

Abstract
Single channel and macroscopic current recording was used to investigate block of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel pore by the permeant anion 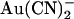 . Block was 1–2 orders of magnitude stronger when
. Block was 1–2 orders of magnitude stronger when 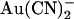 was added to the intracellular versus the extracellular solution, depending on membrane potential. A point mutation within the pore, T-338A, strongly decreased the asymmetry of block, by weakening block by intracellular
was added to the intracellular versus the extracellular solution, depending on membrane potential. A point mutation within the pore, T-338A, strongly decreased the asymmetry of block, by weakening block by intracellular 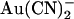 and at the same time strengthening block by external
and at the same time strengthening block by external 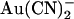 . Block of T-338A, but not wild-type, was strongest at the current reversal potential and weakened by either depolarization or hyperpolarization. In contrast to these effects, the T-338A mutation had no impact on block by the impermeant
. Block of T-338A, but not wild-type, was strongest at the current reversal potential and weakened by either depolarization or hyperpolarization. In contrast to these effects, the T-338A mutation had no impact on block by the impermeant 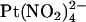 ion. We suggest that the CFTR pore has at least two anion binding sites at which
ion. We suggest that the CFTR pore has at least two anion binding sites at which 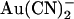 and
and 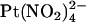 block Cl− permeation. The T-338A mutation decreases a barrier for
block Cl− permeation. The T-338A mutation decreases a barrier for 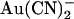 movement between different sites, leading to significant changes in its blocking action. Our finding that apparent blocker binding affinity can be altered by mutagenesis of a residue which does not contribute to a blocker binding site has important implications for interpreting the effects of mutagenesis on channel blocker effects.
movement between different sites, leading to significant changes in its blocking action. Our finding that apparent blocker binding affinity can be altered by mutagenesis of a residue which does not contribute to a blocker binding site has important implications for interpreting the effects of mutagenesis on channel blocker effects.
INTRODUCTION
Understanding the mechanism of action of ion channels has been given a tremendous boost in recent years by the generation of direct structural information in the form of channel crystal structures (1,2). One of the most striking features of these structures has been the identification of multiple discrete permeant ion binding sites within the pores of both cation selective (3,4) and anion selective channels (5). Permeant ion binding within the pore is thought to be key to the function of ion channels, providing the link between ionic selectivity and rapid ion transport both in cation channels (1,6) and anion channels (7,8).
Although direct structural evidence for ion binding sites inside channel pores is a recent breakthrough, the existence of such sites has been postulated for decades based on biophysical evidence (9). Most useful in these studies have been high affinity probes of ion binding sites, such as Ba2+ in K+ channels (10,11), Cd2+ in Ca2+ channels (6), and SCN− in Cl− channels (12). For example, SCN− has been used to probe the functional properties of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel pore (12,13) and also to identify key pore-forming amino acid residues that potentially contribute to anion binding sites inside the pore (14–17). More recently, pseudohalide anions (such as 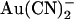 and
and 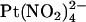 ) that inhibit Cl− permeation at relatively low concentrations have been used to probe the CFTR pore (8,18–21). The apparent affinity of
) that inhibit Cl− permeation at relatively low concentrations have been used to probe the CFTR pore (8,18–21). The apparent affinity of 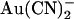 binding is affected by point mutations throughout the putative pore region (21–23); however, since
binding is affected by point mutations throughout the putative pore region (21–23); however, since 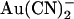 is highly permeant in CFTR (18) it is difficult to separate the effects of mutagenesis on
is highly permeant in CFTR (18) it is difficult to separate the effects of mutagenesis on 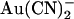 binding and permeation.
binding and permeation. 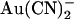 has also recently been used to modify irreversibly cysteine residues introduced into the CFTR pore by site-directed mutagenesis (24). The divalent pseudohalide
has also recently been used to modify irreversibly cysteine residues introduced into the CFTR pore by site-directed mutagenesis (24). The divalent pseudohalide 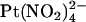 has also been used effectively to probe the CFTR pore, with one important distinction from
has also been used effectively to probe the CFTR pore, with one important distinction from 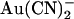 being that
being that 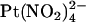 is impermeant (19). Since
is impermeant (19). Since 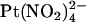 is able to enter the pore and interfere with Cl− permeation when applied to either side of the membrane, it has been suggested that distinct anion binding sites must exist on either side of some barrier inside the pore that prevents
is able to enter the pore and interfere with Cl− permeation when applied to either side of the membrane, it has been suggested that distinct anion binding sites must exist on either side of some barrier inside the pore that prevents 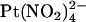 movement (19,25).
movement (19,25).
One amino acid residue that has been implicated in 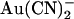 binding in the CFTR pore is T-338 in the sixth transmembrane region (TM6). Thus, the point mutation T-338A significantly reduces the apparent affinity with which intracellular
binding in the CFTR pore is T-338 in the sixth transmembrane region (TM6). Thus, the point mutation T-338A significantly reduces the apparent affinity with which intracellular 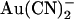 inhibits Cl− current through the channel (21). A number of studies have implicated this threonine residue as being involved in determining the single channel conductance (26) and anion selectivity (26,27) of the pore, perhaps by contributing to the narrowest region of the pore (8,26). Initially it was suggested, based on substituted cysteine accessibility mutagenesis, that the amino acid side chain at this position was not in contact with the aqueous lumen of the pore (28); however, more recent studies have shown that the side chain does in fact line the pore (24,29,30). There is some evidence that mutagenesis of T-338 affects the binding of both permeant (17,21,27) and blocking anions (31,32) within the pore. It has been suggested that T-338 contributes to the narrow, central region of the CFTR pore, which is the main determinant of selectivity between different permeant anions (reviewed by Linsdell (8)).
inhibits Cl− current through the channel (21). A number of studies have implicated this threonine residue as being involved in determining the single channel conductance (26) and anion selectivity (26,27) of the pore, perhaps by contributing to the narrowest region of the pore (8,26). Initially it was suggested, based on substituted cysteine accessibility mutagenesis, that the amino acid side chain at this position was not in contact with the aqueous lumen of the pore (28); however, more recent studies have shown that the side chain does in fact line the pore (24,29,30). There is some evidence that mutagenesis of T-338 affects the binding of both permeant (17,21,27) and blocking anions (31,32) within the pore. It has been suggested that T-338 contributes to the narrow, central region of the CFTR pore, which is the main determinant of selectivity between different permeant anions (reviewed by Linsdell (8)).
This work investigates the interaction between permeant and impermeant blocking ions and the CFTR pore. The complex effects of the T-338A mutant on channel block suggest that this important threonine residue forms a barrier to permeant anion movement inside the pore and that the existence of this barrier underlies asymmetric anion binding properties of the wild-type channel pore. In particular, our finding that mutagenesis of T-338 drastically alters the potency of 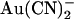 block apparently without directly affecting an anion binding site has important implications for studies that seek to identify blocker binding sites within ion channel pores using mutagenesis.
block apparently without directly affecting an anion binding site has important implications for studies that seek to identify blocker binding sites within ion channel pores using mutagenesis.
MATERIALS AND METHODS
Experiments were carried out on baby hamster kidney cells stably transfected with human wild-type or T-338A-CFTR, prepared as described previously (17). Macroscopic and single channel patch clamp recordings were made from inside out patches excised from these cells, as described in detail previously (21–33). After patch excision and recording of background currents, CFTR channels were activated by exposure to protein kinase A (PKA) catalytic subunit plus MgATP (1 mM) in the cytoplasmic (bath) solution. As in a previous study (22), single channel currents were recorded after weak PKA stimulation (1–10 nM), whereas all macroscopic CFTR currents were recorded after maximal PKA stimulation (∼20 nM) and subsequent treatment with sodium pyrophosphate (PPi, 2 mM) to “lock” channels in the open state. Because single channel activity was recorded under conditions of weak PKA stimulation, patches contained a large number of CFTR channels with very low open probability, and as a result no information is contained in the relative open probability under different ionic conditions. Both intracellular (bath) and extracellular (pipette) solutions were based on one containing (in mM): 150 NaCl, 10 N-Tris[hydroxymethyl]methyl-2-aminoethanesulfonate (TES), 2 MgCl2, pH 7.4, to which 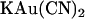 (3 μM–10 mM),
(3 μM–10 mM), 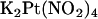 (1 mM), or NaSCN (10 mM) was added as necessary. To study the effects of changing Cl− concentration on block by
(1 mM), or NaSCN (10 mM) was added as necessary. To study the effects of changing Cl− concentration on block by 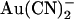 (see Fig. 9), the NaCl concentration in either the intracellular or extracellular solution was reduced to 20 mM by replacement with glucose. In these experiments, membrane voltages were corrected for liquid junction potentials calculated using pCLAMP9 software (Molecular Devices, Sunnyvale, CA). All chemicals were from Sigma (Oakville, ON, Canada) except PKA (Promega, Madison, WI) and K2Pt(NO2)4 (Strem Chemicals, Newburyport, MA).
(see Fig. 9), the NaCl concentration in either the intracellular or extracellular solution was reduced to 20 mM by replacement with glucose. In these experiments, membrane voltages were corrected for liquid junction potentials calculated using pCLAMP9 software (Molecular Devices, Sunnyvale, CA). All chemicals were from Sigma (Oakville, ON, Canada) except PKA (Promega, Madison, WI) and K2Pt(NO2)4 (Strem Chemicals, Newburyport, MA).
FIGURE 9.

