

Abstract
An enkephalin analogue coupled to ‘aminofentanyl’ has been synthesized and tested for biological activities at the μ and δ opioid receptors. Aminofentanyl which represents a structural derivative of fentanyl has been synthesized by acylation of 1-(2-phenethyl)-4-(N-anilino)piperidine with phthaloyl protected β-alaninyl chloride in the presence of DIPEA, followed by deprotection with hydrazine hydrate. Aminofentanyl has also been successfully acylated with ethyl isocyanate, various acid anhydrides, to further investigate structure–activity relationships of these new fentanyl derivatives. Among the new derivatives compound 7 which carries a Tyr-D-Ala-Gly-Phe opioid message sequence showed good opioid affinity (1 nM at both δ and μ opioid receptors) and bioactivity (34.9 nM in MVD and 42 nM in GPI/LMMP bioassays).
Keywords: Fentanyl derivatives, Opioid agonists, Enkephalin analogue
Depending on their nature all opiates can be broadly divided into two categories: non-peptide and peptide based. Morphine which occurs in nature represents one of the most efficient analgesic drugs among the non-peptide based opiates. Endogenous opioid peptides such as endomorphins, enkephalins, and dynorphins occurring naturally in the brain represent the second class of opiates. The latter function as both neuromodulators and hormones, and are responsible for a broad spectrum of physiological effects. A common feature of these two classes of opiates is their binding to the three recognized opioid receptor types: μ, δ,and κ. A tremendous amount of synthetic work was done to alter the potency, selectivity, and bioavailability of these both classes of opioids.
The class of compounds known as 4-anilidopiperidines represent the most powerful synthetic analgesics, which include fentanyl and related compounds.1-13 Fentanyl is a well-known μ-selective synthetic analgesic (ED50 0.011 mg/kg) which is 50–100 times more potent than morphine, has a short duration of action and an onset of action almost immediately after intravenous administration. Fentanyl, sufentanyl, and alfentanyl currently represent the three most popular compounds used for analgesia in clinical practice despite the fact that their use results in side effects such as respiratory depression, physical dependence, and rapid tolerance.14
An evident gap in the literature regarding incorporation of amino acids and peptides into fentanyl chemistry has encouraged us to investigate replacement of the propionyl fragment of fentanyl with various amino acids. We therefore sought to design a hybrid molecule based on two distinct classes of opioid ligands: the peptide portion derived from enkephalins and the non-peptide moiety from a 4-anilidopiperidine series. This novel strategy would be analogous to that used to create molecules such as biphalin [(Tyr-d-Ala-Gly-Phe-NH)2], a dimeric analogue of enkephalin with two peptide based pharmacophores linked by a hydrazide bridge. The original idea of incorporating 1- and 2-substituted fentanyl analogues into peptides based on the structural analogy between the aromatic rings of fentanyl and the Tyr1 and Phe4 residues of the opioid peptides goes back to the early 1980 with the resulting fentanyl analogues showing very weak or no opioid activity, and that study was limited to 1- and 2-substituted analogues.15 The ability to incorporate and retain the characteristic high opioid activity of 4-anilidopiperidines into peptides holds numerous possibilities in the area of drug design and their medicinal applications. For example, attachment of the corresponding unit would allow rather simple introduction of opioid activity into any peptide of interest, which can be fruitful for designing multiple ligands. The inherent μ-opioid receptor selectivity of fentanyl and its analogues would allow, for instance, greater μ-selectivity of opioid analogues.16 Considering recent achievements in converting opioid agonists into antagonists,17 a combination of peptide plus non-peptide opioid has a potential of developing an antagonist of matching potency to fentanyl. We expect that the highly lipophilic character of the 4-anilidopiperidine moiety will increase cell permeability of attached peptides, and consequently their bioavailability.18 Furthermore, we anticipate that replacement of the propionyl moiety of fentanyl with amino acids and peptide residues will decrease the toxicity associated with this class of analgesics.
