

Abstract
Time-resolved small-angle x-ray scattering was used to measure the radius of gyration of cytochrome c after initiation of folding by a pH jump. Submillisecond time resolution was obtained with a microfabricated diffusional mixer and synchrotron radiation. The results show that the protein first collapses to compact denatured structures before folding very fast to the native state.
The stability and folding speed of a protein depend on the structures of the denatured as well as the native state, raising the question: do proteins fold more rapidly from a denatured state of expanded structures or from one of compact structures? Lattice simulations of simplified representations of proteins suggest that slow folding amino acid sequences collapse to compact structures with non-native topologies before folding, while fast folders collapse and fold simultaneously (1–4). We have begun to address this question experimentally with submillisecond small-angle x-ray scattering (SAXS) using synchrotron radiation and a microfabricated diffusional mixer to rapidly initiate folding. SAXS yields the radius of gyration, the most unambiguous measure of compactness. Here we show that, in contrast to the simulations, one of the fastest-folding proteins (cytochrome c: τfolding = 400 μs) first collapses to compact structures before forming the final native state.
X-ray scattering by proteins in solution is sensitive to spatial variations in electron density. Scattering at the smallest angles yields the radius of gyration, Rg, which in conjunction with the protein molecular weight provides a measure of the compactness of globular proteins. Additional structural information can be obtained from scattering at larger angles, which reflects electron density correlations on length scales shorter than Rg. For a compact polymer, such as the native protein or compact denatured structures, I(q)q2 increases at low q, goes through a maximum, and decreases at large q where I(q)αq−4 [I(q) is the scattered intensity; q = 4πsin θ/λ, is the momentum transfer; λ is the x-ray wavelength, 1.54 Å; and 2θ is the scattering angle] (5). In contrast, for a polymer chain undergoing a random walk in space, as can occur for an unfolded protein under strongly denaturing conditions, I(q)q2 first increases, then plateaus (6), and, at large q, where I(q)αq−1, increases linearly (7). In this way, using Kratky plots [I(q)q2 vs. q], one can distinguish easily between random coil and compact conformations, making time-resolved SAXS a powerful tool for investigating structure in protein folding experiments (8–10).
Because of their simpler kinetic behavior, small single-domain proteins have been the focus of both experimental and theoretical investigations of the mechanism of protein folding. Folding times of these proteins in the absence of denaturant are often less than ≈10 ms (11), which is too fast to be studied with the mixing technologies used in previous time-resolved SAXS studies (8–10). We have increased the time resolution by almost 2 orders of magnitude by using a lithographically microfabricated mixing device. Its design is based on the principle that diffusional mixing times scale as the square of the linear dimension of the mixing volume. Short mixing times are achieved by hydrodynamic focusing, which creates a micron or submicron stream of protein solution in contact with a surrounding flowing reservoir (12). We fabricated an x-ray compatible device at the Cornell Nanofabrication Facility (Fig. 1). Synchrotron radiation was necessary for these experiments because of the combination of micro sample volumes and the normally weak scattering of protein solutions. Time-resolved scattering was obtained by passing a micro-collimated x-ray beam through the sample at fixed locations in the flowing stream of protein solution. Relative time was determined from the velocity of the protein stream and the position of the x-ray beam.
Figure 1.

A schematic of the diffusive mixer.
MATERIALS AND METHODS
Fig. 1 shows a cross-sectional view of the central portion of the diffusive mixer. The channels are 100 μm wide and 390 μm deep. In operation, 2.5 mM of cytochrome c in HCl at pH 2 is injected into the inlet port (Fig. 1 Left). We chose cytochrome c (104 aa, Sigma catalog no. C7752) for our initial study using this method because its folding kinetics to times as short as 50 μs already had been characterized (13–18). A 0.1 M phosphate buffer at pH 7 containing 0.2 M imidazole, introduced into the orthogonal channels at a higher pressure, hydrodynamically focuses the low pH, protein-containing solution into a thin stream (12). This focused protein stream, sheathed by the pH 7 buffer, shoots out the fourth exit port of the device (Fig. 1 Right). Rapid increases in pH occur by diffusion of protons out of and buffer into the focused protein solution. Although the stream eventually reaches pH 7, equilibrium studies indicate that cytochrome c is almost fully native above pH 3 (19). Once the pH of the focused protein solution is above 3, the protein folds as it flows. Scattering data at different times can be obtained by moving the device relative to the fixed position of the x-ray beam.
The x-ray experiments were conducted at the Cornell High Energy Synchrotron Source’s D-line, using a multilayer monochromator to enhance the bending magnet beam. The beam at the sample had a flux of 109 x-rays/s into an area that was 40 μm along the width of the protein stream and 120 μm along the direction of flow. A charge-coupled device area detector (20) was used to record the scattering patterns.
