

Abstract
The rate of glucose transport across the plasma membrane of the bloodstream form of Trypanosoma brucei was modulated by titration of the hexose transporter with the inhibitor phloretin, and the effect on the glycolytic flux was measured. A rapid glucose uptake assay was developed to measure the transport activity independently of the glycolytic flux. Phloretin proved a competitive inhibitor. When the effect of the intracellular glucose concentration on the inhibition was taken into account, the flux control coefficient of the glucose transporter was between 0.3 and 0.5 at 5 mM glucose. Because the flux control coefficients of all steps in a metabolic pathway sum to 1, this result proves that glucose transport is not the rate-limiting step of trypanosome glycolysis. Under physiological conditions, transport shares the control with other steps. At glucose concentrations much lower than physiological, the glucose carrier assumed all control, in close agreement with model predictions.
Trypanosoma brucei is a unicellular, eukaryotic parasite that causes African sleeping sickness in humans. For its carbon and free-energy metabolism, this organism depends entirely on glycolysis. Drugs against this organism may therefore be targeted at its glycolytic enzymes. One of the main challenges is the design of selective drugs, which do not only kill the parasite, but leave the host cells unharmed. We have proposed that Metabolic Control Analysis can be used as a tool to increase the selectivity of drugs (1, 2). The idea is that inhibiting an enzyme that controls the flux in the parasite to a great extent, but that does not, or to a lesser extent, control the flux in the host cells, should increase the selectivity of drugs.
Metabolic Control Analysis (3–6) offers a method to determine quantitatively to what extent an enzyme or transporter controls a metabolic flux. The control exerted by an enzyme on a steady-state flux can be determined by modulating the activity of that enzyme, at constant activities of all other enzymes. The flux control coefficient is operationally defined as the relative change of the flux divided by the relative change of the modulated enzyme activity (cf. Eq. 1, below). If the flux changes proportionally to the enzyme activity, this enzyme is truly rate-limiting, and its flux control coefficient is 1. If an enzyme has a flux control coefficient of 0, it does not control the flux at all. Experimentally determined flux control coefficients have been anywhere between 1 and 0 (7–9) and, in some cases, in branched pathways, −1 (3). In an ideal metabolic pathway, the sum of the flux control coefficients of all enzymes is always 1 (10, 11). From the perspective of Metabolic Control Analysis, an enzyme is a promising drug target if it has a high flux control coefficient in the parasite but a low flux control coefficient in host cells.
The transport of glucose into the cell has been viewed as the rate-limiting step of T. brucei glycolysis (12–15). However, compelling evidence is lacking (1). When the more precise Metabolic Control Analysis was used, it became clear that the concept of a single “rate-limiting step” does not apply to trypanosome glycolysis (2).
Calculations, based on the available enzyme kinetics, showed that glucose transport need not be the rate-limiting step of glycolysis in the bloodstream form of T. brucei (2). At low glucose concentrations (i.e., <4 mM), the glycolytic flux was indeed controlled by glucose transport. When going from 4 to 8 mM blood glucose, however, a proportion of control shifted to other enzymes in the glycolytic pathway (2). At the normal blood glucose concentration of 5 mM, the calculated flux control coefficient of the glucose transporter depended strongly on the kinetic parameters of the transporter and the glycolytic enzymes. Because these are known to finite accuracy, it was impossible to predict at which glucose concentration the transporter lost the control of the flux and how control was distributed under physiological conditions. Therefore, we here measured the flux control coefficient of the T. brucei glucose transporter directly.
In the bacteria Escherichia coli and Salmonella typhimurium, the glucose transport activity has been varied by genetic means around wild-type level. It did not control the rate of glucose oxidation (8, 16). In no eukaryotic cell has the control exerted by a glucose transporter on the glycolytic flux been determined directly: i.e., by varying the amount or activity of the transporter. In perfused rat heart, the control of glucose use was determined indirectly from in vivo concentrations of glycolytic intermediates and in vitro enzyme kinetics. The glucose transporter (Glut4) was thus predicted to have substantial flux control in the absence of insulin but to lose control on stimulation by this hormone (17). In muscle cells, the rate of glycogen synthesis was controlled by either glucose transport or hexokinase, or by these steps together (18). Direct measurement of the control exerted by glucose transport is complicated by the multiplicity of glucose-transporter genes in most eukaryotes (19, 20) and by the fact that manipulation of their expression is more difficult than in enteric bacteria. Furthermore, inhibitors of glucose transport, if available, are often competitive, and their direct effect on the transporter does not only depend on the extracellular glucose concentration but also on the intracellular glucose concentration.