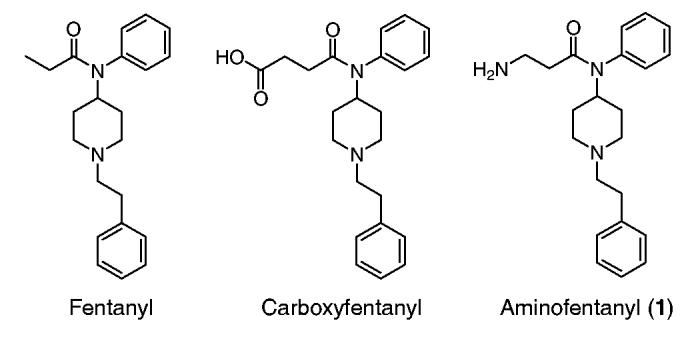
The high pharmaceutical potential of the anilidopiperidines, as well as the fact that aminofentanyl in contrast to its commercially available counterpart, carboxyfentanyl (BIOMOL International®, PA),19 is not described in the literature, have prompted us to find a synthetic strategy that would allow us to couple β-amino acids with 1-(2-phenethyl)-4-(N-anilino)-piperidine (2). This β-alanine derivative would differ from the original structure of fentanyl only by an amino group attached to the third carbon of propionyl moiety.
Our initial attempts to employ traditional peptide coupling reagents such as HBTU and DIC in the presence of HOBt proved to be unsuccessful for coupling amino acids to compound 2.20 On the other hand, attempts to couple the corresponding Fmoc acid chloride in anhydrous (DCM and TEA) or biphasic (DCM/5% NaHCO3 in water) media gave low coupling yields and partial cleavage of the Nβ-Fmoc group. We have found that the designed compound can be readily prepared by coupling the phthaloyl protected acid chloride of β-alanine with compound 2 in the presence of DIPEA or TEA, followed by deprotection with hydrazine hydrate in refluxing ethanol (Scheme 1). In contrast to α-phthaloyl amino acids, attempted deprotection of phthaloyl group of compound 3 with ethanolamine in refluxing ethanol gave multiple products with only 35% yield of target product 1. Thionyl chloride in refluxing toluene was used to convert the phthaloyl protected β-alanine to its acid chloride. The precursor of this reaction, compound 3,21 was obtained as a solid due to its low solubility in organic solvents such as acetone or methanol which significantly simplified the corresponding purification. We also found that compound 122 could be prepared faster via a single pot procedure: in this case synthesis and hydrolysis of compound 3 were carried out in one flask, and the compound 1 was purified by crystallization from EtOAc. Compound 1 was advanced to the derivatives 4a–c by treatment with the corresponding acid anhydrides.23 The urea derivative 4d was accessed by the treatment of 1 with EtNCO.24 Phthaloyl amino acid chlorides were chosen in the synthesis of an enkephalin analogue, even though deprotection of phthaloyl group required rather harsh conditions which are routinely avoided in peptide chemistry. We found that the employment of phthaloylphenylalanyl chloride prepared by treatment with thionyl chloride in refluxing toluene followed by coupling in the presence of TEA gave us a completely racemized product.25 Although no racemization of phthaloyl protected phenylalanine was reported under less drastic conditions, such as activation with oxalyl chloride at room temperature,26 Boc-Phe-OH was used for corresponding coupling with DCC as a condensing reagent. The reaction of 5 with Ac2O afforded 6.27 We were pleased to obtain the desired peptides in good yields after coupling the phthaloyl protected Nα-amino acid chlorides in the presence of DIPEA followed by deprotection with ethanolamine, extraction with DCM or EtOAc,28 drying over MgSO4, and evaporation of the solvent. In the case of phthaloyl protected α-amino acid chlorides, the duration of coupling was very fast as monitored by TLC and a reaction time of 20–30 min was sufficient and maintained throughout the synthesis. The coupling of phthaloyl d-alanyl chloride was free of racemization as confirmed by HPLC and NMR analysis. Compounds 5 and intermediate peptides were used for peptide synthesis without purification to obtain the final product 7.29 Peptides 6 and 7 were used in biological tests without further purification because their purity, as determined by HPLC analysis, was of 95%. It is notable that isolation and purification of our compounds were done conveniently by extracting with Et2O(4a–d, 5, and 6) or 1/4 mixture of EtOAc/Et2O (7),28 filtering through Celite (to remove salts or DCU in the case of 5) and precipitating by passing dry HCl through the solvent.
Scheme 1.