Voltage dependence of block of T-338A-CFTR by internal 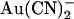 ions depends on the transmembrane Cl− gradient. (A) Example leak-subtracted I/V relationships recorded from inside out membrane patches. Currents were recorded before and after addition of 1 mM
ions depends on the transmembrane Cl− gradient. (A) Example leak-subtracted I/V relationships recorded from inside out membrane patches. Currents were recorded before and after addition of 1 mM 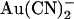 to the intracellular solution. Chloride concentration was reduced from 154 mM to 24 mM in either the intracellular (left) or extracellular (right) solution by partial replacement of NaCl by glucose. (B) Mean fractional current remaining (i/i0) after addition of this concentration of
to the intracellular solution. Chloride concentration was reduced from 154 mM to 24 mM in either the intracellular (left) or extracellular (right) solution by partial replacement of NaCl by glucose. (B) Mean fractional current remaining (i/i0) after addition of this concentration of 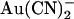 to the intracellular solution, with symmetrical 154 mM Cl− (•), 24 mM intracellular Cl− (○), or 24 mM extracellular Cl− (□). Mean of data from 4–6 patches.
to the intracellular solution, with symmetrical 154 mM Cl− (•), 24 mM intracellular Cl− (○), or 24 mM extracellular Cl− (□). Mean of data from 4–6 patches.
Current traces were filtered at 50 Hz (for single channel currents) or 100–200 Hz (for macroscopic currents) using an eight-pole Bessel filter, digitized at 250 Hz–1 kHz, and analyzed using pCLAMP9 software (Molecular Devices). Single channel current amplitudes were estimated from all-points amplitude histograms (e.g., Fig. 1, B and C). Macroscopic current-voltage (I/V) relationships were constructed using depolarizing voltage ramp protocols, with a rate of change of voltage of 100–125 mV s−1 (34,35). Background (leak) currents recorded before the addition of PKA were subtracted digitally, leaving uncontaminated CFTR currents (19,35). Macroscopic chord conductance (G) was estimated at different voltages by dividing the current by the driving force (membrane potential minus current reversal potential; the reversal potential was 0 mV in all cases) and is plotted as a fraction of maximal chord conductance (GMAX) (see Fig. 5 B).
FIGURE 1.

Asymmetric block of wild-type CFTR by 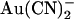 ions. (A) Example single channel currents recorded from inside out patches at membrane potentials of +50 mV and −50 mV, as indicated. In each case the closed state of the channel is indicated by the line on the far left. Currents were recorded in the absence of
ions. (A) Example single channel currents recorded from inside out patches at membrane potentials of +50 mV and −50 mV, as indicated. In each case the closed state of the channel is indicated by the line on the far left. Currents were recorded in the absence of 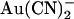 (control) or with
(control) or with 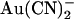 present in the intracellular solution (100 μM) or the extracellular solution (10 mM). In both cases
present in the intracellular solution (100 μM) or the extracellular solution (10 mM). In both cases 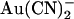 causes a reduction in unitary current amplitude, as further demonstrated from amplitude histograms prepared from these current traces at +50 mV (B) and −50 mV (C) and also from mean unitary current-voltage relationships recorded under these conditions (D and E). Mean of data from 3–5 patches in D and E.
causes a reduction in unitary current amplitude, as further demonstrated from amplitude histograms prepared from these current traces at +50 mV (B) and −50 mV (C) and also from mean unitary current-voltage relationships recorded under these conditions (D and E). Mean of data from 3–5 patches in D and E.
FIGURE 5.

Macroscopic currents demonstrate the unusual voltage dependence of 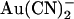 block of T-338A-CFTR. (A) Example leak-subtracted current-voltage (I/V) relationships recorded from inside out membrane patches after maximal current stimulation with PKA and PPi. Currents were recorded in the absence of
block of T-338A-CFTR. (A) Example leak-subtracted current-voltage (I/V) relationships recorded from inside out membrane patches after maximal current stimulation with PKA and PPi. Currents were recorded in the absence of 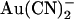 (left), with 2 mM
(left), with 2 mM 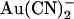 present in the intracellular solution (center), or with 1 mM
present in the intracellular solution (center), or with 1 mM 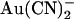 in the extracellular solution (right). (B) Quantification of the shape of the I/V curve under these conditions. The chord conductance at different voltages, relative to the maximum chord conductance (G/GMAX), was estimated as described in Materials and Methods. Mean of data from 4–5 patches.
in the extracellular solution (right). (B) Quantification of the shape of the I/V curve under these conditions. The chord conductance at different voltages, relative to the maximum chord conductance (G/GMAX), was estimated as described in Materials and Methods. Mean of data from 4–5 patches.
Concentration-inhibition relationships were fitted by the equation
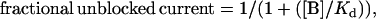 |
(1) |
where [B] is the blocker concentration and Kd its apparent dissociation constant.
Energy barrier profiles based on transition rate theory were constructed using the AJUSTE computer program developed by Alvarez et al. (36) and used previously to model anion permeation and block in the CFTR pore (13,37). Two models were developed, in which the pore was assumed to have either one or two binding sites for permeant anions (Cl− and 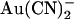 ). Adjustable parameters during the fitting procedure were the energies for both Cl− and
). Adjustable parameters during the fitting procedure were the energies for both Cl− and 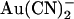 ions at the energy minima (wells) and maxima (peaks). For simplicity and to minimize the number of adjustable parameters, wells and peaks in the one-site model were fixed at values described in a previous study (13), and in the two-site model wells and peaks were fixed at regular intervals within the transmembrane electric field. Other details of the modeling procedure are as described previously (13,36,37).
ions at the energy minima (wells) and maxima (peaks). For simplicity and to minimize the number of adjustable parameters, wells and peaks in the one-site model were fixed at values described in a previous study (13), and in the two-site model wells and peaks were fixed at regular intervals within the transmembrane electric field. Other details of the modeling procedure are as described previously (13,36,37).
Throughout, mean data are presented graphically with error bars reflecting mean ± SE; where no error bars are shown, this error is smaller than the size of the symbol. Statistical comparisons between groups of data were carried out using Student's two-tailed t-test, with P < 0.05 being considered statistically significant.
RESULTS
Asymmetric block of wild-type CFTR by Au(CN)2− ions
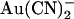 ions have previously been shown by our lab (19,38) and others (24) to cause a reduction in the amplitude of unitary CFTR Cl− channel current, due to its ability to bind within the pore with high affinity and interrupt the flow of Cl− ions. However, although
ions have previously been shown by our lab (19,38) and others (24) to cause a reduction in the amplitude of unitary CFTR Cl− channel current, due to its ability to bind within the pore with high affinity and interrupt the flow of Cl− ions. However, although 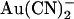 is highly permeant in CFTR (18,21) and can inhibit CFTR Cl− currents from either side of the membrane (24), its inhibitory effects have previously been studied at the single channel level only when present in the intracellular solution (19,24,38). Fig. 1 shows the block of unitary CFTR Cl− currents recorded in inside out membrane patches by both intracellular and extracellular
is highly permeant in CFTR (18,21) and can inhibit CFTR Cl− currents from either side of the membrane (24), its inhibitory effects have previously been studied at the single channel level only when present in the intracellular solution (19,24,38). Fig. 1 shows the block of unitary CFTR Cl− currents recorded in inside out membrane patches by both intracellular and extracellular 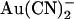 . As reported previously (24,38), intracellular
. As reported previously (24,38), intracellular 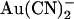 caused a voltage-dependent block of unitary Cl− current at concentrations below 1 mM (Fig. 1, A–D). However, very much higher concentrations of
caused a voltage-dependent block of unitary Cl− current at concentrations below 1 mM (Fig. 1, A–D). However, very much higher concentrations of 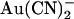 were required in the extracellular solution to produce comparable levels of inhibition (Fig. 1, A–C and E). This asymmetry of blocking action is seen more clearly from single channel concentration-inhibition curves shown in Fig. 2 A. These curves further demonstrate that, unlike intracellular
were required in the extracellular solution to produce comparable levels of inhibition (Fig. 1, A–C and E). This asymmetry of blocking action is seen more clearly from single channel concentration-inhibition curves shown in Fig. 2 A. These curves further demonstrate that, unlike intracellular 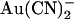 , extracellular
, extracellular 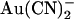 exhibits little or no voltage dependence of block. Fits to these curves by Eq. 1 were used to estimate the apparent affinity of
exhibits little or no voltage dependence of block. Fits to these curves by Eq. 1 were used to estimate the apparent affinity of 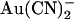 block; results of these fits are shown in Fig. 2 B.
block; results of these fits are shown in Fig. 2 B. 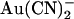 inhibits unitary Cl− current with significantly (∼1–2 orders of magnitude) lower affinity when present in the extracellular versus the intracellular solution, depending on the membrane potential. Thus, despite its high permeability in the CFTR pore,
inhibits unitary Cl− current with significantly (∼1–2 orders of magnitude) lower affinity when present in the extracellular versus the intracellular solution, depending on the membrane potential. Thus, despite its high permeability in the CFTR pore, 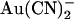 shows highly asymmetric blocking effects on Cl− movement through the pore.
shows highly asymmetric blocking effects on Cl− movement through the pore.
FIGURE 2.