As a result of the large dimensions of these channels, low driving pressures for the fluids are involved and both the width and speed of the focused protein solution vary significantly as a function of depth in the channel. At the surface of the device, the protein stream was close to 40 μm wide; its width rapidly decreased with depth and there was a large section with a width of 2.5 μm. The pH of this thin section rose above 3 within about 200 μs as determined by confocal microscopy with a pH-sensitive fluorescent dye (the corresponding position in the device is defined to be t = 0 for the analysis). This technique provides a direct measure of proton diffusion. The protein stream also broadens as a result of diffusion; however, because of the larger mass of the macromolecules, they diffuse much more slowly than the protons. At the exit of the outlet channel, the average final protein concentration is expected to be a factor of two less than the initial concentration. The mean speed of the focused protein solution for the work described in this paper was 30 cm/s.
RESULTS AND DISCUSSION
Kratky plots of the data from three positions in the device are shown in Fig. 2. For the initial state, measured at a position before mixing, the product Iq2 increases nearly linearly at large q, indicating that the average structure is expanded and random-coil like. It is, however, not possible to extract an Rg from the data at low q because of interference caused by the high concentration of a highly charged protein (21) (2.5 mM corresponds to an average separation of ≈10 nm; in a high concentration of chemical denaturant at pH 7, but at low protein concentration and therefore with little interference from interparticle scattering, the Rg is 30–32 Å and the Kratky plot indicates random-coil like scattering; ref. 22). At the second position, which is roughly indicated in Fig. 1 and corresponds to a range of times between 150 and 500 μs after mixing, the decrease of Iq2 at large q is characteristic of a compact structure. Fitting the data at low q with the function K q2exp(−Rg2q2/3) (K is an instrumental constant) yields an apparent Rg of 15.9 ± 0.7 Å. The Kratky plot for the position corresponding to an average time of 10 msec after mixing is identical to that of the native protein at equilibrium and yields an Rg = 13.9 ± 0.4 Å, the same value measured in previous equilibrium SAXS experiments (22, 23). The data were fit for q < 0.15, a range that yields an excellent fit to better-quality, static data.
Figure 2.

Time-resolved Kratky plots. (a) A Kratky plot of the protein in the inlet channel, to the left of the cross in Fig. 1, is shown, indicating that the initial (pH 2) state is expanded and random coil-like. (b) The SAXS pattern after the device had been translated to illuminate the protein stream as indicated in Fig. 1, 200 μm from the center of the cross. This x-ray exposure averaged over a time interval from 150 to 500 μs, assuming that the protein stream traveled at its mass averaged velocity. Roughly 90% of the volume of the protein solution was above pH 3 in this interval. We have subtracted 10% of the initial pH 2 state from the observed Kratky plot at this position to generate the curve shown in b. A simple calculation based on these average numbers and using the rate constants for a pH jump to 4.5 (15, 18), indicates that 45% of the protein above pH 3 at this location was in the denatured state and 55% was in the native state. The measured Rg from the data of b (Rgapp = 15.9 ± 0.7 Å) is related to that of the denatured (RgD) and native (RgN) states as follows: (Rgapp)2 = fD(RgD)2 + fN(RgN)2, where fD/N represents the fraction of the sample in the denatured/native state (22). Our calculation of fD is a lower limit (hence our calculated RgD = 18.1 ± 0.9 Å is an upper limit) for the following reasons: (i) We have ignored the contribution from the expanded denatured state in calculating the Rg of the compact denatured state from Rgapp. (ii) We expect the folding to occur more slowly with a final pH of 3 than for the calculated final pH of 4.5. (iii) Imidazole may not completely bind before non-native histidines produce misfolded structures that fold more slowly than the imidazole complex (13–15). (iv) The calculation assumes that all of the protein is mixed at t = 0, the position at which the thinnest, 2.5-μm section is mixed. The mixing is actually a continuous process, with thicker sections mixing at later times. (c) The Kratky plot at a position that corresponds to a time of ≈10 msec after mixing (red, noisy curve). This data suggests that the protein had folded completely to the native state. A Kratky plot of a much larger sample of native cytochrome c in equilibrium at pH 7 is shown as the green (smooth) curve plotted on top of the data. The best fit to the data in b and c are shown as dashed, blue lines.