Reagents and conditions: (i) Pht-AA-Cl (β-Ala), DIPEA , DCM, 0 °C, 92%; (ii) N2H4·nH2O, ethanol, reflux, 63%; (iii) EtNCO or acid anhydride, DIPEA, DCM, 65–97%; (iv) Boc-Phe-OH, DCC, DIPEA, DCM; (v) TFA, DCM, 94% (two steps); (vi) Ac2O, DIPEA, 72%; (vii) stepwise chain elongation using (i) and (ii) (ethanolamine instead of N2H4·nH2O) methods for Gly (80%) and d-Ala (94%), and (iv) and (v) for Tyr (65%).
Opioid binding affinities (see Table 1) of the new analogues for the human δ-opioid receptor (hDOR) or the rat μ-opioid receptor (rMOR) were determined by radioligand competition analysis using DPDPE to label the δ-opioid receptor and DAMGO to label the μ-opioid receptor in cell membrane preparations from transfected cells that stably express the respective receptor type.30
Table 1.
Binding affinities
The functional bioactivity profiles of selected ligands (see Table 2) were determined in MVD and GPI/LMMP smooth muscle preparations as described previously.33 IC50 values, relative potency estimates, and their associated errors were determined by fitting the data to the Hill equation by a computerized non-linear least-square method.
Table 2.
In vitro functional activity
As reported in Table 1, compounds 1–6 showed weak binding affinity at the opioid receptors. Compound 1 tested as a free base also showed weak in vitro activity. Interestingly, although carboxyfentanyl was used to raise antibodies against fentanyl, no data were available in the literature about its opioid binding affinity. Our tests showed that the binding affinity of carboxyfentanyl is of the same magnitude as that of aminofentanyl except the latter has higher μ-opioid receptor selectivity and bioactivity. Compound 7 which carries a Tyr-d-Ala-Gly-Phe opioid message sequence showed good opioid affinity and bioactivity. It is interesting to note that this compound did not show any affinity for κ-receptors and was binding reversibly when tested in its neutral form. In vivo studies of compound 7 are currently underway and will be a subject of a separate report. This study reveals that attachment of a β-aminoacid and its N-acetylated analogues leads to substantial drop in opioid binding affinity of corresponding fentanyl derivatives. Thus, it appears that attachment of a peptide bond to the propionyl moiety of fentanyl encounters unfavorable interactions at the μ- and particularly at δ-opioid binding sites. A comparison of the affinities of compounds presented by Essawi and Portoghese,15 Montero et al.,34 and ours (4b, 4e, and 7) indicates that propionyl moiety together with phenethyl part of fentanyl play a very important role in opioid receptor binding and activation.
In summary, we were able to identify some interesting structure–activity relationships around the propionyl moiety of fentanyl. Compound 7 which carries a Tyr-d-Ala-Gly-Phe opioid message sequence showed good opioid affinity and bioactivity suggesting that a novel class of analgesics can be further developed utilizing this approach. Aminofentanyl was synthesized in good yield by coupling of Nb-phthaloylalanyl chloride with 1-(2-phenethyl)-4-(N-anilino)piperidine followed by hydrolysis with hydrazine hydrate. Phthaloyl amino acid chlorides proved to be highly efficient for coupling to a sterically and electronically demanding amine. Amenability of phthaloyl protected amino acid chlorides for efficient solution-phase synthesis of enantiomerically pure peptides was also demonstrated.
Acknowledgments
This work was supported by grants from USPHS and NIDA.
