, and M. A. Hemminga. 2006. Motional restrictions of membrane proteins: A site-directed spin labeling study. Biophys. J 91:3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marvin, D. A. 1998. Filamentous phage structure, infection and assembly. Curr. Opin. Struct. Biol. 8:150–158. [DOI] [PubMed] [Google Scholar]
- 30.Fernandes, F., L. M. S. Loura, R. B. M. Koehorst, R. B. Spruijt, M. A. Hemminga, and M. Prieto. 2004. Quantification of protein-lipid selectivity using FRET: application to the M13 major coat protein. Biophys. J. 87:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]