The result from our experiments is the x-ray scattering pattern at the intermediate time interval of 150–500 μs. During this period, however, there is contribution to the observed scattering from structures of more than one state of the protein. Previous submillisecond fluorescence kinetic studies can be used to determine the relative contributions (15, 18). In those studies folding was monitored by the decay of tryptophan fluorescence, which results from increased quenching by the heme via Forster excitation energy transfer as the heme-tryptophan distance decreases. Under the conditions of our experiment the fluorescence decay is biphasic, with ≈80% of the amplitude decaying in a 50-μs relaxation (18) and ≈20% in the subsequent formation of the native structure occurring in a 400-μs relaxation (15, 18). From a three-state analysis of the amplitudes and relaxation rates, it was concluded that an equilibrium mixture of two denatured states exists at the end of the 50-μs relaxation, corresponding to compact and expanded denatured structures in a roughly 5:1 ratio (18). Compactness was assumed because of the low quantum yield of fluorescence, estimated to be <5%, corresponding to a mean heme-tryptophan distance <2.0 nm (compared with ≈1 nm in the native structure and ≈4 nm in the acid denatured structures). According to this model, then, our observed Kratky plot in the intermediate time range, at 150–500 μs, is primarily a linear combination of Kratky plots from two states: compact denatured and native. Because of incomplete mixing, there is also a small contribution from denatured protein at pH 2. Using the populations from the fluorescence kinetics we have calculated an upper limit for the Rg of the compact denatured state, 18.1 ± 0.9 Å. The Rg of the compact denatured structures is therefore no more than 30% larger than that of the native structure. This is the same ratio observed for the denatured state formed by the addition of salt at pH 2, which collapses the structure because of counterion shielding of the electrostatic repulsion among the 24 positively charged residues (23). Thus, in the absence of denaturant, one of the fastest folding proteins known first collapses to form a relatively compact denatured state.
It is interesting to compare this result with lattice simulations (1–4) of simplified representations of proteins, which suggest that proteins fold faster if they do not first collapse to form compact structures. In the simulations these collapsed denatured structures are misfolded. The subsequent step to form the native topology is slowed because it involves breaking many non-native contacts. If, on the other hand, folding and collapse occur simultaneously, fewer non-native contacts form, and folding in the simulations is faster. An interesting explanation for the apparent disagreement between our experimental result and the simulations is suggested by the rate expression for folding, considered as diffusion of the polypeptide on a partially rough, free energy landscape (24, 25). In this description the folding time, τf, is given by (24):
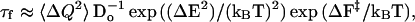 |
1 |
where ΔQ2 is the mean square fluctuation in the configurational coordinate in the denatured state, ΔE2 is the roughness of the energy landscape, Do is the diffusion constant in the absence of roughness, and ΔF‡ is the free energy barrier separating the denatured and native states. If the compact denatured state has a native-like rather than misfolded topology, Eq. 1 predicts that folding is faster because all three terms in this expression for τf are expected to be smaller. These considerations suggest that one contribution to fast folding in cytochrome c is a native-like topology of the denatured state (30). There is experimental evidence to support this hypothesis from amide exchange experiments, which show that the compact denatured state contains segments of native helical structure (26). It would, of course, be quite important if a native-like topology were a general property of compact denatured states (27). The “folding problem” at the level of overall topology then would be solved for this class of proteins in the initial rapid collapse of the polypeptide chain (τcollapse = 50 μs for cytochrome c; ref. 18). Rapid collapse, moreover, to native-like topologies with burial of hydrophobic residues may have a biological advantage by reducing aggregation and proteolysis (28).
The current work suggests that it will be important to obtain systematic data on folding kinetics and time-resolved SAXS for a number of proteins. Such studies will be greatly aided by the brilliant third-generation synchrotron sources that have micron-sized microfocused beams 100-fold more intense than those used here. Together with faster flows and thinner streams time resolution an order of magnitude faster will be possible. Acquisition of higher signal-to-noise scattering data also should permit the determination of additional structural information on the compact denatured state (29).
Acknowledgments
We thank E. Fontes, M. Novak, M. Skvarla, P. Infante, W. Zipfel, and R. Williams for valuable assistance, and J. Hofrichter, V. Muñoz, A. Szabo, S. J. Hagen, J. Trewhella, and D. Shalloway for helpful discussions. This work used Cornell High Energy Synchrotron Source, a National User Facility supported by the National Science Foundation; the Cornell Nanofabrication Facility, supported by the National Science Foundation, Cornell University, and industrial affiliates; and the Developmental Resource for Biophysical Imaging and Opto-Electronics at Cornell, a National Institutes of Health-National Center for Research Resources facility. This work was supported by grants from the National Science Foundation and the Department of Energy.