Abbreviations
- Ac
acetyl
- Ac2O
acetic anhydride
- Boc
tert-butyloxycarbonyl
- DCC
N,N-dicyclohexylcarbodiimide
- DCM
dichloromethane
- DCU
N,N-dicyclohexylurea
- DEPT
decoupled edited phase transfer
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylform-amide
- DMSO
dimethylsulfoxide
- hDOR
human δ-opioid receptor
- DPDPE
c[d-Pen2,d-Pen5]enkephalin
- DAMGO
[d-Ala2, NMePhe4, Gly5-ol]enkephalin
- Et
ethyl
- Et2O
diethyl ether
- EtOAc
ethyl acetate
- EtNCO
ethylisocyanate
- GPI
guinea pig ileum
- HOBt
1-hydroxybenzotriazole
- LMMP
longitudinal muscle myenteric plexus
- rMOR
rat μ-opioid receptor
- MVD
mouse vas deferens
- Pht
phthaloyl
- TEA
triethylamine
- TFA
trifluoroacetic acid
References and notes
- 1.(a) Janssen PAJ, Gardocki JF. U.S. Patent 3141823 1964; (b) Janssen PAJ. Fr. Patent M2430 1964
- 2.Riley TN, Hale DB, Wilson MC. J. Pharm. Sci. 1973;62:983. doi: 10.1002/jps.2600620627. [DOI] [PubMed] [Google Scholar]
- 3.Janssen PAJ, Van Daele GHP. Deutch. Patent 2610228 1976
- 4.Leysen JE, Laduron PM, Niemegeers CJE. In: Characteristics and Function of Opioids. Van Ree J, Terenius L, editors. Elsevier; North Holland: 1978. pp. 479–482. [Google Scholar]
- 5.(a) Van Bever WFM, Niemegeers CJE, Schellekens KHL, Janssen PAJ. Arzneim.-Forsch. 1976;26:1548. [PubMed] [Google Scholar]; (b) Janssen PAJ, Van Daele GHP. U.S. Patent 4179569 1979
- 6.Fang SN, Ge BJ, Dai QY, Li QZ, Zhou DH, Ni CH, Wu RQ, Huang ZM. Acta Pharm. Sin. 1983;18:823. [PubMed] [Google Scholar]
- 7.Huang BS, Deutsche KH, Lalinde NL, Terrell RC, Kudzma LV. Eur. Patent 160422 1985
- 8.Janssens F, Torremans J, Janssen PAJ. J. Med. Chem. 1986;29:2290. doi: 10.1021/jm00161a027. [DOI] [PubMed] [Google Scholar]
- 9.Kudzma LV, Spencer HK, Severnak SA. U.S. Patent 4791121 1988
- 10.(a) Vartanyan SA, Vartanyan RS, Zhamagortsyan VN, Martirosyan VO, Vlasenko V, Azlivyan AS, Durgaryan LK. U.S. Patent 736583 1985; (b) Vartanyan RS, Martirosyan VO, Vartanyan SA, Vlasenko EV, Durgaryan LK, Azlivyan AS. Khim.-Farmatsevticheskii Zh. 1989;23:562. [Google Scholar]; (c) Vartanyan RS, Martirosyan VO, Vartanyan SA, Engoyan AP, Vlasenko EV, Durgaryan LK, Azlivyan AS, Val'dman AV. Khim.-Farmatsevticheskii Zh. 1989;23:573. [Google Scholar]
- 11.(a) Bagley JR, Spencer KH. Eur. Patent 277794 1988; (b) Bagley JR, Wynn RL, Rudo FG, Doorley BM, Spencer HK, Spaulding T. J. Med. Chem. 1989;32:663. doi: 10.1021/jm00123a028. [DOI] [PubMed] [Google Scholar]; (c) Rothman RB, Xu H, Seggel M, Jacobson AE, Rice KC, Brine GA, Carroll FI. Life Sci. 1991;48:111. doi: 10.1016/0024-3205(91)90346-d. [DOI] [PubMed] [Google Scholar]
- 12.Lalinde N, Moliterni J, Wright D, Spencer HK, Ossipov MH, Spaulding TC, Rudo FG. J. Med. Chem. 1990;33:2876. doi: 10.1021/jm00172a032. [DOI] [PubMed] [Google Scholar]
- 13.(a) Feldman PL, James MK, Brackeen MF, Bilotta JM, Schuster SV, Lahey AP, Lutz MW, Johnson MR, Leighton HJ. J. Med. Chem. 1991;34:2202. doi: 10.1021/jm00111a041. [DOI] [PubMed] [Google Scholar]; (b) Feldman PL, James MK, Brackeen MF, Johnson MR, Leighton HJ. Eur. Patent 383579 1990
- 14.Bowdle TA. Drug Saf. 1998;19:173. doi: 10.2165/00002018-199819030-00002. [DOI] [PubMed] [Google Scholar]
- 15.Essawi MYH, Portoghese PS. J. Med. Chem. 1983;26:348. doi: 10.1021/jm00357a007. [DOI] [PubMed] [Google Scholar]