Concentration and voltage dependence of block by 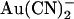 ions. (A) Fractional unitary current remaining (i/i0) after addition of different concentrations of
ions. (A) Fractional unitary current remaining (i/i0) after addition of different concentrations of 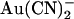 to the intracellular or extracellular solution: (•) internal
to the intracellular or extracellular solution: (•) internal 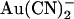 , −50 mV; (○) internal
, −50 mV; (○) internal 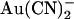 , +50 mV; (▾) external
, +50 mV; (▾) external 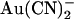 , −50 mV; (∇) external
, −50 mV; (∇) external 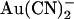 , +50 mV. Each of these four sets of data has been fitted by Eq. 1. (B) Mean Kd calculated from curves such as those shown in (A) for both internal (•) and external (○)
, +50 mV. Each of these four sets of data has been fitted by Eq. 1. (B) Mean Kd calculated from curves such as those shown in (A) for both internal (•) and external (○) 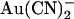 . Mean of data from 3–5 patches. Asterisks indicate a statistically significant difference from the Kd for internal
. Mean of data from 3–5 patches. Asterisks indicate a statistically significant difference from the Kd for internal 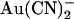 .
.
Block of T-338A-CFTR by Au(CN)2−
We also investigated 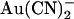 block of unitary currents in T-338A-CFTR. As described in the introduction, this mutant has been shown to exhibit elevated unitary Cl− conductance (26), increased
block of unitary currents in T-338A-CFTR. As described in the introduction, this mutant has been shown to exhibit elevated unitary Cl− conductance (26), increased 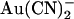 permeability (21), and weakened block by intracellular
permeability (21), and weakened block by intracellular 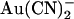 at the macroscopic current level (21,22). The effects of intracellular and extracellular
at the macroscopic current level (21,22). The effects of intracellular and extracellular 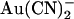 on wild-type and T-338A-CFTR single channel currents are compared in Fig. 3. These traces indicate that, whereas the inhibitory effects of 100 μM intracellular
on wild-type and T-338A-CFTR single channel currents are compared in Fig. 3. These traces indicate that, whereas the inhibitory effects of 100 μM intracellular 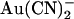 are weakened in T-338A, the inhibitory effects of 1 mM extracellular
are weakened in T-338A, the inhibitory effects of 1 mM extracellular 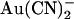 are enhanced in this mutant.
are enhanced in this mutant.
FIGURE 3.

Both internal and external 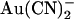 block are altered in T-338A-CFTR. Example single channel currents for wild-type (A) and T-338A-CFTR (B) recorded from inside out patches at membrane potentials of +50 mV and −50 mV, as indicated. In each case the closed state of the channel is indicated by the line on the far left. Currents were recorded in the absence of
block are altered in T-338A-CFTR. Example single channel currents for wild-type (A) and T-338A-CFTR (B) recorded from inside out patches at membrane potentials of +50 mV and −50 mV, as indicated. In each case the closed state of the channel is indicated by the line on the far left. Currents were recorded in the absence of 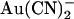 (control) or with
(control) or with 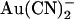 present in the intracellular solution (100 μM) or the extracellular solution (1 mM).
present in the intracellular solution (100 μM) or the extracellular solution (1 mM).
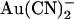 block of T-338A-CFTR unitary currents is explored in more detail in Fig. 4. Unitary current-voltage relationships recorded in the presence of the same concentration of
block of T-338A-CFTR unitary currents is explored in more detail in Fig. 4. Unitary current-voltage relationships recorded in the presence of the same concentration of 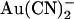 (1 mM) in either the intracellular or extracellular solution are shown in Fig. 4 A. The unitary current-voltage relationship recorded in the presence of extracellular
(1 mM) in either the intracellular or extracellular solution are shown in Fig. 4 A. The unitary current-voltage relationship recorded in the presence of extracellular 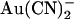 shows an unusual “N”-shape, suggesting that block is stronger close to 0 mV than at either strongly hyperpolarized or strongly depolarized membrane potentials (Fig. 4 A). In the presence of intracellular
shows an unusual “N”-shape, suggesting that block is stronger close to 0 mV than at either strongly hyperpolarized or strongly depolarized membrane potentials (Fig. 4 A). In the presence of intracellular 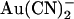 , the current-voltage relationship appears linear at hyperpolarized voltages but outwardly rectified at depolarized voltages (Fig. 4 A). The concentration dependence of
, the current-voltage relationship appears linear at hyperpolarized voltages but outwardly rectified at depolarized voltages (Fig. 4 A). The concentration dependence of 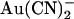 block of T-338A single channel currents is shown in Fig. 4, B and C. The apparent affinity of block is similar for both internal and external
block of T-338A single channel currents is shown in Fig. 4, B and C. The apparent affinity of block is similar for both internal and external 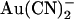 , and in both cases the apparent Kd is minimal close to 0 mV and increases with either hyperpolarization or depolarization of the membrane potential (Fig. 4 C). The unusual “U”-shaped voltage dependence of apparent blocker affinity for external
, and in both cases the apparent Kd is minimal close to 0 mV and increases with either hyperpolarization or depolarization of the membrane potential (Fig. 4 C). The unusual “U”-shaped voltage dependence of apparent blocker affinity for external 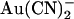 is a consequence of the “N”-shaped unitary current-voltage relationship (Fig. 4 A), and the slight “U”-shape also seen with internal
is a consequence of the “N”-shaped unitary current-voltage relationship (Fig. 4 A), and the slight “U”-shape also seen with internal 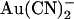 (Fig. 4 C) hints at a similar, perhaps less pronounced, biphasic voltage dependence of block. To confirm this unusual current-voltage relationship shape, we recorded macroscopic T-338A current-voltage relationships over extended voltage ranges (−100 to +100 mV) in the absence of
(Fig. 4 C) hints at a similar, perhaps less pronounced, biphasic voltage dependence of block. To confirm this unusual current-voltage relationship shape, we recorded macroscopic T-338A current-voltage relationships over extended voltage ranges (−100 to +100 mV) in the absence of 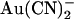 or with
or with 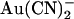 in the internal (2 mM) or external (1 mM) solutions, after maximal channel activation with PKA and ATP and subsequent channel “locking open” with PPi. As shown in Fig. 5 A, under these conditions T-338A-CFTR exhibits an almost completely linear macroscopic current-voltage relationship over the voltage range −100 to +100 mV, which, due to the locking of channels in the open state, should reflect the open channel unitary current-voltage relationship. In the presence of
in the internal (2 mM) or external (1 mM) solutions, after maximal channel activation with PKA and ATP and subsequent channel “locking open” with PPi. As shown in Fig. 5 A, under these conditions T-338A-CFTR exhibits an almost completely linear macroscopic current-voltage relationship over the voltage range −100 to +100 mV, which, due to the locking of channels in the open state, should reflect the open channel unitary current-voltage relationship. In the presence of 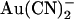 in either the intracellular or extracellular solution; however, the macroscopic current-voltage relationship shows an “N”-shape (Fig. 5 A) reminiscent of the shape of the unitary current-voltage relationships in the presence of
in either the intracellular or extracellular solution; however, the macroscopic current-voltage relationship shows an “N”-shape (Fig. 5 A) reminiscent of the shape of the unitary current-voltage relationships in the presence of 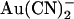 (Fig. 4 A). Quantification of the relative chord conductance at different membrane potentials indicates that, under control conditions, conductance in T-338A is maximal around 0 mV and decreases very slightly at strongly hyperpolarized and strongly depolarized membrane potentials (Fig. 5 B). In contrast, in the presence of both intracellular and extracellular
(Fig. 4 A). Quantification of the relative chord conductance at different membrane potentials indicates that, under control conditions, conductance in T-338A is maximal around 0 mV and decreases very slightly at strongly hyperpolarized and strongly depolarized membrane potentials (Fig. 5 B). In contrast, in the presence of both intracellular and extracellular 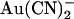 conductance is minimal close to 0 mV and increases greatly when the membrane potential is hyperpolarized or depolarized (Fig. 5 B). By analogy with the single channel current-voltage relationships and voltage dependence of Kd shown in Fig. 4, this reflects strong block by both intracellular and extracellular
conductance is minimal close to 0 mV and increases greatly when the membrane potential is hyperpolarized or depolarized (Fig. 5 B). By analogy with the single channel current-voltage relationships and voltage dependence of Kd shown in Fig. 4, this reflects strong block by both intracellular and extracellular 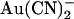 at 0 mV and relief of block by both hyperpolarization and depolarization of the membrane potential.
at 0 mV and relief of block by both hyperpolarization and depolarization of the membrane potential.
FIGURE 4.

Concentration- and voltage dependence of block of T-338A-CFTR by 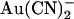 ions. (A) Mean unitary current-voltage relationships for T-338A-CFTR, under control conditions (○) and in the presence of 1 mM
ions. (A) Mean unitary current-voltage relationships for T-338A-CFTR, under control conditions (○) and in the presence of 1 mM 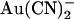 (•) in the intracellular (left) or extracellular (right) solution. (B) Fractional unitary current remaining (i/i0) after addition of different concentrations of
(•) in the intracellular (left) or extracellular (right) solution. (B) Fractional unitary current remaining (i/i0) after addition of different concentrations of 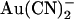 to the intracellular (left) or extracellular (right) solution, at membrane potentials of −50 mV (•) and +50 mV (○). Each of these four sets of data has been fitted by Eq. 1. (C) Mean Kd calculated from curves such as those shown in B for both internal (•) and external (○)
to the intracellular (left) or extracellular (right) solution, at membrane potentials of −50 mV (•) and +50 mV (○). Each of these four sets of data has been fitted by Eq. 1. (C) Mean Kd calculated from curves such as those shown in B for both internal (•) and external (○) 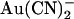 . Mean of data from 3–8 patches in each panel.