ABBREVIATION
- SAXS
small-angle x-ray scattering
References
- 1.Socci N D, Onuchic J N. J Chem Phys. 1995;103:4732–4743. [Google Scholar]
- 2.Mirny L A, Abkevich V, Shakhnovich E I. Folding Design. 1996;1:103–116. doi: 10.1016/S1359-0278(96)00019-3. [DOI] [PubMed] [Google Scholar]
- 3.Klimov D K, Thirumalai D. Phys Rev Lett. 1996;76:4070–4073. doi: 10.1103/PhysRevLett.76.4070. [DOI] [PubMed] [Google Scholar]
- 4.Chan H S, Dill K A. Proteins. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 5.Porod G. In: Small Angle X-Ray Scattering. Glatter O, Kratky O, editors. London: Academic; 1982. pp. 17–52. [Google Scholar]
- 6.Debye P. J Phys Colloid Chem. 1947;51:18–32. doi: 10.1021/j150451a002. [DOI] [PubMed] [Google Scholar]
- 7.Kirste R G, Oberthur R C. In: Small Angle X-Ray Scattering. Glatter O, Kratky O, editors. London: Academic; 1982. pp. 387–432. [Google Scholar]
- 8.Eliezer D, Chiba K, Tsuruta H, Doniach S, Hodgson K O, Kihara H. Biophys J. 1993;65:912–917. doi: 10.1016/S0006-3495(93)81124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semisotnov G V, Kihara H, Kotova N V, Kimura K, Amemiya Y, Wakabayashi K, Serdyuk Y N, Timchenko A A, Chiba K, Nikaido K, et al. J Mol Biol. 1996;262:559–574. doi: 10.1006/jmbi.1996.0535. [DOI] [PubMed] [Google Scholar]
- 10.Segel D J, Bachmann A, Hofrichter J, Hodgson K O, Doniach S, Kiefhaber T. J Mol Biol. 1999;288:489–499. doi: 10.1006/jmbi.1999.2703. [DOI] [PubMed] [Google Scholar]
- 11.Jackson S E. Folding Design. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 12.Knight J B, Vishwanath A, Brody J P, Austin R H. Phys Rev Lett. 1998;80:3863–3866. [Google Scholar]
- 13.Sosnick T R, Mayne L, Hiller R, Englander S W. Nat Struct Biol. 1994;1:149–156. doi: 10.1038/nsb0394-149. [DOI] [PubMed] [Google Scholar]
- 14.Elove G A, Bhuyan A K, Roder H. Biochemistry. 1994;33:6925–6935. doi: 10.1021/bi00188a023. [DOI] [PubMed] [Google Scholar]
- 15.Chan C-K, Hu Y, Takahashi S, Rousseau D L, Eaton W A, Hofrichter J. Proc Natl Acad Sci USA. 1997;94:1779–1784. doi: 10.1073/pnas.94.5.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi S, Yeh S-R, Das T K, Chan C-K, Gottfried D S, Rousseau D L. Nat Struct Biol. 1997;4:44–50. doi: 10.1038/nsb0197-44. [DOI] [PubMed] [Google Scholar]
- 17.Sosnick T R, Shtilerman M D, Mayne L, Englander S W. Proc Natl Acad Sci USA. 1997;94:8545–8550. doi: 10.1073/pnas.94.16.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shastry M C R, Roder H. Nat Struct Biol. 1998;5:385–392. doi: 10.1038/nsb0598-385. [DOI] [PubMed] [Google Scholar]
- 19.Babul J, Stellwagen E. Biochemistry. 1972;11:1195–1200. doi: 10.1021/bi00757a013. [DOI] [PubMed] [Google Scholar]
- 20.Tate M W, Eikenberry E F, Barna S L, Wall M E, Lowrance J L, Gruner S M. J Appl Crystallogr. 1995;28:196–205. [Google Scholar]
- 21.Wu C-F, Chen S-H. Biopolymers. 1988;27:1065–1083. doi: 10.1002/bip.360270703. [DOI] [PubMed] [Google Scholar]
- 22.Segel D, Fink A L, Hodgson K O, Doniach S. Biochemistry. 1998;37:12443–12451. doi: 10.1021/bi980535t. [DOI] [PubMed] [Google Scholar]
- 23.Kataoka M, Hagihara Y, Mihara K, Goto Y. J Mol Biol. 1993;229:591–596. doi: 10.1006/jmbi.1993.1064. [DOI] [PubMed] [Google Scholar]
- 24.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 25.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 26.Sauder J M, Roder H. Folding Design. 1998;3:293–301. doi: 10.1016/S1359-0278(98)00040-6. [DOI] [PubMed] [Google Scholar]
- 27.Ptitsyn O B. Curr Opin Struct Biol. 1995;5:74–78. doi: 10.1016/0959-440x(95)80011-o. [DOI] [PubMed] [Google Scholar]
- 28.Hagen S J, Hofrichter J, Szabo A, Eaton W A. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doniach S, Bascle J, Garel T, Orland H. J Mol Biol. 1995;254:960–967. doi: 10.1006/jmbi.1995.0668. [DOI] [PubMed] [Google Scholar]
- 30.Thirumalai D. J Phys I France. 1995;5:1457–1467. [Google Scholar]