

Abstract
Context
Once-daily HIV treatment regimens are being used in clinical practice with the objective of improving patient acceptance and adherence.
Objective
To evaluate the efficacy and safety of saquinavir-soft-gelatin capsule (SGC)/ritonavir combination (1600 mg/100 mg) vs efavirenz (600 mg) both once daily and combined with 2 nucleoside analogs twice daily.
Setting
Twenty-six centers in the United States, Canada, and Puerto Rico.
Patients
A total of 171 antiretroviral naive HIV-infected individuals were enrolled in a 48-week, phase 3, open-label, randomized study.
Main Outcome Measure
Proportion of patients with HIV-RNA levels < 50 copies/mL. The pharmacokinetic profile of saquinavir-SGC was analyzed in a subset of randomly selected patients.
Results
In the primary intent-to-treat population at week 48, 51% (38/75) and 71% (55/77) of patients in the saquinavir-SGC/ritonavir and efavirenz groups, respectively, achieved HIV-RNA suppression < 50 copies/mL (P = .5392, 95% 1-sided confidence interval [CI] = −33.5%). In the on-treatment (OT) population, 73% (38/52) and 93% (54/58) of patients in the saquinavir-SGC/ritonavir and efavirenz groups, respectively, had effective viral suppression < 50 copies/mL (P = .5015, 95% 1-sided CI = −33.4%). Mean CD4+ cell counts increased by 239 and 204 cells/microliters (mcL), in the saquinavir-SGC/ritonavir and efavirenz groups, respectively, in the OT analysis (P = .058). Both regimens were reasonably well tolerated, although more gastrointestinal adverse events were reported with saquinavir-SGC/ritonavir. Pharmacokinetic profiles in 6 patients showed an observed median Cmin at 24 hours of 429 ng/mL (range, 68-1750 ng/mL).
Conclusions
Once-daily efavirenz was statistically superior to once-daily saquinavir-SGC/ritonavir. Gastrointestinal adverse effects were commonly associated with treatment failure in the saquinavir-SGC/ritonavir arm of the study.
Keywords: saquinavir soft-gelatin capsule/ritonavir, once-daily, efavirenz, efficacy, pharmacokinetics
Readers are encouraged to respond to George Lundberg, MD, Editor of MedGenMed, for the editor's eye only or for possible publication via email: glundberg@medscape.net
Introduction
The introduction of highly active antiretroviral therapy (HAART) has produced dramatic reductions in morbidity and mortality rates associated with HIV-1 infection in the United States.[1,2] In clinical trials HAART has reduced plasma HIV-1 RNA levels to less than 400 copies/mL in 60% to 90% of patients.[3] Strict adherence to HAART is necessary to prevent viral replication and the emergence of drug-resistant viruses, which can compromise the final therapeutic benefit.[4,5] Treatment regimens with improved dosing schedules such as once-daily dosing, are likely to improve patient acceptance and adherence.[6] Furthermore, once-daily dosing of HAART may be particularly beneficial in the implementation of directly observed therapy (DOT) in prisons, at needle-exchange sites, and in drug rehabilitation programs.[4]
To date, 6 antiretroviral (ARV) agents, efavirenz, tenofovir, didanosine, lamivudine, coformulated lamivudine/abacavir, atazanavir and amprenavir boosted with ritonavir are approved for once-daily dosing by the US Food and Drug Administration (FDA). However, in addition to these drugs, there are several agents such as nevirapine and other boosted protease inhibitors (PIs) that are being used once daily in clinical practice based on their half lives. The availability of a wide choice of once-daily treatment regimens has made it easier for HIV-infected patients to find an optimal therapy that suits their lifestyle. While efavirenz appears to be an ideal once-daily treatment option due to its potency and convenience with a low pill burden,[7] adverse events, in particular relating to the central nervous system, have been reported following administration,[8] which may potentially limit its use in some HIV-infected patients. Additionally, efavirenz may not be appropriate in some settings because it may have teratogenic effects.[8]
Ritonavir-boosted PI regimens are widely used in clinical practice,[9,10] because several boosted PI combinations have pharmacokinetic profiles that support once-daily dosing.[11] These include saquinavir/ritonavir,[12] amprenavir/ritonavir,[13] fosamprenavir/ritonavir,[14] lopinavir/ritonavir [15] and atazanavir/ritonavir.[16] In initial studies, positive pharmacokinetic and efficacy data have been observed with the use of once-daily saquinavir/ritonavir in PI-naive and experienced individuals.[12,17] Therefore, to further investigate the saquinavir/ritonavir boosted PI combination as a potential once-daily treatment regimen, we evaluated the efficacy and safety of saquinavir-soft-gelatin capsule (SGC)/ritonavir 1600 mg/100 mg vs efavirenz 600 mg in a prospective, randomized, multicenter clinical trial. Both treatment regimens were administered once daily in addition to 2 nucleoside reverse transcriptase inhibitors (NRTIs) (twice daily) as part of combination HAART therapy regimens to ARV-naive, HIV-infected individuals. As part of this clinical study, the pharmacokinetic profile of saquinavir-SGC was assessed in a subset of patients.