. Mean of data from 3–8 patches in each panel.
The inhibitory effects of intracellular and extracellular 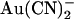 on wild-type and T-338A-CFTR are compared directly in Fig. 6. These data show that the T-338A mutation significantly decreases the apparent affinity of block by intracellular
on wild-type and T-338A-CFTR are compared directly in Fig. 6. These data show that the T-338A mutation significantly decreases the apparent affinity of block by intracellular 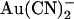 (especially at hyperpolarized membrane potentials where the block of wild-type is strongest) while significantly increasing the apparent affinity of block by extracellular
(especially at hyperpolarized membrane potentials where the block of wild-type is strongest) while significantly increasing the apparent affinity of block by extracellular 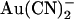 .
.
FIGURE 6.

Comparison of the blocking effects of internal and external 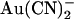 ions between wild-type and T-338A-CFTR. Mean Kds for wild-type (•) are as shown in Fig. 2 B; those for T-338A (○) are as in Fig. 4 C. In each case, asterisks indicate a statistically significant difference from wild-type.
ions between wild-type and T-338A-CFTR. Mean Kds for wild-type (•) are as shown in Fig. 2 B; those for T-338A (○) are as in Fig. 4 C. In each case, asterisks indicate a statistically significant difference from wild-type.
Block of T-338A-CFTR by other small anions
We hypothesized that the dramatic weakening of the asymmetry of 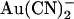 block that we observed in T-338A-CFTR was related to the fact that
block that we observed in T-338A-CFTR was related to the fact that 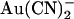 is a permeant blocker of the channel pore (18,21). We therefore wondered if the T-338A mutation would affect the apparent affinity of block by small anions that act as open channel blockers but that are impermeant. One such anion is the divalent pseudohalide ion
is a permeant blocker of the channel pore (18,21). We therefore wondered if the T-338A mutation would affect the apparent affinity of block by small anions that act as open channel blockers but that are impermeant. One such anion is the divalent pseudohalide ion 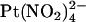 , which is impermeant but can block the channel from either the intracellular and extracellular side of the membrane (19,25). Indeed, we have previously shown by macroscopic current recording that the T-338A mutation does not significantly affect block by intracellular
, which is impermeant but can block the channel from either the intracellular and extracellular side of the membrane (19,25). Indeed, we have previously shown by macroscopic current recording that the T-338A mutation does not significantly affect block by intracellular 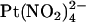 (19). To investigate the blocking effects of extracellular
(19). To investigate the blocking effects of extracellular 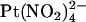 we used single channel recording from inside out membrane patches in the absence or presence of
we used single channel recording from inside out membrane patches in the absence or presence of 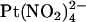 in the extracellular solution (Fig. 7). Unitary current-voltage relationships recorded under these conditions (Fig. 7 B), as well as the fractional current observed in the presence of 1 mM
in the extracellular solution (Fig. 7). Unitary current-voltage relationships recorded under these conditions (Fig. 7 B), as well as the fractional current observed in the presence of 1 mM 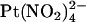 (Fig. 7 C), indicate that
(Fig. 7 C), indicate that 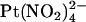 causes a strongly voltage-dependent inhibition of unitary current amplitude that appears similar in both wild-type and T-338A-CFTR.
causes a strongly voltage-dependent inhibition of unitary current amplitude that appears similar in both wild-type and T-338A-CFTR.
FIGURE 7.

Block by external 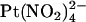 ions. (A) Example single channel currents for wild-type and T-338A-CFTR recorded from inside out patches at a membrane potential of +70 mV, under control conditions (left) and in the presence of 1 mM
ions. (A) Example single channel currents for wild-type and T-338A-CFTR recorded from inside out patches at a membrane potential of +70 mV, under control conditions (left) and in the presence of 1 mM 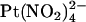 in the extracellular solution (right). (B) Mean unitary current-voltage relationships for wild-type and T-338A-CFTR, under control conditions (○) and in the presence of 1 mM
in the extracellular solution (right). (B) Mean unitary current-voltage relationships for wild-type and T-338A-CFTR, under control conditions (○) and in the presence of 1 mM 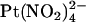 in the extracellular solution (•). (C) Fractional unitary current remaining in the presence of 1 mM extracellular
in the extracellular solution (•). (C) Fractional unitary current remaining in the presence of 1 mM extracellular 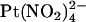 . Mean of data from 3–5 patches in each panel.
. Mean of data from 3–5 patches in each panel.
Unitary Cl− current in wild-type CFTR is also blocked by another permeant anion, SCN− (13), albeit with a considerably lower affinity than that observed for 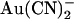 . However, although SCN− shows a fairly similar permeability to
. However, although SCN− shows a fairly similar permeability to 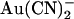 (12,18,21), its blocking effects are quite symmetrical, with an ∼2-fold higher Kd having been reported for extracellular SCN− block of unitary Cl− currents (14.2 mM at 0 mV membrane potential) compared to intracellular SCN− (6.2 mM at 0 mV) (13). Since one of the most striking effects of the T-338A mutation was the weakening of the asymmetry of
(12,18,21), its blocking effects are quite symmetrical, with an ∼2-fold higher Kd having been reported for extracellular SCN− block of unitary Cl− currents (14.2 mM at 0 mV membrane potential) compared to intracellular SCN− (6.2 mM at 0 mV) (13). Since one of the most striking effects of the T-338A mutation was the weakening of the asymmetry of 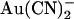 block, we also wondered how this mutation would affect the already quite symmetrical blocking effects of SCN−. As shown in Fig. 8, A and B, 10 mM SCN− had broadly similar blocking effects on T-338A unitary Cl− currents when present in either the intracellular or extracellular solution. As with
block, we also wondered how this mutation would affect the already quite symmetrical blocking effects of SCN−. As shown in Fig. 8, A and B, 10 mM SCN− had broadly similar blocking effects on T-338A unitary Cl− currents when present in either the intracellular or extracellular solution. As with 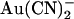 block, the unitary current-voltage relationship in the presence of SCN− was noticeably “N”-shaped (Fig. 8 B). As a result, the voltage dependence of block, like that of
block, the unitary current-voltage relationship in the presence of SCN− was noticeably “N”-shaped (Fig. 8 B). As a result, the voltage dependence of block, like that of 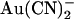 , showed a “U”-shape, with strongest inhibition close to 0 mV (Fig. 8, C and D). Whereas SCN− caused a similar degree of inhibition of wild-type CFTR (13) (Fig. 8, C and D), the voltage dependence of block was more conventional, with intracellular SCN− blocking most strongly at hyperpolarized membrane potentials (Fig. 8 C) and extracellular SCN− blocking most strongly at depolarized potentials (Fig. 8 D).
, showed a “U”-shape, with strongest inhibition close to 0 mV (Fig. 8, C and D). Whereas SCN− caused a similar degree of inhibition of wild-type CFTR (13) (Fig. 8, C and D), the voltage dependence of block was more conventional, with intracellular SCN− blocking most strongly at hyperpolarized membrane potentials (Fig. 8 C) and extracellular SCN− blocking most strongly at depolarized potentials (Fig. 8 D).
FIGURE 8.

Block of T-338A-CFTR by internal and external SCN− ions. (A) Example single channel currents for T-338A-CFTR, recorded from inside out patches at membrane potentials of +50 mV or −50 mV, under control conditions and in the presence of 10 mM SCN− in the intracellular or extracellular solution, as indicated. (B) Mean unitary current-voltage relationships for T-338A-CFTR, under control conditions (○) and in the presence of 10 mM SCN− (•) in the intracellular (left) or extracellular (right) solution. Mean of data from 3–10 patches. (C) Fractional unitary current remaining in the presence of 10 mM intracellular SCN− for wild-type (•) and T-338A (○). (D) Fractional unitary current remaining in the presence of 10 mM extracellular SCN− for wild-type (•) and T-338A (○). In C and D, data for wild-type are taken from Linsdell (13) under identical ionic conditions.
Origin of the voltage dependence of Au(CN)2− block of T-338A-CFTR
A striking feature of T-338A-CFTR block by both 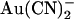 and SCN− is the “N”-shaped current-voltage relationship in the presence of blocker (Figs. 4 A, 5 A, and 8 B), reflecting a blocking event that is strongest close to 0 mV and is weakened by both hyperpolarization and depolarization. To investigate whether this reflected dependence of block on voltage itself or on the rate of Cl− movement through the pore, we investigated block of T-338A-CFTR macroscopic currents by intracellular
and SCN− is the “N”-shaped current-voltage relationship in the presence of blocker (Figs. 4 A, 5 A, and 8 B), reflecting a blocking event that is strongest close to 0 mV and is weakened by both hyperpolarization and depolarization. To investigate whether this reflected dependence of block on voltage itself or on the rate of Cl− movement through the pore, we investigated block of T-338A-CFTR macroscopic currents by intracellular 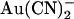 in the presence of different transmembrane Cl− concentration gradients (Fig. 9). Whether Cl− was reduced to 20 mM in either the intracellular or extracellular solution, current inhibition by addition of 1 mM
in the presence of different transmembrane Cl− concentration gradients (Fig. 9). Whether Cl− was reduced to 20 mM in either the intracellular or extracellular solution, current inhibition by addition of 1 mM 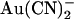 was still associated with an “N”-shaped current-voltage relationship (Fig. 9 A). As a result, maximal current inhibition occurred close to the current reversal potential (Fig. 9 B) under all ionic conditions studied. This suggests that voltage-dependent Cl− movement through the pore—rather than membrane potential per se—is responsible for the weakening of
was still associated with an “N”-shaped current-voltage relationship (Fig. 9 A). As a result, maximal current inhibition occurred close to the current reversal potential (Fig. 9 B) under all ionic conditions studied. This suggests that voltage-dependent Cl− movement through the pore—rather than membrane potential per se—is responsible for the weakening of 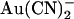 inhibition at extreme membrane potentials that results in the “N”-shaped current-voltage relationships observed.
inhibition at extreme membrane potentials that results in the “N”-shaped current-voltage relationships observed.