- 16.Schiller PW, Nguyen TMD, Chung NN, Lemieux C. J. Med. Chem. 1989;32:698. doi: 10.1021/jm00123a035. [DOI] [PubMed] [Google Scholar]
- 17.Schiller PW, Weltrowska G, Nguyen TMD, Lemieux C, Chung NN, Lu Y. Life Sci. 2003;73:691. doi: 10.1016/s0024-3205(03)00389-8. [DOI] [PubMed] [Google Scholar]
- 18.Lee K, Jung W-H, Park CW, Hong CY, Kim IC, Kim S, Oh YS, Kwon OH, Lee S-H, Park HD, Kim SW, Lee YH, Yoo Y. J. Bioorg. Med. Chem. Lett. 1998;18:2563. doi: 10.1016/s0960-894x(98)00456-9. [DOI] [PubMed] [Google Scholar]
- 19.McConnell RI, Benchikh EO, Fitzgerald SP, Lamont JV. Eur. Patent 1312923 2003
- 20.Maryanoff BE, Simon EJ, Gioannini T, Gorissens H. J. Med. Chem. 1982;25:913. doi: 10.1021/jm00350a006. [DOI] [PubMed] [Google Scholar]
- 21. Compound 2 (10.0 g, 35.7 mmol) and DIPEA (9.2 g, 71.3 mmol) were dissolved in 100 mL of dry DCM. The mixture was cooled in an ice-water bath. A solution of phthaloyl β-alanine chloride (12.7 g, 53.5 mmol) in 100 mL DCM was added dropwise. The reaction mixture was stirred overnight. The mixture was then diluted with 100 mL DCM and washed with 5% aqueous K2CO3. The aqueous phase was extracted with DCM (2× 50 mL). The combined organic layers were washed with brine and dried over anhydrous magnesium sulfate. The product solidified after solvent removal in vacuo. The resulting solid was washed with small amounts of acetone and dried in vacuo. Compound 3: yield: 15.8 g, mp 205 °C, and as a hydrochloric salt 260 °C (from Et2O); 1H NMR (500 MHz, CDCl3): δ 1.42 (qd, 2H, J1 = 12.1 Hz, J2 = 3.3 Hz), 1.80 (br d, 2H, J = 12.1 Hz), 2.14 (br t, 2H, J = 11.5 Hz) 2.32 (t, 2H, J = 7.4 Hz), 2.54 (m, 2H), 2.72 (m, 2H), 2.99 (br d, 2H, J = 11.5 Hz), 3.91 (t, 2H, J = 7.5 Hz), 4.64 (tt, 1H, J1 = 12.1 Hz, J2 = 3.8 Hz), 7.14 (m, 5H), 7.25 (m, 2H), 7.34 (m, 3H), 7.66 (m, 2H), 7.77 (m, 2H); 13C NMR (500 MHz, CDCl3): δ 30.3, 33.4, 33.7, 34.3, 52.1, 52.9, 60.3, 123.0, 125.9, 128.2, 128.4, 128.5, 129.4, 130.3, 132.0, 133.7, 138.1, 140.1, 167.9, 169.4. MS: m/z 481.9 [M+H]+; HRMS calcd: 481.2365; found: 481.2437.
- 22. Compound 3 (15.6 g, 32.4 mmol) was heated at reflux with 50 mL of hydrazine hydrate and 200 mL of ethanol for 2 h. The reaction mixture was then cooled to 0 °C. A bulky precipitate, which appeared upon cooling, was removed by filtration. The filtrates were concentrated in vacuo to remove ethanol, taken into EtOAc, and washed with saturated NaHCO3 solution. The aqueous phase was extracted with EtOAc (2× 50 mL). The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo to a volume ca. 30 mL. The product crystallized shortly after being placed in a freezer. The resulting crystals were filtered and dried in vacuo. Compound 1: yield: 7.2 g, mp 98–99 °C. 1H NMR (600 MHz, CDCl3): δ 1.44 (qd, 2H, J1 = 12.2 Hz, J2 = 3.6 Hz), 1.80 (br d, 2H, J = 12.2 Hz), 1.8 (s, 2H, NH2), 2.07 (t, 2H, J = 6.1 Hz), 2.15 (t, 2H, J = 11.5 Hz), 2.53 (m, 2H), 2.72 (m, 2H), 2.87 (t, 2H, J = 6.0 Hz), 2.99 (br d, 2H, J = 11.5 Hz), 4.67 (tt, 1H, J1 = 12.1 Hz, J2 = 3.7 Hz), 7.09 (d, 2H, J = 8.0 Hz), 7.15 (m, 3H), 7.24 (t, 2H, J = 7.5 Hz), 7.37 (m, 3H); 13C NMR (600 MHz, CDCl3): δ 30.4, 33.6, 37.9, 38.2, 52.1, 52.9, 60.2, 125.8, 128.1, 128.2, 128.4, 129.2, 130.3, 138.4, 140.0, 171.3. MS: m/z 352.1 [M+H]+; HRMS calcd: 351.2311; found: 351.2375.