Glucose enters bloodstream-form T. brucei by facilitated diffusion (21, 22). Many inhibitors of this process are available (13, 15). To determine the flux control exerted by the transporter, the inhibition of the transport rate and of the steady-state flux have to be measured in separate, independent experiments. In previous studies, the steady-state glucose consumption flux was used to characterize the kinetics of the glucose transporter (15, 23). The assumption underlying these studies was that glucose transport is the rate-limiting step of trypanosome glycolysis. Because precisely this assumption is the object of our investigations, this method could not be used here. Ter Kuile and Opperdoes avoided this assumption when they measured the uptake of radiolabeled d-glucose over 7.5 seconds by centrifugation through oil (14). The measured zero-trans uptake rate, however, was lower than the steady-state glucose consumption flux (14, 15), demonstrating that these measurements underestimated the zero-trans-uptake rate. Perhaps the increase of the intracellular substrate concentration during the uptake assay inhibited further influx or caused efflux (24–26). This problem may be avoided by measuring the uptake of glucose over a shorter time period (25).
Here we have used phloretin, a competitive inhibitor of glucose transport into trypanosomes, to investigate experimentally whether the glucose transporter fully controls the glycolytic flux under physiological conditions or whether it shares the control with other steps. To deal with the aforementioned complications, a new, more rapid assay of glucose transport has been used, and the effect of intracellular glucose has been analyzed quantitatively. We show that the flux control coefficient of the T. brucei glucose transporter is between 0.3 and 0.5 at 5 mM of blood glucose and even increases at lower glucose concentrations. This makes glucose transport a promising target for antitrypanosomal drugs.
MATERIALS AND METHODS
Chemicals.
d-[U-14C] glucose was from Amersham Pharmacia. Phloretin (3-[4-hydroxyphenyl]-1-[2,4,6-trihydroxyphenyl]-1-propanone) was from Sigma. A phloretin stock solution was prepared in 70% ethanol. The final ethanol concentration in cell suspensions was <1% and did not affect the results.
Cultivation of Trypanosomes.
All experiments were performed on the bloodstream form of T. brucei 427. Male, 300-g Wistar rats were infected with T. brucei, and, after 4 days, the parasites were isolated from the blood by DEAE-cellulose chromatography (DE52, Whatman) (27). The cells were washed by centrifugation, were resuspended in a 90 mM Tris⋅HCl buffer containing 2.5 mM KCl, 77.5 mM NaCl, 5 mM MgCl2, 2 mM Na2HPO4, and 50 mM glucose at pH 7.5 (to be referred to as buffer A, modified from ref. 28), and were stored on ice. In this buffer, the cells maintained a constant motility and oxygen consumption capacity for at least 7 hours after isolation.
Glucose Transport.
The rate of glucose transport was measured by a filtration technique modified from a method to measure glucose uptake in Saccharomyces cerevisiae (29). An aliquot of cells was washed three times by centrifugation at 4°C and was resuspended in a 90 mM Tris⋅HCl buffer, which had been thermostated at 37°C and contained 3.1 mM KCl, 96.9 mM NaCl, 5 mM MgCl2, 2 mM Na2HPO4, and 2 mM glycerol at pH 7.5. The cells were incubated at assay temperature and were aerated for 1 minute. The trypanosomes suspension (50 μl) and d-[U-14C] glucose (12.5 μl, at 5× the final concentration) were mixed and incubated for ∼5 seconds (measured accurately to 0.1 second). Uptake was terminated by quenching 50 μl in 10 ml of buffer A, maintained below 0°C on a salt-ice mixture. Cells were rapidly collected by filtration on a glass microfibre filter (Whatman GF/C). They were not washed after filtration to prevent the loss of the contents of broken cells. The filters were transferred immediately to scintillation vials containing 10 ml of scintillant, and radioactivity was measured in a Beckman liquid scintillation counter. Blanks were measured by adding the labeled glucose directly to the quenching buffer before the cells were added separately. The blanks were further treated as samples. The glucose concentration in the stock solution was measured by NADH-linked enzymatic analysis (30) in an automatic analyzer (COBAS FARA, Roche, Gipf-Oberfrick, Switzerland). Each aliquot of washed cells was used for duplicate experiments. The cells entered the scintillant within 3 minutes after resuspension. If the washed cells were used for triplicate experiments, the transport rate in the third experiment was lower because of a deterioration of the cells after depletion of the carbon source, glycerol, from the suspension. The protein concentration was measured according to Lowry et al. (31) in each aliquot of cells (typically 5–7 mg/ml). Because the buffer weakly interfered with the protein assay, the same amount of buffer as present in the samples was added to the BSA standards. To allow comparison of the obtained results to literature data, the number of cells per milligram of protein was counted under the microscope: 1.94 × 108 cells per mg protein.