Rate theory models of Au(CN)2− interaction with wild-type and T-338A-CFTR
To illustrate how the T-338A mutation might decrease the apparent affinity of block by internal 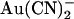 and yet increase the apparent affinity of block by external
and yet increase the apparent affinity of block by external 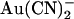 , we used the single channel I/V relationships shown in Figs. 1 and 4 to develop simple rate theory models of the CFTR pore. As described in the Discussion, our results could reflect either one or two
, we used the single channel I/V relationships shown in Figs. 1 and 4 to develop simple rate theory models of the CFTR pore. As described in the Discussion, our results could reflect either one or two 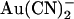 binding sites in the pore, and to reflect this we have developed two models, with either one or two anion binding sites (energy wells) within the pore. Best fit energy profiles for both Cl− and
binding sites in the pore, and to reflect this we have developed two models, with either one or two anion binding sites (energy wells) within the pore. Best fit energy profiles for both Cl− and 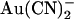 were simultaneously optimized by the fitting procedure, and the effects of the T-338A mutation were modeled by altering a single parameter—i.e., by lowering one energy barrier for both Cl− and
were simultaneously optimized by the fitting procedure, and the effects of the T-338A mutation were modeled by altering a single parameter—i.e., by lowering one energy barrier for both Cl− and 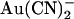 movement in the pore (see Discussion). Relative energies for the two different anions are shown graphically as energy barrier profiles in Fig. 10, A–D. The predictions of the models (Fig. 10, E–H) indicate that both the single binding site model (Fig. 10, A and B) and the two binding site model (Fig. 10, C and D) are able to replicate the most salient features observed experimentally—strong block of wild-type by internal
movement in the pore (see Discussion). Relative energies for the two different anions are shown graphically as energy barrier profiles in Fig. 10, A–D. The predictions of the models (Fig. 10, E–H) indicate that both the single binding site model (Fig. 10, A and B) and the two binding site model (Fig. 10, C and D) are able to replicate the most salient features observed experimentally—strong block of wild-type by internal 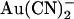 , weak block of wild-type by external
, weak block of wild-type by external 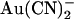 , weakened block of T-338A by internal
, weakened block of T-338A by internal 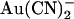 , and yet strengthened block of this mutant by external
, and yet strengthened block of this mutant by external 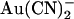 .
.
FIGURE 10.

Rate theory modeling of Cl− permeation and 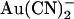 block. (A–D) Best fit energy profiles for Cl− (solid lines) and
block. (A–D) Best fit energy profiles for Cl− (solid lines) and 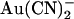 (dashed lines) movement in the CFTR pore. (A) One-site model for wild-type. (B) One-site model for T-338A. (C) Two-site model for wild-type. (D) Two-site model for T-338A. To avoid wrongly implying physical meaning to the parameters described by the model, energy is plotted in relative units only. (E–H) Predictions of the models shown in A–D. Data points for wild-type are taken from Fig. 1, D and E, and for T-338A from Fig. 4 A. In each panel, lines represent the predictions of the model shown above that panel.
(dashed lines) movement in the CFTR pore. (A) One-site model for wild-type. (B) One-site model for T-338A. (C) Two-site model for wild-type. (D) Two-site model for T-338A. To avoid wrongly implying physical meaning to the parameters described by the model, energy is plotted in relative units only. (E–H) Predictions of the models shown in A–D. Data points for wild-type are taken from Fig. 1, D and E, and for T-338A from Fig. 4 A. In each panel, lines represent the predictions of the model shown above that panel.
DISCUSSION
Au(CN)2− is an asymmetric blocker of the wild-type CFTR channel pore
Inhibition of wild-type CFTR by 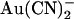 has been investigated in many previous studies (18,21,24,33,38). When applied to the cytoplasmic face of inside out membrane patches,
has been investigated in many previous studies (18,21,24,33,38). When applied to the cytoplasmic face of inside out membrane patches, 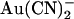 has two inhibitory effects on CFTR Cl− currents—a high affinity reduction in channel open probability, and a lower affinity, voltage-dependent reduction in unitary current amplitude (38). This second effect appears to reflect binding of
has two inhibitory effects on CFTR Cl− currents—a high affinity reduction in channel open probability, and a lower affinity, voltage-dependent reduction in unitary current amplitude (38). This second effect appears to reflect binding of 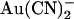 within the open channel pore, where it impedes Cl− movement through the pore. External
within the open channel pore, where it impedes Cl− movement through the pore. External 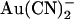 has been reported to inhibit wild-type CFTR with a broadly similar potency and probably by the same mechanism (18,24); however, these effects have been observed only at the whole cell current level. It is somewhat surprising, therefore, that the effects of
has been reported to inhibit wild-type CFTR with a broadly similar potency and probably by the same mechanism (18,24); however, these effects have been observed only at the whole cell current level. It is somewhat surprising, therefore, that the effects of 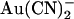 on CFTR single channel current amplitude show such strong asymmetry—with external
on CFTR single channel current amplitude show such strong asymmetry—with external 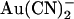 being 1–2 orders of magnitude lower affinity than intracellular
being 1–2 orders of magnitude lower affinity than intracellular 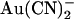 (Fig. 2). Thus the inhibitory effects of extracellular
(Fig. 2). Thus the inhibitory effects of extracellular 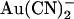 that we observe at the single channel level (Fig. 1)—which suggest a Kd of ∼10 mM (Fig. 2)—appear to be much weaker than those reported using whole cell recording (18,24). This discrepancy may reflect multiple inhibitory effects of extracellular
that we observe at the single channel level (Fig. 1)—which suggest a Kd of ∼10 mM (Fig. 2)—appear to be much weaker than those reported using whole cell recording (18,24). This discrepancy may reflect multiple inhibitory effects of extracellular 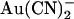 that contribute to the overall inhibition observed at the whole cell level—a situation analogous to the multiple inhibitory effects of intracellular
that contribute to the overall inhibition observed at the whole cell level—a situation analogous to the multiple inhibitory effects of intracellular 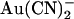 that contribute to its blocking actions on macroscopic Cl− currents (38). Thus it is possible (but not investigated) that external
that contribute to its blocking actions on macroscopic Cl− currents (38). Thus it is possible (but not investigated) that external 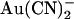 could affect the open probability of the channel; it has been shown previously that CFTR gating is sensitive to extracellular anions including Cl− itself (39). It is less likely that
could affect the open probability of the channel; it has been shown previously that CFTR gating is sensitive to extracellular anions including Cl− itself (39). It is less likely that 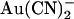 is permeating the channel to inhibit channel open probability at an intracellular site of action, since the inhibitory effects of external
is permeating the channel to inhibit channel open probability at an intracellular site of action, since the inhibitory effects of external 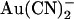 on whole cell currents have been shown to be readily reversible (24). Irrespective of the reasons for these discrepancies, the strikingly different effects of
on whole cell currents have been shown to be readily reversible (24). Irrespective of the reasons for these discrepancies, the strikingly different effects of 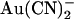 on single channel current amplitude suggest that
on single channel current amplitude suggest that 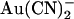 ions bind much more tightly inside the pore when added to the intracellular versus the extracellular solution.
ions bind much more tightly inside the pore when added to the intracellular versus the extracellular solution.