- 23. Selected data. Compound 4a: 1H NMR (500 MHz, CDCl3) β 1.45 (qd, 2H, J1 = 12.3 Hz, J2 = 3.7 Hz), 1.80 (br d, 2H, J = 12.6 Hz), 1.93 (s, 3H), 2.10 (t, 2H, J = 5.7 Hz), 2.15 (td, 2H, J1 = 12.0 Hz, J2 = 1.8 Hz), 2.54 (m, 2H), 2.72 (m, 2H), 3.01 (br d, 2H, J = 11.7 Hz), 3.39 (dt, 2H, J1 = 5.9 Hz, J2 = 5.3 Hz), 4.63 (tt, 1H, J1 = 12.2 Hz, J2 = 3.9 Hz), 6.52 (t, 1H, J = 5.3 Hz), 7.06 (m, 2H), 7.16 (m, 3H), 7.25 (t, 2H, J = 7.4 Hz), 7.38 (m, 3H); 13C NMR (500 MHz, CDCl3): δ 23.3, 30.4, 33.7, 34.9, 35.1, 52.4, 52.9, 60.3, 125.9, 128.2, 128.4, 128.5, 129.4, 130.1, 137.8, 139.9, 169.7, 171.5. MS: m/z 394.2 [M+H]+; HRMS calcd: 393.2416; found: 393.2509. Compound 4b: 1H NMR (500 MHz, CDCl3): δ 1.13 (t, 3H, J = 7.6 Hz), 1.45 (qd, 2H, J1 = 12.1 Hz, J2 = 3.7 Hz), 1.80 (br d, 2H, J = 12.1 Hz), 2.10 (t, 2H, J = 5.7 Hz), 2.16 (m, 2H), 2.17 (q, 2H, J = 7.6 Hz), 2.54 (m, 2H), 2.73 (m, 2H), 3.01 (br d, 2H, J = 11.7 Hz), 3.40 (dt, 2H, J1 = 5.8 Hz, J2 = 5.4 Hz), 4.63 (tt, 1H, J1 = 12.1 Hz, J2 = 3.9 Hz), 6.53 (t, 1H, J = 5.4 Hz, NH), 7.05 (m, 2H), 7.16 (m, 3H), 7.25 (t, 2H, J = 7.5 Hz), 7.38 (m, 3H); 13C NMR (500 MHz, CDCl3): δ 9.7, 29.6, 30.3, 33.6, 34.9, 52.3, 52.8, 60.2, 125.9, 128.2, 128.4, 128.5, 129.4, 129.9, 137.8, 139.9, 171.6, 173.4. MS: m/z 408.1 [M+H]+; HRMS calcd: 407.2573; found: 407.2659. Compound 4c: 1H NMR (500 MHz, CDCl3): δ 1.48 (qd, 2H, J1 = 12.2 Hz, J2 = 3.3 Hz), 1.81 (br d, 2H, J = 12.2 Hz), 2.17 (t, 2H, J = 5.5 Hz), 2.19 (t, 2H, J = 11.5 Hz), 2.56 (m, 2H), 2.74 (m, 2H), 3.04 (br d, 2H, J = 11.5 Hz), 3.50 (dt, 2H, J1 = 5.8 Hz, J2 = 5.4 Hz), 4.62 (tt, 1H, J1 = 12.2 Hz, J2 = 3.9 Hz), 7.07 (m, 2H), 7.16 (m, 3H), 7.25 (m, 2H), 7.41 (m, 3H), 7.69 (br t, 1H, J = 5.4 Hz, NH); 13C NMR (500 MHz, CDCl3): δ 30.2, 33.6, 33.9, 35.6, 52.6, 52.8, 60.2, 126.0, 128.3, 128.5, 128.8, 129.6, 129.9, 137.6, 139.9, 156.7, 156.9, 171.1. MS: m/z 448.1 [M+H]+; HRMS calcd: 447.2134; found: 447.2221.