In the new assay, a low concentration of glycerol (2 mM) was provided as a carbon source during incubation of the cells before the addition of labeled glucose. This was important to maintain viability in the absence of glucose. The low glycerol concentration should not affect glucose transport significantly because no direct interaction of glycerol with the transporter has been demonstrated (13, 14). The most important modification of the rapid assay from the way it has been applied to yeast (29) was that the filter on which the cells were collected was not washed. Previous attempts to measure glucose uptake by filtration failed because the trypanosomes were damaged in the procedure (32). Because trypanosomes can stand vortexing and centrifugation, our hypothesis was that the damage occurred on drying of the filter. If the filter was washed, the measured “uptake rate” decreased by 90% compared to if the filter was not washed. Consistently, cell counting revealed that only 10% of the cells could be recovered intact from the filters. For measuring glucose uptake into trypanosomes, washing away of adherent glucose is not essential. First, glucose does not seem to adhere to the trypanosome’s surface coat, as it does to the yeast’s cell wall. Secondly, their glucose uptake system has a higher affinity for glucose, so that lower concentrations were used than with yeast. At lower glucose concentrations, the signal-to-blank ratio became higher. Any extracellular glucose adhering to the filter or to the cells was corrected for by the blanks. The lowest signal to blank ratio (at 20 mM glucose) was 2.
For comparison, the silicone-oil-centrifugation assay of transport also was performed (14). The method of Ter Kuile and Opperdoes (14) was used, except that the cells were incubated for only 5 seconds with radioactive glucose, rather than for 7.5 seconds or longer. When the inhibition of glucose transport by phloretin was measured, the inhibitor was added together with the radiolabeled glucose.
Flux Measurements.
An aliquot of cells was washed three times by centrifugation at 4°C and was resuspended in a 90 mM Tris⋅HCl buffer (pH 7.5) containing 3.1 mM KCl, 96.9 mM NaCl, 5 mM MgCl2, 2 mM Na2HPO4, and the indicated concentration of glucose at 37°C. The rate of oxygen consumption was monitored in a thermostated vessel with a Clark electrode. Part of the cells was used to measure oxygen consumption, and the rest was used for protein determination, as described above. Phloretin was added to cells when they were consuming oxygen at steady state. A new steady state was obtained rapidly. For determination of the concentrations of other metabolites, the cells were kept in an open, thermostated vessel and were aerated by bubbling with water-saturated pressurized air. To check that the cell suspensions remained aerobic, the oxygen concentration was monitored with a Clark electrode throughout the experiment. Samples were taken by injecting 75 μl of the cell suspension into 37.5 μl of 15% perchloric acid. Samples were vortexed, were centrifuged (5 minutes, Eppendorf table centrifuge, full speed), were neutralized by mixing 80 μl of supernatant with 45 μl of 1 M K2CO3, and were centrifuged again. The pH of the resulting supernatants was checked with pH paper (pH 7–8). They were stored at −20°C until further analysis within a week. Glucose, pyruvate, and glycerol were measured by NADH-linked analysis (30) in an automatic analyzer (COBAS FARA, Roche).
Control Coefficients.
The flux control coefficient of a step i is defined as
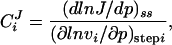 |
1 |
in which J is the steady-state flux, p is a parameter that only affects the activity of step i, and vi is the activity of step i.
To measure the flux control coefficient of the glucose transporter, a specific inhibitor of this step, phloretin, was used. The effect on the oxygen consumption flux was measured, and the effect on the transport rate was calculated from measured inhibition kinetics at constant concentrations of all other metabolites (in particular, at constant intracellular glucose). The flux was plotted against the transport activity (as a fraction of the uninhibited value), and the flux control coefficient was calculated as the slope of the curve at 100% (i.e., uninhibited) transport activity. When the effect of phloretin was studied at a low concentration of glucose, the analysis was complicated by substantial decrease of the glucose concentration during the measurements, causing decrease of the flux. To calculate the effect of phloretin on the flux, a reference trace was recorded in the absence of the inhibitor. Because phloretin competed with glucose, the inhibition of the transporter depended on the actual glucose concentration. The glucose concentration was measured at the start of the experiment, and the actual concentration at the moment of addition of the inhibitor was calculated based on the fact that trypanosomes consume one molecule of glucose per molecule of oxygen (33).
RESULTS
The Kinetics of Glucose Transport.
The glucose transport assay used in this paper was based on filtration of the cells, allowing a shorter incubation of the cells with the radiolabeled glucose and a more accurate timing than in the silicone-oil-centrifugation assay (14). The Vmax measured after a 5-second incubation exceeded the Vmax for a 7.5-second silicone-oil-centrifugation assay (14). It was high enough to explain the steady-state flux at saturating substrate concentration (Table 1). The Km was lower in the new, more rapid filtration assay than in the 7.5-second centrifugation assay (Table 1), implying that the underestimation of the rate by the 7.5-second assay is most significant at low glucose concentrations. This lower Km corresponds better to the inhibition constant of glucose (0.90 ± 0.04 mM) with respect to the transport of the nonmetabolizable analogue 6-deoxy-d-glucose (13). For comparison, the silicone-oil centrifugation method was performed at 5-second time scale. The results of this more rapid centrifugation assay corresponded to those of the new filtration assay (Table 1).
Table 1.