Why is 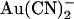 block of Cl− permeation through the pore so strongly asymmetric? It is well known that large organic anions usually block the CFTR channel pore only from the inside, due primarily to their interaction with a positively charged amino acid side chain (K-95 in TM1) in the wide inner vestibule of the pore (40–42). These large substances are thought to be excluded from the extracellular mouth of the pore for reasons related to their size (43). In contrast,
block of Cl− permeation through the pore so strongly asymmetric? It is well known that large organic anions usually block the CFTR channel pore only from the inside, due primarily to their interaction with a positively charged amino acid side chain (K-95 in TM1) in the wide inner vestibule of the pore (40–42). These large substances are thought to be excluded from the extracellular mouth of the pore for reasons related to their size (43). In contrast, 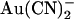 is highly permeant in CFTR (in fact, with a higher permeability than Cl− itself) (18,21), such that it is expected to have access to binding sites throughout the pore when present in either the intracellular or the extracellular solution. Nevertheless, its interactions with binding sites within the pore appear to be very different depending on which side of the membrane it is applied from. This could reflect either a single
is highly permeant in CFTR (in fact, with a higher permeability than Cl− itself) (18,21), such that it is expected to have access to binding sites throughout the pore when present in either the intracellular or the extracellular solution. Nevertheless, its interactions with binding sites within the pore appear to be very different depending on which side of the membrane it is applied from. This could reflect either a single 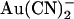 binding site in the pore that is more accessible from the intracellular solution or separate binding sites that underlie block by intracellular and extracellular
binding site in the pore that is more accessible from the intracellular solution or separate binding sites that underlie block by intracellular and extracellular 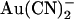 , with the intracellular site showing a greater affinity for
, with the intracellular site showing a greater affinity for 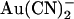 . Currently we do not have enough information to be able to discriminate between these two models, and as a result we have developed rate theory models having either one or two binding sites (energy wells). Indeed, the energy profiles generated for wild-type CFTR reflect the general features described above; the single site model (Fig. 10 A) shows a lower energy barrier (resulting in easier access) for
. Currently we do not have enough information to be able to discriminate between these two models, and as a result we have developed rate theory models having either one or two binding sites (energy wells). Indeed, the energy profiles generated for wild-type CFTR reflect the general features described above; the single site model (Fig. 10 A) shows a lower energy barrier (resulting in easier access) for 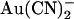 entry from the intracellular side of the membrane, whereas the two-site model (Fig. 10 C) shows a deeper energy well (resulting in tighter binding) for the more cytoplasmically located binding site. Although we find that the single binding site model is the simplest that can reproduce our single channel data (Fig. 10, D and E), there are several factors that we consider as favoring the second model with two binding sites. First, we believe that there are blocker binding sites in both the outer and inner mouths of the pore, since the impermeant anion
entry from the intracellular side of the membrane, whereas the two-site model (Fig. 10 C) shows a deeper energy well (resulting in tighter binding) for the more cytoplasmically located binding site. Although we find that the single binding site model is the simplest that can reproduce our single channel data (Fig. 10, D and E), there are several factors that we consider as favoring the second model with two binding sites. First, we believe that there are blocker binding sites in both the outer and inner mouths of the pore, since the impermeant anion 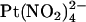 can block Cl− permeation when added to either side of the membrane, apparently by binding to different sites (19,25). This is also consistent with the presence of a blocker binding site in the outer pore vestibule proposed by Muanprasat et al. (44). Second, point mutations in both the extracellular and intracellular mouths of the pore affect
can block Cl− permeation when added to either side of the membrane, apparently by binding to different sites (19,25). This is also consistent with the presence of a blocker binding site in the outer pore vestibule proposed by Muanprasat et al. (44). Second, point mutations in both the extracellular and intracellular mouths of the pore affect 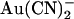 block, and this may reflect disruption of separate binding sites. Thus,
block, and this may reflect disruption of separate binding sites. Thus, 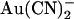 block is greatly weakened by mutagenesis of R-334 (in TM6) (22,23), a positively charged residue which is involved in attracting extracellular Cl− ions into the outer mouth of the pore (45), and of K-95 (in TM1) (22), the positive charge of which attracts intracellular Cl− ions into the wide inner pore vestibule (40). Finally, block by extracellular
block is greatly weakened by mutagenesis of R-334 (in TM6) (22,23), a positively charged residue which is involved in attracting extracellular Cl− ions into the outer mouth of the pore (45), and of K-95 (in TM1) (22), the positive charge of which attracts intracellular Cl− ions into the wide inner pore vestibule (40). Finally, block by extracellular 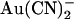 is apparently voltage independent (Fig. 2 B); although the voltage dependence of intracellular
is apparently voltage independent (Fig. 2 B); although the voltage dependence of intracellular 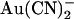 block is complex in origin (33), this lack of voltage dependence would at first glance appear inconsistent with extracellular
block is complex in origin (33), this lack of voltage dependence would at first glance appear inconsistent with extracellular 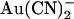 ions moving deeply into the pore to block at a binding site located in the inner pore vestibule (Fig. 10 A).
ions moving deeply into the pore to block at a binding site located in the inner pore vestibule (Fig. 10 A).
Au(CN)2− block of the T-338A-CFTR channel pore is much more symmetrical
Previously we showed, using macroscopic current recording, that block by intracellular 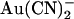 was greatly weakened in T-338A-CFTR and suggested that T-338 may contribute to a lyotropic anion binding site in the pore (21,22). However, the results here at the single channel level indicate that whereas this mutation does weaken block by intracellular
was greatly weakened in T-338A-CFTR and suggested that T-338 may contribute to a lyotropic anion binding site in the pore (21,22). However, the results here at the single channel level indicate that whereas this mutation does weaken block by intracellular 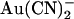 , it actually strengthens block by extracellular
, it actually strengthens block by extracellular 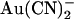 (Fig. 6). This apparently contradictory effect of a single mutation is difficult to reconcile with disruption of an
(Fig. 6). This apparently contradictory effect of a single mutation is difficult to reconcile with disruption of an 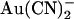 binding site in the pore. Instead, we now propose that the T-338A mutation alters the movement of
binding site in the pore. Instead, we now propose that the T-338A mutation alters the movement of 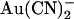 within the pore, changing its ability to access one or more binding sites the properties of which are not directly affected by the mutation. Consistent with this idea, block by impermeant
within the pore, changing its ability to access one or more binding sites the properties of which are not directly affected by the mutation. Consistent with this idea, block by impermeant 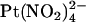 ions is not affected by the T-338A mutation, whether
ions is not affected by the T-338A mutation, whether 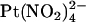 is added to the intracellular (19) or extracellular solution (Fig. 7). This suggests that
is added to the intracellular (19) or extracellular solution (Fig. 7). This suggests that 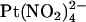 ions are restricted to binding sites on the same side of the membrane to which they are applied and that the T-338A mutation has no impact either on the
ions are restricted to binding sites on the same side of the membrane to which they are applied and that the T-338A mutation has no impact either on the 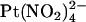 binding affinity of these sites or on
binding affinity of these sites or on 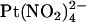 movement between these sites.
movement between these sites.
The ways in which the T-338A mutation affects 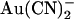 inhibition can be interpreted in terms of either the single or double binding site models shown in Fig. 10. If
inhibition can be interpreted in terms of either the single or double binding site models shown in Fig. 10. If 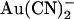 is considered as acting at a single site, then the T-338A mutation could increase the rate of
is considered as acting at a single site, then the T-338A mutation could increase the rate of 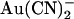 movement between this site and the extracellular solution. This would have the effect of weakening
movement between this site and the extracellular solution. This would have the effect of weakening 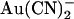 block from the inside (since
block from the inside (since 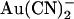 would now better be able to escape its binding site to the extracellular solution, relieving block) and at the same time strengthening block from the outside (since
would now better be able to escape its binding site to the extracellular solution, relieving block) and at the same time strengthening block from the outside (since 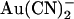 would now better be able to reach its binding site). In the two binding site model, the effects of the T-338A mutation could result from an increase in the rate of
would now better be able to reach its binding site). In the two binding site model, the effects of the T-338A mutation could result from an increase in the rate of 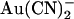 movement between the two sites. The effect on
movement between the two sites. The effect on 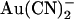 block is very similar to that described above for the single site model; intracellular
block is very similar to that described above for the single site model; intracellular 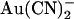 block is weakened because of enhanced unblock by
block is weakened because of enhanced unblock by 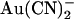 permeation to the outside, whereas extracellular
permeation to the outside, whereas extracellular 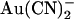 block is strengthened because of increased access to the higher affinity, internal binding site. Note that in both of these suggested models, the effect of the T-338A mutation is to lower a barrier to
block is strengthened because of increased access to the higher affinity, internal binding site. Note that in both of these suggested models, the effect of the T-338A mutation is to lower a barrier to 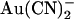 movement within the pore, rather than to affect an
movement within the pore, rather than to affect an 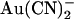 binding site. Indeed, for the energy barrier profiles developed (Fig. 10), the effects of the T-338A mutation are reproduced by lowering the height of a single energy barrier—either the barrier between the binding site and the extracellular solution in the one-site model (Fig. 11 A) or the barrier between the two binding sites in the two-site model (Fig. 11 B).
binding site. Indeed, for the energy barrier profiles developed (Fig. 10), the effects of the T-338A mutation are reproduced by lowering the height of a single energy barrier—either the barrier between the binding site and the extracellular solution in the one-site model (Fig. 11 A) or the barrier between the two binding sites in the two-site model (Fig. 11 B).
FIGURE 11.

Modeling of the effects of the T-338A mutation. Energy profiles shown in Fig. 10 are redrawn to show the effect of the T-338A mutation on Cl− and 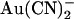 movement in the pore, both for the one-site model (top) and the two-site model (bottom). In each case, solid lines represent the energy profile for wild-type, and dashed lines the changes made to model T-338A. The effects of the mutation are modeled as (top) lowering the energy barrier that exists between the single anion binding site and the extracellular solution, or (bottom) lowering the energy barrier that exists between the two anion binding sites. As illustrated in Fig. 10, these localized changes in energy profile were sufficient to model the salient differences in
movement in the pore, both for the one-site model (top) and the two-site model (bottom). In each case, solid lines represent the energy profile for wild-type, and dashed lines the changes made to model T-338A. The effects of the mutation are modeled as (top) lowering the energy barrier that exists between the single anion binding site and the extracellular solution, or (bottom) lowering the energy barrier that exists between the two anion binding sites. As illustrated in Fig. 10, these localized changes in energy profile were sufficient to model the salient differences in 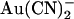 interactions between wild-type and T-338A.
interactions between wild-type and T-338A.
The effects of the T-338A mutation are consistent with either the one-site or two-site model (Fig. 11). Indeed, a single binding site model is the simplest that can reproduce the effects of 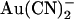 observed at the single channel level (Fig. 10 F). Nevertheless, we would interpret the lack of effect of this mutation on block by either intracellular or extracellular
observed at the single channel level (Fig. 10 F). Nevertheless, we would interpret the lack of effect of this mutation on block by either intracellular or extracellular 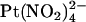 block as supporting the two-site model, since these data are consistent with the idea that there are blocker binding sites located both external and internal to the location of T-338 within the pore, as illustrated in the two-site model (Fig. 11 B).
block as supporting the two-site model, since these data are consistent with the idea that there are blocker binding sites located both external and internal to the location of T-338 within the pore, as illustrated in the two-site model (Fig. 11 B).