- 24. Selected data. Compound 4d: 1H NMR (500 MHz, CDCl3): δ 1.10 (t, 3H, J = 7.2 Hz), 1.46 (qd, 2H, J1 = 12.0 Hz, J2 = 2.9 Hz), 1.79 (br d, 2H, J = 11.9 Hz), 2.11 (t, 2H), 2.14 (br t, 2H, J = 11.4 Hz), 2.55 (m, 2H), 2.73 (m, 2H), 3.02 (br d, 2H, J = 11.4 Hz), 3.16 (m, 1H), 3.34 (dt, 2H, J1 = 5.8 Hz, J2 = 5.4 Hz), 4.61 (tt, 1H, J1 = 12.1 Hz, J2 = 3.8 Hz), 5.04 (br s, 1H, NH), 5.54 (br s, 1H, NH), 7.06 (m, 2H), 7.16 (m, 3H), 7.26 (m, 2H), 7.38 (m, 3H); 13C NMR (500 MHz, CDCl3): δ 15.4, 30.3, 33.6, 35.1, 35.9, 52.3, 52.9, 60.2, 125.9, 128.2, 128.4, 129.4, 130.1, 137.9, 139.9, 158.3, 171.9. MS: m/z 423.1 [M+H]+; HRMS calcd: 422.2682; found: 422.2752.
- 25.Petrov RR, Vardanyan RS, Nichol GS, Carducci MD, Ma S-W, Lai JY, Hruby VJ. Acta Cryst. 2006;E62:o2815. doi: 10.1107/S1600536806021817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephen WH. Org. Proc. Dev. 1999;3:241. [Google Scholar]
- 27. Selected data. Compound 6: 1H NMR (500 MHz, CDCl3): δ 1.41 (qd, 2H, J1 = 12.2 Hz, J2 = 4 Hz), 1.77 (br d, 2H, J = 12.6 Hz), 1.89 (m (part. overlap.), 1H), 1.93 (s, 1H), 2.02 (ddd, 1H, J1 = 16.9 Hz, J2 = 6.7 Hz, J3 = 4.9 Hz), 2.13 (br dd, 2H, J1 = 11.9 Hz, J2 = 4.3 Hz), 2.53 (m, 2H), 2.71 (m, 2H), 2.98 (m (part. overlap), 2H), 3.0 (br d (part. overlap.), 2H, J = 7 Hz), 3.28 (m, 1H), 3.36 (m, 1H), 4.58 (tt, 1H, J1 = 12.1 Hz, J2 = 3.8 Hz), 4.61 (dt, 1H, J1 = 7.5 Hz, J2 = 7.3 Hz), 6.68 (d, 1H, J = 8.0 Hz, NH), 6.75 (t, 1H, J = 5.9 Hz, NH), 7.04 (m, 2H), 7.14–7.28 (m, 10H), 7.37 (m, 3H); 13C (assigned using 13C DEPT-90 and DEPT-135) NMR (500 MHz, CDCl3): δ 22.9 (CH3), 30.3 (CH2), 33.7 (CH2), 34.6 (CH2), 35.0 (CH2), 38.7 (CH2), 52.3 (CH), 52.9 (CH2), 54.4 (CH), 60.3 (CH2), 125.8 (CH), 126.6 (CH), 128.2 (CH), 128.3 (CH), 128.4 (CH), 129.1 (CH), 129.3 (CH), 130.1 (CH), 136.6 (Cq), 137.8 (Cq), 139.9 (Cq), 169.6 (C=O), 170.5 (C=O), 170.8 (C=O). MS: m/z 541.2 [M+H]+; HRMS calcd: 540.3100; found: 540.3157.
- 28. The aqueous solution was adjusted to pH 8 with NaHCO3.