The glucose transport characteristics as determined with different methods at 37°C
| Method | Apparent Vmax, nmol⋅min−1⋅mg⋅protein−1 | Apparent Km, mM | |
|---|---|---|---|
| Transport | Filtration, 5 seconds | 167 ± 11 (n = 6) | 1.2 ± 0.2 (n = 6) |
| Centrifugation through oil, 5 seconds | 151 ± 7 (n = 6) | n.d. | |
| Centrifugation through oil, 7.5 seconds | 91.6* | 2.0* | |
| Flux | Steady-state O2-consumption rate | 170 ± 6 (n = 15) |
For comparison, the maximal steady-state O2-consumption rate at saturating (50 mM) glucose concentration at 37°C is given. The standard errors of the mean in the filtration experiment and the oxygen consumption experiment are based on n completely independent experiments, with cells isolated from different rats. In each independent replicate of the filtration experiment, the Vmax and the Km were obtained by nonlinear regression from a data-set of at least six points, between 0.1 and 20 mM extracellular glucose, measured in duplicate. Average ± standard error of the mean of n such independent Vmax and Km determinations are given. The Vmax of the 5-second centrifugation experiment was measured as the rate of glucose transport at 5.2 mM glucose. Here, n replicates were performed with cells isolated from a single rat. n.d., not determined.
From ref. 14.
Phloretin Inhibits Glucose Transport Competitively.
The activity of the glucose transporter was modulated by titration with phloretin, a general inhibitor of facilitated diffusion transporters. This compound inhibited the erythrocyte glucose carrier competitively (34). On the contrary, phloretin has been reported to be a noncompetitive inhibitor of glucose transport in bloodstream form T. brucei. However, this conclusion was based on steady-state measurements rather than on initial transport experiments, and no numbers were reported (15). We reexamined this issue by using the 5-second glucose transport assay. Phloretin increased the apparent Km for glucose without any effect on the Vmax (Fig. 1). We conclude that phloretin is a competitive inhibitor of the trypanosome glucose transporter, with a Ki of 21 μM (Fig. 2).
Figure 1.

Phloretin inhibits zero-trans-glucose influx competitively. Glucose transport was measured rapidly (5 seconds) in the absence (●) and presence (■) of 45 μM (A) or 91 μM (B) phloretin. The full lines represent the fitted Michaelis–Menten curves. A, no phloretin: Vmax = 169 ± 2 nmol⋅min−1⋅mg⋅protein−1 and Km = 0.73 ± 0.01 mM. A, with phloretin: Vmax = 169 ± 9 nmol⋅min−1⋅mg⋅protein−1 and Km ± 2.99 ± 0.36 mM. B, no phloretin: Vmax = 198 ± 12 nmol⋅min−1⋅mg⋅protein−1 and Km = 1.85 ± 0.38 mM. B, with phloretin: Vmax = 193 ± 5 nmol⋅min−1⋅mg⋅protein−1 and Km = 6.35 ± 0.39 mM. In the absence of phloretin, ethanol was added to correct for any solvent effect. The data in A were obtained with one batch of cells, isolated from a single rat. The data in B were measured with another batch of cells, obtained from a different rat.
Figure 2.

The inhibitor constant of phloretin is 21 μM. The apparent Km of zero-trans-glucose influx depended linearly on the inhibitor concentration. The inhibitor constant Ki was derived from this curve by using the equation Kmapp = Km(1 + [phloretin]/Ki). Each data point was obtained by linear regression of a complete Michaelis–Menten curve, containing at least 6 points between 0.1 and 20 mM glucose (or 40 mM glucose in the presence of phloretin), measured in duplicate. The standard deviation was obtained from the nonlinear regression procedure. Independent experiments with different isolates were treated as individual data points.
Inhibition of the Glycolytic Flux by Phloretin.
Subsequently, the effect of modulation of the glucose transport activity on the glycolytic flux was measured. Already low concentrations of phloretin decreased the steady-state rate at which trypanosomes consumed oxygen (Fig. 3). Under aerobic conditions, bloodstream-form trypanosomes convert all glucose to pyruvate, with the concomitant consumption of one molecule of oxygen per molecule of glucose (33). Therefore, the rate of oxygen consumption is a measure of the steady-state glycolytic flux. Trypanosomes can, however, convert part of the glucose to glycerol without using oxygen. Normally this only happens under anaerobic conditions. Indeed, also in the presence of phloretin, the glucose was still fully converted to pyruvate (Fig. 4).
Figure 3.

The steady-state oxygen consumption flux as a function of the phloretin concentration at 0.36 (■, □) and at 5 mM glucose (●, ○). The open and closed symbols represent independent experiments that were carried out with two batches of trypanosomes, isolated from different rats. Each datapoint represents average ± standard deviation of at least three replicates.
Figure 4.