Voltage and chloride dependence of Au(CN)2− block
The voltage dependence of 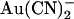 block is also strongly altered in T-338A-CFTR. In wild-type CFTR, block by intracellular
block is also strongly altered in T-338A-CFTR. In wild-type CFTR, block by intracellular 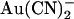 shows a conventional voltage dependence, with block by this negatively charged substance being strengthened by hyperpolarization of the membrane potential (Figs. 1 D and 2 B). Block by external
shows a conventional voltage dependence, with block by this negatively charged substance being strengthened by hyperpolarization of the membrane potential (Figs. 1 D and 2 B). Block by external 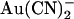 appears to be voltage insensitive (Figs. 1 E and 2 B), suggesting that the blocker binding site may be outside of the transmembrane electric field. Although some similarities are seen in T-338A (for example, block by internal
appears to be voltage insensitive (Figs. 1 E and 2 B), suggesting that the blocker binding site may be outside of the transmembrane electric field. Although some similarities are seen in T-338A (for example, block by internal 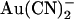 is stronger at hyperpolarized than at depolarized voltages; Fig. 4 C), the most striking aspect of the voltage dependence of block in this mutant is the “U”-shaped apparent affinity-voltage relationship that characterizes block by both intracellular and extracellular
is stronger at hyperpolarized than at depolarized voltages; Fig. 4 C), the most striking aspect of the voltage dependence of block in this mutant is the “U”-shaped apparent affinity-voltage relationship that characterizes block by both intracellular and extracellular 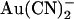 (Figs. 4 C and 5). This unusual relationship between apparent blocker affinity and voltage suggests that block is relieved by both hyperpolarization and by depolarization of the membrane potential, which most likely reflects the ability of the blocking ion to exit either to the extracellular or the intracellular solution. A similar voltage dependence of block is also observed in T-338A-CFTR by the permeant blocking ion SCN− (Fig. 8) but not the impermeant
(Figs. 4 C and 5). This unusual relationship between apparent blocker affinity and voltage suggests that block is relieved by both hyperpolarization and by depolarization of the membrane potential, which most likely reflects the ability of the blocking ion to exit either to the extracellular or the intracellular solution. A similar voltage dependence of block is also observed in T-338A-CFTR by the permeant blocking ion SCN− (Fig. 8) but not the impermeant 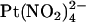 , which shows a conventional voltage dependence of block when present in the extracellular (Fig. 7) or intracellular solution (19).
, which shows a conventional voltage dependence of block when present in the extracellular (Fig. 7) or intracellular solution (19).
Block of wild-type CFTR by intracellular 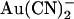 is strongly influenced by the Cl− concentration. The affinity of block is greatly decreased and the voltage dependence of block greatly increased as the concentration of Cl− in the extracellular solution is increased, suggesting that movement of intracellular
is strongly influenced by the Cl− concentration. The affinity of block is greatly decreased and the voltage dependence of block greatly increased as the concentration of Cl− in the extracellular solution is increased, suggesting that movement of intracellular 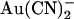 and extracellular Cl− ions are coupled (33). In contrast, intracellular Cl− has no apparent effect on block by intracellular
and extracellular Cl− ions are coupled (33). In contrast, intracellular Cl− has no apparent effect on block by intracellular 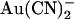 (33). In T-338A-CFTR, both intracellular and extracellular Cl− appear to have a similar impact on block by intracellular
(33). In T-338A-CFTR, both intracellular and extracellular Cl− appear to have a similar impact on block by intracellular 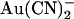 , shifting the voltage dependence of block such that the maximum apparent blocker affinity appears to follow the current reversal potential (Fig. 9). This unusual result suggests that
, shifting the voltage dependence of block such that the maximum apparent blocker affinity appears to follow the current reversal potential (Fig. 9). This unusual result suggests that 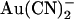 block in this mutant is, in fact, more sensitive to the voltage-dependent flow of Cl− ions than to the transmembrane voltage itself. Again, this is consistent with the idea that blocking
block in this mutant is, in fact, more sensitive to the voltage-dependent flow of Cl− ions than to the transmembrane voltage itself. Again, this is consistent with the idea that blocking 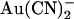 ions can exit the pore in either direction; at hyperpolarized voltages, rapid Cl− efflux will sweep blocking
ions can exit the pore in either direction; at hyperpolarized voltages, rapid Cl− efflux will sweep blocking 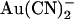 ions from the pore into the extracellular solution, whereas at depolarized potentials Cl− ions will flow into the intracellular solution and carry blocking
ions from the pore into the extracellular solution, whereas at depolarized potentials Cl− ions will flow into the intracellular solution and carry blocking 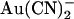 ions along with them.
ions along with them.
Role of T-338 as a barrier to anion movement in the pore
Previously we suggested that T-338 contributes to an important lyotropic anion binding site in the pore, based on the finding that the T-338A mutation weakened open channel block by intracellular 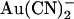 ions (21,22). However, this work suggests that this mutation does not in fact affect an
ions (21,22). However, this work suggests that this mutation does not in fact affect an 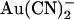 binding site, but instead alters the ability of
binding site, but instead alters the ability of 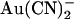 to move between different sites within the channel pore. The finding that a point mutation in the pore can have such a large effect on the apparent affinity of a blocker (either increase or decrease) depending on the exact experimental protocol and our suggestion that this occurs without an actual modification of blocker binding sites, therefore, offers an important caveat for site-directed mutagenesis studies that seek to identify blocker binding sites within ion channel pores. Given the different effects of the T-338A mutation on the blocking effects of
to move between different sites within the channel pore. The finding that a point mutation in the pore can have such a large effect on the apparent affinity of a blocker (either increase or decrease) depending on the exact experimental protocol and our suggestion that this occurs without an actual modification of blocker binding sites, therefore, offers an important caveat for site-directed mutagenesis studies that seek to identify blocker binding sites within ion channel pores. Given the different effects of the T-338A mutation on the blocking effects of 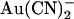 and
and 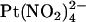 , this caveat might be particularly germane when investigating permeant blockers.
, this caveat might be particularly germane when investigating permeant blockers.
We suggest that the mutation T-338A removes or reduces a barrier to permeant anion movement inside the CFTR channel pore (Fig. 11). The existence of this barrier within the wild-type channel pore appears to play an important role in underlying asymmetric interactions between permeant ions and the pore that result in the strongly asymmetric blocking effects of 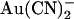 (Fig. 2). Removal of this barrier also allows Cl− ions to effectively sweep blocking
(Fig. 2). Removal of this barrier also allows Cl− ions to effectively sweep blocking 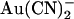 ions from the pore, resulting in a blocking action that is more dependent on voltage-dependent Cl− flux than on transmembrane voltage itself (Fig. 9). We propose that the barrier associated with T-338 effectively separates the outer and inner pore vestibules, both of which contain permeant anion binding sites with different properties.
ions from the pore, resulting in a blocking action that is more dependent on voltage-dependent Cl− flux than on transmembrane voltage itself (Fig. 9). We propose that the barrier associated with T-338 effectively separates the outer and inner pore vestibules, both of which contain permeant anion binding sites with different properties.
It is tempting to speculate that the lyotropic anion binding sites situated external and internal to the T-338 “barrier” involve the pore-lining positive charges of R-334 and K-95, respectively. Indeed, of those amino acids that have been studied by site-directed mutagenesis, these two residues have the greatest impact on apparent 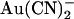 binding affinity (22). Furthermore, current models of the pore place these two residues on the extracellular and intracellular side of T-338, respectively (8). Unfortunately, mutagenesis of either of these two positively charged residues causes a considerable decrease in unitary Cl− conductance (22,45,46), making single channel studies on the effect of internal and external
binding affinity (22). Furthermore, current models of the pore place these two residues on the extracellular and intracellular side of T-338, respectively (8). Unfortunately, mutagenesis of either of these two positively charged residues causes a considerable decrease in unitary Cl− conductance (22,45,46), making single channel studies on the effect of internal and external 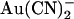 ions on these mutants unfeasible. Most other substitutions of T-338 also lead to a large reduction in single channel conductance (26).
ions on these mutants unfeasible. Most other substitutions of T-338 also lead to a large reduction in single channel conductance (26).
CONCLUSIONS
Our results suggest that T-338 contributes to a barrier to anion movement inside the pore. For Cl− permeation, the consequence of lowering this barrier is an increase in unitary conductance. For anions that interact more strongly with binding sites inside the pore, such as 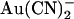 , the existence of a barrier divides the pore into intracellularly and extracellularly accessible compartments, resulting in strongly side-dependent interactions between anions and the channel. Lowering of the barrier, as occurs in the T-338A mutant, results in a channel that interacts much more symmetrically with high affinity permeant anions.
, the existence of a barrier divides the pore into intracellularly and extracellularly accessible compartments, resulting in strongly side-dependent interactions between anions and the channel. Lowering of the barrier, as occurs in the T-338A mutant, results in a channel that interacts much more symmetrically with high affinity permeant anions.
Acknowledgments
We thank Dr. Osvaldo Alvarez for providing the AJUSTE computer program used for rate theory modeling, and Elizabeth VandenBerg for technical assistance.
This work was supported by the Canadian Institutes of Health Research.