- 29. Selected data. Compound 7: 1H NMR (600 MHz, DMSO-d6): δ 1.16 (d, 3H, J = 6.9 Hz), 1.19 (br q, 2H, J = 12.0 Hz), 1.69 (br d, 2H, J = 10.8 Hz), 1.9–2.04 (m, 4H), 2.44 (m, 2H), 2.58–2.66 (m, 3H), 2.80 (dd, 1H, J1 = 13.3 Hz, J2 = 9.4 Hz), 2.86–2.97 (m, 4H), 3.17 (m, 1H), 3.25 (m, 1H), 3.5 (br s, 2H, NH2), 3.56 (t, 1H, J = 6.2 Hz), 3.60 (dd, 1H, J1 = 16.8 Hz, J2 = 5.5 Hz), 3.72 (dd, 2H, J1 = 6.1 Hz, J2 = 16.6 Hz), 4.22 (m, 1H), 4.36 (dt, 1H, J1 = 8.6 Hz, J2 = 5.2 Hz), 4.45 (tt, 1H, J1 = 12.0 Hz, J2 = 3.2 Hz), 6.71 (d, 2H, J = 8.3 Hz), 7.02 (d, 2H, J = 8.2 Hz), 7.12–7.21 (m, 10H), 7.23 (t, 2H, J = 7.6 Hz), 7.43 (dd, 1H, J1 = 7.0 Hz, J2 = 6.8 Hz), 7.47 (t, 2H, J = 7.1 Hz), 7.89 (t, 1H, J = 5.1 Hz), 7.94 (d, 1H, J = 8.2 Hz), 8.29 (br s, 1H), 8.32 (br s, 1H); 13C (part. assigned using 13C DEPT-90 and DEPT-135) NMR (500 MHz, CDCl3): δ 17.8 (CH3), 30.0 (CH2), 32.9 (CH2), 34.3 (CH2), 35.2 (CH2), 37.6 (CH2), 42.1 (CH2), 48.5 (CH), 51.9(CH), 52.5 (CH2), 54.3 (CH), 55.8 (CH), 59.5 (CH2), 115.0 (CH), 117.8 (Cq), 125.7 (CH), 126.1 (CH), 127.9 (CH), 128.1 (CH), 128.2 (CH), 128.5 (CH), 129.0 (CH), 129.3 (CH), 130.1 (CH), 130.3 (CH), 137.8 (Cq), 138.4 (Cq), 140.4 (Cq), 155.9 (Cq), 168.5 (C=O), 169.4 (C=O), 170.7 (C=O), 172.5 (C=O), 173.4 (C=O); MS: m/z 790.3 [M+H]+; HRMS calcd: 789.4214; found: 789.4271.
- 30.(a) Polt R, Porreca F, Szabo L, Bilsky EJ, Davis TP, Horvath R, Abbruscato TJ, Yamamura HJ, Hruby VJ. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7114. doi: 10.1073/pnas.91.15.7114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Misicka A, Lipkowski AW, Horvath R, Davis P, Kramer TH, Yamamura HI, Hruby VJ. Life Sci. 1992;51:1025. doi: 10.1016/0024-3205(92)90501-f. [DOI] [PubMed] [Google Scholar]
- 31.Yeadon M, Kitchen I. Neuropharmacology. 1988;27:345. doi: 10.1016/0028-3908(88)90141-4. [DOI] [PubMed] [Google Scholar]
- 32.Lipkowski AW, Misicka A, Davis P, Stropova D, Janders J, Lachwa M, Porreca F, Yamamura HI, Hruby VJ. Bioorg. Med. Chem. Lett. 1999;9:2763. doi: 10.1016/s0960-894x(99)00464-3. [DOI] [PubMed] [Google Scholar]
- 33.Kramer TH, Davis P, Hruby VJ, Burks TF, Porreca F. J. Pharmacol. Exp. Ther. 1993;266:577. [PubMed] [Google Scholar]
- 34.Montero A, Goya P, Jagerovic N, Callado LF, Meana JJ, Giron R, Goicoechea C, Martin MI. Bioorg. Med. Chem. 2002;10:1009. doi: 10.1016/s0968-0896(01)00356-x. [DOI] [PubMed] [Google Scholar]