Glucose is fully converted to pyruvate under aerobic conditions in the presence of phloretin. Glucose (▴), pyruvate (■), and glycerol (●) have been monitored both in the absence (A) and presence (B) of 100 μM phloretin. The sum of the concentrations of these compounds (2 [glucose] + [pyruvate] + [glycerol], indicated by ×) slightly increased, probably because of the evaporation of water from the incubation vessel. The cell densities were 3.3 mg protein/ml (A) and 2.5 mg protein/ml (B), respectively.
The Glucose Transporter Partly Controls the Glycolytic Flux at a Physiological Glucose Concentration.
To determine the flux control coefficient, the direct effect of the inhibitor on the steady-state glucose transport rate must be known at constant concentrations of all other reactants. The only known effector of the glucose transporter is the intracellular glucose concentration. If there were no free intracellular glucose, the effect of the inhibitor on the steady-state glucose transport rate should equal the effect on the zero-trans influx, and the flux control by the carrier should be complete. At low concentrations, intracellular glucose might reduce the steady-state flux to below the zero-trans rate and reduce the flux control by the transporter to below 1 but not yet affect the inhibition of the carrier by phloretin. In a first approximation, this was assumed to be the case. At low extracellular glucose concentration (0.36 mM), the glucose consumption flux was almost proportional to the glucose transport activity (Fig. 5). The slope of this curve should represent the flux control coefficient of the glucose transporter. At a physiological extracellular glucose concentration (5 mM), the flux was much less sensitive to inhibition of the glucose transporter (Fig. 5), implying that, in this case, the flux control coefficient was lower. When the slopes of the curves in Fig. 5 were evaluated at 100%, the calculated flux control coefficients were 1.2 and 0.42 at 0.5 and 5 mM of glucose, respectively.
Figure 5.

At a low extracellular glucose concentration, the flux is more sensitive to inhibition of glucose transport than at a physiological glucose concentration. The activity of the glucose transporter was varied by addition of various amounts of phloretin. The rate of glucose transport was calculated, based on phloretin acting as a competitive inhibitor of zero-trans influx with a Ki of 21 μM and assuming that the intracellular glucose concentration was zero. At 5 mM of glucose (●, ○), the flux was less sensitive to inhibition of glucose transport than at 0.36 mM of glucose (■, □). Open and closed symbols represent independent experiments with trypanosomes isolated from different rats.
In a second approximation, the possible effect of the intracellular glucose concentration on the inhibition by phloretin was taken into account in the determination of the flux control coefficient. At extracellular glucose concentrations of up to 5 mM, the measured intracellular glucose concentration was proportional to the extracellular glucose concentration (14).
An equation describing the effect of phloretin on the steady-state transport kinetics was derived by using the King–Altman method (42, 43). According to the 4-state carrier model (24), the net steady-state uptake rate is
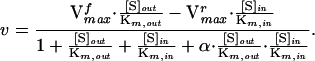 |
2 |
in which [S]out and [S]in are the extracellular and intracellular glucose concentrations, respectively. Vmaxf and Vmaxr are the forward and reverse Vmax values. Km,out and Km,in are the Km’s for influx and efflux of glucose, respectively, and α is a constant factor that distinguishes the kinetics of the 4-state carrier from reversible Michaelis–Menten kinetics.
A competitive inhibitor that binds to the carrier on the outside affects not only Km,out, but also Vmaxr, Km,in, and α. This inhibition is described by two inhibition constants: Ki1 which was measured above, and Ki2. Ki2 was not measured. Theoretically, the ratio of these inhibition constants, Ki2/Ki1, can assume any value between 0.5 and infinity. Details are published as supplemental data on the PNAS web site, www.pnas.org.
The derived equation contains one unknown parameter (Ki2/Ki1). This parameter was varied between its theoretical boundaries. Again, the effect of phloretin on the glucose transport rate was calculated, but now at the measured, steady-state intracellular glucose concentration, according to the kinetic equations derived in the Appendix (which is published as supplemental data on the PNAS web site, www.pnas.org). JO2 was plotted against the rate of glucose transport, and the slope of this curve at 100% represented the corrected flux control coefficient. The results are listed in Table 2. It turned out that the uncertainty in Ki2/Ki1 was not very important for the determination of the control coefficient at low extracellular glucose concentrations. At 5 mM of extracellular glucose, the uncertainty led to an uncertainty in the control coefficient of ∼0.2. This is barely significant as compared to the experimental error, which is always high in control coefficients, by their nature of being derivatives. Importantly, the difference between the control coefficients at 0.5 and 5 mM of extracellular glucose clearly exceeds these differences. Because the flux control coefficients of all steps in a pathway sum up to one (3), the above results imply that the glucose transporter shared the control of the flux with other steps at 5 mM glucose. At 0.36 mM glucose, it assumed all control.
Table 2.