Chantal N. St. Aubin's present address is Dept. of Pharmacology, University of Alberta, Edmonton, Alberta, Canada.
References
- 1.MacKinnon, R. 2003. Potassium channels. FEBS Lett. 555:62–65. [DOI] [PubMed] [Google Scholar]
- 2.Dutzler, R. 2004. Structural basis for ion conduction and gating in ClC chloride channels. FEBS Lett. 564:229–233. [DOI] [PubMed] [Google Scholar]
- 3.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 4.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+-channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 5.Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- 6.Sather, W. A., and E. W. McCleskey. 2003. Permeation and selectivity in calcium channels. Annu. Rev. Physiol. 65:133–159. [DOI] [PubMed] [Google Scholar]
- 7.Chen, T.-Y. 2005. Structure and function of CLC channels. Annu. Rev. Physiol. 67:809–839. [DOI] [PubMed] [Google Scholar]
- 8.Linsdell, P. 2006. Mechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. Exp. Physiol. 91:123–129. [DOI] [PubMed] [Google Scholar]
- 9.Hille, B. 2001. Ion Channels of Excitable Membranes, 3rd ed. Sinauer Associates, Sunderland, MA.
- 10.Neyton, J., and C. Miller. 1988. Potassium blocks barium permeation through a calcium-activated potassium channel. J. Gen. Physiol. 92:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neyton, J., and C. Miller. 1988. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+-activated K+ channel. J. Gen. Physiol. 92:569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawson, D. C., S. S. Smith, and M. K. Mansoura. 1999. CFTR: mechanism of anion conduction. Physiol. Rev. 79:S47–S75. [DOI] [PubMed] [Google Scholar]
- 13.Linsdell, P. 2001. Thiocyanate as a probe of the cystic fibrosis transmembrane conductance regulator chloride channel pore. Can. J. Physiol. Pharmacol. 79:573–579. [PubMed] [Google Scholar]
- 14.Tabcharani, J. A., J. M. Rommens, Y.-X. Hou, X.-B. Chang, L.-C. Tsui, J. R. Riordan, and J. W. Hanrahan. 1993. Multi-ion pore behaviour in the CFTR chloride channel. Nature. 366:79–82. [DOI] [PubMed] [Google Scholar]
- 15.Mansoura, M. K., S. S. Smith, A. D. Choi, N. W. Richards, T. V. Strong, M. L. Drumm, F. S. Collins, and D. C. Dawson. 1998. Cystic fibrosis transmembrane conductance regulator (CFTR) anion binding as a probe of the pore. Biophys. J. 74:1320–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta, J., A. Evagelidis, J. W. Hanrahan, and P. Linsdell. 2001. Asymmetric structure of the cystic fibrosis transmembrane conductance regulator chloride channel pore suggested by mutagenesis of the twelfth transmembrane region. Biochemistry. 40:6620–6627. [DOI] [PubMed] [Google Scholar]
- 17.Linsdell, P. 2001. Relationship between anion binding and anion permeability revealed by mutagenesis within the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Physiol. 531:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith, S. S., E. D. Steinle, M. E. Meyerhoff, and D. C. Dawson. 1999. Cystic fibrosis transmembrane conductance regulator. Physical basis for lyotropic anion selectivity patterns. J. Gen. Physiol. 114:799–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong, X., and P. Linsdell. 2003. Mutation-induced blocker permeability and multiion block of the CFTR chloride channel pore. J. Gen. Physiol. 122:673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, X., S. S. Smith, and D. C. Dawson. 2003. CFTR: what's it like inside the pore? J. Exp. Zool. 300A:69–75. [DOI] [PubMed] [Google Scholar]
-
21.Gong, X., S. M. Burbridge, E. A. Cowley, and P. Linsdell. 2002. Molecular determinants of
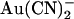 binding and permeability within the cystic fibrosis transmembrane conductance regulator Cl− channel pore. J. Physiol. 540:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
binding and permeability within the cystic fibrosis transmembrane conductance regulator Cl− channel pore. J. Physiol. 540:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar] - 22.Ge, N., C. N. Muise, X. Gong, and P. Linsdell. 2004. Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 279:55283–55289. [DOI] [PubMed] [Google Scholar]
- 23.Gong, X., and P. Linsdell. 2003. Molecular determinants and role of an anion binding site in the external mouth of the CFTR chloride channel pore. J. Physiol. 549:387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano, J. R., X. Liu, E. R. Borg, C. S. Alexander, C. F. Shaw, and D. C. Dawson. 2006. CFTR: ligand exchange locks a permeant anion in the pore. Biophys. J. 91:1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge, N., and P. Linsdell. 2006. Interactions between impermeant blocking ions in the cystic fibrosis transmembrane conductance regulator chloride channel pore: evidence for anion-induced conformational changes. J. Membr. Biol. 210:31–42. [DOI] [PubMed] [Google Scholar]
- 26.Linsdell, P., S.-X. Zheng, and J. W. Hanrahan. 1998. Non-pore lining amino acid side chains influence anion selectivity of the human CFTR Cl− channel expressed in mammalian cell lines. J. Physiol. 512:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarty, N. A., and Z.-R. Zhang. 2001. Identification of a region of strong discrimination in the pore of CFTR. Am. J. Physiol. 281:L852–L867. [DOI] [PubMed] [Google Scholar]
- 28.Cheung, M., and M. H. Akabas. 1996. Identification of cystic fibrosis transmembrane conductance regulator channel-lining residues in and flanking the M6 membrane-spanning segment. Biophys. J. 70:2688–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu, X., Z.-R. Zhang, M. D. Fuller, J. Billingsley, N. A. McCarty, and D. C. Dawson. 2004. CFTR: a cysteine at position 338 in TM6 senses a positive electrostatic potential in the pore. Biophys. J. 87:3826–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, X., C. Alexander, J. Serrano, E. Borg, and D. C. Dawson. 2006. Variable reactivity of a cysteine at position 338 in cystic fibrosis transmembrane conductance regulator reflects different chemical states of the thiol. J. Biol. Chem. 281:8275–8285. [DOI] [PubMed] [Google Scholar]
- 31.Gong, X., S. M. Burbridge, A. C. Lewis, P. Y. D. Wong, and P. Linsdell. 2002. Mechanism of lonidamine inhibition of the CFTR chloride channel. Br. J. Pharmacol. 137:928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta, J., and P. Linsdell. 2002. Point mutations in the pore region directly or indirectly affect glibenclamide block of the CFTR chloride channel. Pflugers Arch. 443:739–747. [DOI] [PubMed] [Google Scholar]
- 33.Gong, X., and P. Linsdell. 2003. Coupled movement of permeant and blocking ions in the CFTR chloride channel pore. J. Physiol. 549:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linsdell, P., and J. W. Hanrahan. 1996. Disulphonic stilbene block of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a mammalian cell line and its regulation by a critical pore residue. J. Physiol. 496:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linsdell, P., and J. W. Hanrahan. 1998. Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J. Gen. Physiol. 111:601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvarez, O., A. Villarroel, and G. Eisenman. 1992. Calculation of ion currents from energy profiles and energy profiles from ion currents in multibarrier, multisite, multioccupancy channel model. Methods. Enzymol. 207:816–854. [DOI] [PubMed] [Google Scholar]
- 37.Linsdell, P., J. A. Tabcharani, and J. W. Hanrahan. 1997. Multi-ion mechanism for ion permeation and block in the cystic fibrosis transmembrane conductance regulator chloride channel. J. Gen. Physiol. 110:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
38.Linsdell, P., and X. Gong. 2002. Multiple inhibitory effects of
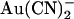 ions on cystic fibrosis transmembrane conductance regulator Cl− channel currents. J. Physiol. 540:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
ions on cystic fibrosis transmembrane conductance regulator Cl− channel currents. J. Physiol. 540:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar] - 39.Wright, A. M., X. Gong, B. Verdon, P. Linsdell, A. Mehta, J. R. Riordan, B. E. Argent, and M. A. Gray. 2004. Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J. Biol. Chem. 279:41658–41663. [DOI] [PubMed] [Google Scholar]
- 40.Linsdell, P. 2005. Location of a common inhibitor binding site in the cytoplasmic vestibule of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 280:8945–8950. [DOI] [PubMed] [Google Scholar]
- 41.Sheppard, D. N., and K. A. Robinson. 1997. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J. Physiol. 503:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou, Z., S. Hu, and T.-C. Hwang. 2002. Probing an open CFTR pore with organic anion blockers. J. Gen. Physiol. 120:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCarty, N. A. 2000. Permeation through the CFTR chloride channel. J. Exp. Biol. 203:1947–1962. [DOI] [PubMed] [Google Scholar]
- 44.Muanprasat, C., N. D. Sonawane, D. Salinas, A. Taddei, L. J. V. Galietta, and A. S. Verkman. 2004. Discovery of glycine hydrazide pore-occluding CFTR inhibitors. Mechanism, structure-activity analysis, and in vivo efficacy. J. Gen. Physiol. 124:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith, S. S., X. Liu, Z.-R. Zhang, F. Sun, T. E. Kriewall, N. A. McCarty, and D. C. Dawson. 2001. CFTR: covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J. Gen. Physiol. 118:407–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gong, X., and P. Linsdell. 2004. Maximization of the rate of chloride conduction in the CFTR channel pore by ion-ion interactions. Arch. Biochem. Biophys. 426:78–82. [DOI] [PubMed] [Google Scholar]