The flux control coefficient of the glucose transporter (Cglucose transportJO2) depends on the extracellular glucose concentration
| [Glc]out, mM | [Glc]in, mM | Assumed Ki2/Ki1 | Cglucose transportJ02 |
|---|---|---|---|
| 0.36 | 0 | — | 1.2 ± 0.2 |
| 0.36 | 0.036 | 0.5 | 1.2 ± 0.2 |
| 0.36 | 0.036 | 1000 | 1.2 ± 0.2 |
| 5 | 0 | — | 0.42 ± 0.09 |
| 5 | 0.4 | 0.5 | 0.35 ± 0.08 |
| 5 | 0.4 | 1000 | 0.50 ± 0.11 |
The flux control coefficient was calculated from the data in Fig. 5, either neglecting intracellular glucose or taking into account its measured concentration (14) as described in the Appendix (see supplemental data at www.pnas.org). For the unknown parameter Ki2/Ki1, two extreme values were assumed. Irrespective of the assumptions, the transporter had a substantially lower control at 5 mM glucose than at 0.36 mM glucose. A further increase of Ki2/Ki1 did not affect the values calculated for the flux control coefficients. When zero intracellular glucose was assumed, flux control coefficients were calculated by linear regression of the linear parts of Fig. 5, i.e., without the highest phloretin concentration (lowest flux). Both independent data sets were pooled. Fitted flux control coefficients (slopes in Fig. 5) are given plus or minus standard error, as obtained from linear regression. When intracellular glucose was taken into account, the rate of glucose transport was recalculated as a function of phloretin at fixed intracellular glucose concentration, as described in the Appendix. Again, JO2 was plotted against the glucose uptake rate, and flux control coefficients were calculated as described above.
DISCUSSION
The flux control coefficient of the glucose transporter in bloodstream-form T. brucei was measured at 5 mM glucose, the most common concentration in the blood (35, 36): It was between 0.3 and 0.5. At a low glucose concentration (0.36 mM), the flux control coefficient of the glucose transporter was measured to be 1.2, in reasonable agreement with model calculations, which predicted 1 at this concentration (2). In contrast to earlier speculations (12–15), these results prove that glucose transport is not the rate-limiting step of trypanosome glycolysis. Its flux control coefficient depends on the conditions, and, at a physiological glucose concentration, the glucose transporter exerts only some 40% of the total control.
Previously, the flux control coefficient of the glucose carrier was calculated as a function of the extracellular glucose concentration by using a detailed kinetic model of trypanosome glycolysis (2). Qualitatively, these calculations agree with the measured control coefficients: At low glucose concentrations, the glucose transporter exerted all control, but, in the physiological range of glucose concentrations, it lost part of the control. At which concentration this shift of control would occur could not be predicted reliably because this modeling result was highly sensitive to the kinetic parameters. According to the model, the glucose transporter had a flux control coefficient of 0.9 at 5 mM glucose (2) whereas a value between 0.3 and 0.5 was measured. Apart from the strong sensitivity of this flux control coefficient to almost all uncertainties in kinetic parameters, there is one more specific explanation for the discrepancy: In the model, the Km of the glucose transporter was 2 mM (2, 37), as had been measured by Ter Kuile and Opperdoes (14), although our present measurements have indicated that it is more likely to be 1 mM (Table 1). When a Km of 1 mM was inserted in the model, the calculated flux control coefficient of the glucose transporter was 0.45, which agrees with the measured value.
Theoretically, the flux control by the glucose transporter should approach 1 at low glucose concentrations (2), but a control coefficient of 1.2 was measured. The experimental determination of the control coefficient is more error-prone at low concentrations of glucose than at high concentrations. At the lower concentration, the decrease of the glucose concentration during the flux measurement was relatively large, and, therefore, extensive corrections were required. It is also possible, however, that the Ki of phloretin was slightly overestimated because of the presence of glycerol in the glucose-uptake assay. Under many conditions, glycerol did not influence the uptake of glucose or glucose analogues (13, 14). Nevertheless, we found that glycerol inhibited the apparent uptake rate at high glucose concentrations (result not shown). By using the model of trypanosome glycolysis (2, 37), it was demonstrated that this inhibition may be fully explained by metabolic effects, notably the competition of glucose and glycerol for glycosomal ATP. According to the model, the glycosomal ATP concentration dropped by >90% during the preincubation with glycerol, which led to an underestimation of the apparent glucose transport rate over the first 10 seconds. The underestimation of the uptake rate was less at lower uptake rates. Then, the metabolic capacity sufficed to prevent accumulation of intracellular glucose. This should lead to an overestimation of the inhibition constant and, consequently, also to a (slight) overestimation of the flux control coefficient.
The measurement of the control by the glucose transporter was complicated by the presence of intracellular glucose, which influenced the direct effect of phloretin on this transporter. The analysis that was developed to deal with intracellular glucose (see the Appendix published as supplemental data on www.pnas.org) yielded two inhibition constants, of which only one was measured and the other was estimated. This left us with a minor uncertainty in the control coefficients. In principle, the analysis can be completed by measuring the inhibition kinetics in efflux experiments. The method developed in this paper might be applied more widely to measure the control exerted by other symmetrical facilitated diffusion transporters, given that an uncertainty of 20% in a flux control coefficient is generally considered acceptable.
An application of this study is the optimization of the design of antitrypanosomal drugs. Inhibition of an enzyme or transporter with a higher flux control coefficient in the parasite than in its host should reduce the glycolytic flux in the former to a greater extent than in the latter (1). Although glucose transport turned out not to be the sole rate-limiting step, the finding that it exerts part of the flux control warrants a stronger emphasis on the design of inhibitors of this process. Structural differences between the mammalian glucose transporters and the trypanosome glucose transporter should allow the design of compounds that selectively inhibit the parasite transporter (38). Because the sum of the flux control coefficients remains one, the fact that the control by the glucose transporter is lower than the model predicted (2) implies that other enzymes should exert more control. According to that model, control should shift to aldolase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, and glycerol-3-phosphate dehydrogenase, when the transporter lost it (2). Inhibitors of T. brucei glyceraldehyde-3-phosphate dehydrogenase are available already (39, 40), and inhibitors of T. brucei phosphoglycerate kinase are forthcoming, now that the crystal structure of this enzyme is known (41). If indeed these enzymes exert more control than was calculated previously, the perspectives of killing the trypanosomes by inhibiting these enzymes have improved. On the other hand, in view of the results of this paper, the design of inhibitors of glucose transport seems equally, or even more, urgent. First, because the remaining 60% of control is approximately equally shared among four enzymes, their individual flux control coefficients are substantially lower (on average 0.15) than that of the glucose transporter. Secondly, we have demonstrated experimentally that the flux control coefficient of the glucose transporter increases when the transport activity is decreased because of a decrease of the extracellular glucose concentration. The same is likely to happen when inhibitors are applied. These findings make the glucose transporter a very promising target for drugs against Trypanosomiasis.
Supplementary Material
Acknowledgments
We would like to thank M. Hendrix in Amsterdam and J. van Roy in Brussels for animal care and trypanosome cultivation. J. Diderich and B. Teusink helped with some of the experiments. This study was supported by the Netherlands Organization for Scientific Research (NWO) and the Netherlands Association of Biotechnology Research Schools (ABON).
References
- 1.Bakker B M, Westerhoff H V, Michels P A M. J Bioenerg Biomembr. 1995;27:513–525. doi: 10.1007/BF02110191. [DOI] [PubMed] [Google Scholar]
- 2.Bakker B M, Michels P A M, Opperdoes F R, Westerhoff H V. J Biol Chem. 1999;274:14551–14559. doi: 10.1074/jbc.274.21.14551. [DOI] [PubMed] [Google Scholar]
- 3.Westerhoff H V, Van Dam K. Thermodynamics and Control of Biological Free-Energy Transduction. Amsterdam: Elsevier; 1987. [Google Scholar]
- 4.Fell D A. Biochem J. 1992;286:313–330. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinrich R, Schuster S. The Regulation of Cellular Systems. New York: Chapman & Hall; 1996. [Google Scholar]
- 6.Fell D A. Understanding the Control of Metabolism. London: Portland Press; 1997. [Google Scholar]
- 7.Groen A K, Wanders R J A, Westerhoff H V, Van der Meer R, Tager J M. J Biol Chem. 1982;257:2754–2757. [PubMed] [Google Scholar]
- 8.Ruijter G J G, Postma P W, Van Dam K. J Bacteriol. 1991;173:6184–6191. doi: 10.1128/jb.173.19.6184-6191.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snoep J L, Arfman N, Yomano L P, Westerhoff H V, Conway T, Ingram L O. Biotechnol Bioeng. 1996;51:190–197. doi: 10.1002/(SICI)1097-0290(19960720)51:2<190::AID-BIT8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 10.Kacser H, Burns J A. Symp Soc Exp Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 11.Kholodenko B N, Westerhoff H V. Trends Biochem Sci. 1995;20:52–54. doi: 10.1016/s0968-0004(00)88955-0. [DOI] [PubMed] [Google Scholar]
- 12.Gruenberg J, Sharma P R, Deshusses J. Eur J Biochem. 1978;89:461–469. doi: 10.1111/j.1432-1033.1978.tb12549.x. [DOI] [PubMed] [Google Scholar]
- 13.Eisenthal R, Game S, Holman G D. Biochim Biophys Acta. 1989;985:81–89. doi: 10.1016/0005-2736(89)90107-7. [DOI] [PubMed] [Google Scholar]
- 14.Ter Kuile B H, Opperdoes F R. J Biol Chem. 1991;266:857–862. [PubMed] [Google Scholar]
- 15.Seyfang A, Duszenko M. Eur J Biochem. 1991;202:191–196. doi: 10.1111/j.1432-1033.1991.tb16362.x. [DOI] [PubMed] [Google Scholar]
- 16.Van der Vlag J, Van’t Hof R, Van Dam K, Postma P W. Eur J Biochem. 1995;230:170–182. doi: 10.1111/j.1432-1033.1995.0170i.x. [DOI] [PubMed] [Google Scholar]
- 17.Kashiwaya Y K, Sato K, Tsuchiya N, Thomas S, Fell D A, Veech R L, Passonneau J V. J Biol Chem. 1994;269:25502–25514. [PubMed] [Google Scholar]
- 18.Shulman R G, Bloch G, Rothman D L. Proc Natl Acad Sci USA. 1995;92:8535–8542. doi: 10.1073/pnas.92.19.8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldwin S A. Biochim Biophys Acta. 1993;1154:17–49. doi: 10.1016/0304-4157(93)90015-g. [DOI] [PubMed] [Google Scholar]
- 20.Kruckeberg A L. Arch Microbiol. 1996;166:283–292. doi: 10.1007/s002030050385. [DOI] [PubMed] [Google Scholar]
- 21.Ter Kuile B H. J Bioenerg Biomembr. 1994;26:167–172. doi: 10.1007/BF00763065. [DOI] [PubMed] [Google Scholar]
- 22.Tetaud E, Barrett M P, Bringaud F, Baltz T. Biochem J. 1997;325:569–580. doi: 10.1042/bj3250569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wille U, Seyfang A, Duszenko M. Eur J Biochem. 1996;236:228–233. doi: 10.1111/j.1432-1033.1996.00228.x. [DOI] [PubMed] [Google Scholar]
- 24.Stein W D. Transport and Diffusion Across Cell Membranes. London: Academic; 1986. [Google Scholar]
- 25.Walsh M C, Smits H P, Van Dam K. Yeast. 1994;10:1553–1558. doi: 10.1002/yea.320101204. [DOI] [PubMed] [Google Scholar]
- 26.Teusink B, Diderich J A, Westerhoff H V, Van Dam K, Walsh M C. J Bacteriol. 1998;180:556–562. doi: 10.1128/jb.180.3.556-562.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanham S M. Nature (London) 1968;218:1273–1274. doi: 10.1038/2181273a0. [DOI] [PubMed] [Google Scholar]
- 28.Kiaira J K, Njogu M R. Biotechnol Appl Biochem. 1994;20:347–356. doi: 10.1111/j.1470-8744.1994.tb00322.x. [DOI] [PubMed] [Google Scholar]
- 29.Walsh M C, Smits H P, Scholte M, Van Dam K. J Bacteriol. 1994;176:953–958. doi: 10.1128/jb.176.4.953-958.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergmeyer H U. Methods of Enzymatic Analysis. Weinheim, Germany: Verlag Chemie; 1974. [Google Scholar]
- 31.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 32.Munoz-Antonia T, Richards F F, Ullu E. Mol Biochem Parasitol. 1991;47:73–82. doi: 10.1016/0166-6851(91)90149-z. [DOI] [PubMed] [Google Scholar]
- 33.Ryley J F. Biochem J. 1956;62:215–222. doi: 10.1042/bj0620215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krupka R M, Devés R. Biochim Biophys Acta. 1980;598:134–144. doi: 10.1016/0005-2736(80)90271-0. [DOI] [PubMed] [Google Scholar]
- 35.Frankel S, Reitman S. Gradwohl’s Clinical Laboratory Methods and Diagnosis. St. Louis: Mosby; 1963. [Google Scholar]
- 36.Henry R J, Cannon D C, Winkelman J W. Clinical Chemistry Principles and Techniques. Baltimore: Harper & Row; 1974. [Google Scholar]
- 37.Bakker B M, Michels P A M, Opperdoes F R, Westerhoff H V. J Biol Chem. 1997;272:3207–3215. doi: 10.1074/jbc.272.6.3207. [DOI] [PubMed] [Google Scholar]
- 38.Walmsley A R, Barret M P, Bringaud F, Gould G W. Trends Biochem Sci. 1998;23:476–481. doi: 10.1016/s0968-0004(98)01326-7. [DOI] [PubMed] [Google Scholar]
- 39.Verlinde C L M J, Callens M, Van Calenbergh S, Van Aerschot A, Herdewijn P, Hannaert V, Michels P A M, Opperdoes F R, Hol W G J. J Med Chem. 1994;37:3605–3613. doi: 10.1021/jm00047a017. [DOI] [PubMed] [Google Scholar]
- 40.Aronov A M, Suresh S, Buckner F S, Van Voorhis W C, Verlinde C L M J, Opperdoes F R, Hol W G J, Gelb M H. Proc Natl Acad Sci USA. 1999;96:4273–4278. doi: 10.1073/pnas.96.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstein B E, Michels P A M, Hol W G. Nature (London) 1997;385:275–278. doi: 10.1038/385275a0. [DOI] [PubMed] [Google Scholar]
- 42.King E L, Altman C. J Phys Chem. 1956;60:1375–1378. [Google Scholar]
- 43.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. Cambridge, U.K.: Univ. Press; 1995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}