Materials and Methods
Study Design
This was a phase 3, open-label, randomized, multicenter study conducted at 26 centers in the United States, Canada, and Puerto Rico. Antiretroviral-naive, HIV-infected adults were randomized to receive either saquinavir-SGC/ritonavir (1600 mg/100 mg, 9 pills) or efavirenz (600 mg, 3 pills) once daily, both in combination with 2 NRTIs twice daily. An interim analysis was planned at week 24, with a follow-up extension to week 48. Once patients reached 48 weeks, they had the option of continuing in the study until a common study closure (CSC) date or of withdrawing at that time. The CSC date was the date on which the last randomized patient reached 48 weeks of treatment.
The NRTIs allowed in the study included the following agents: zidovudine (AZT, Retrovir), lamivudine (3TC, Epivir), 3TC/AZT (Combivir), stavudine (d4T, Zerit), or didanosine (ddI, Videx). The NRTIs were selected by the investigators following consultation with the study participants, with the exception of didanosine, which was dosed twice daily according to the FDA approval status at the time this study was conducted. Patients were randomized via stratification of the screening plasma HIV-RNA value (5000-75,000 copies/mL vs > 75,000 copies/mL). Saquinavir-SGC and ritonavir were to be taken at the same time and with food. The study was designed and monitored in accordance with Good Clinical Practices. Prior approval was obtained from institutional review boards/local ethics committees of the participating centers and written informed consent was obtained from all study participants.
Patients
The defined study population consisted of male or female (nonpregnant, nonnursing) adults (18 years or older), with HIV-RNA values ≥ 5000 copies/mL and CD4+ cell counts ≥ 75 cells/mcL. All patients were ARV-naive (no more than 2 weeks of previous ARV therapy since HIV diagnosis). Patients with significant laboratory abnormalities, active hepatitis B or hepatitis C, severe hepatic impairment, or malignancy requiring chemotherapy or radiation therapy were excluded from the study.
Study Populations
All efficacy analyses were performed on the primary intent-to-treat (ITT) and on-treatment (OT) populations. The primary ITT population included all patients who received at least 1 dose of study drug after randomization and had at least 1 efficacy evaluation. The OT population included those patients in the study who had an observational value for all efficacy variables and laboratory measurements, at that particular timepoint in the study. The safety-evaluable population included all randomized patients who received at least 1 dose of study medication and had at least 1 postbaseline safety assessment.
Efficacy and Safety Measures
The primary efficacy variable was the proportion of patients with plasma HIV-RNA values < 50 copies/mL using the Ultra-Sensitive assay (Roche Diagnostics), at weeks 24 and 48. Secondary efficacy analyses included the change from baseline in CD4+ cell counts. Primary and secondary variables were assessed at baseline (week 0), every 4 weeks until week 24, and every 8 weeks until week 48, or at study discontinuation.
Adverse events were graded by intensity, and their relationship to the study medications was assessed. Fasting lipid profile was also assessed. Laboratory abnormalities were graded by severity. Marked laboratory shifts were defined as a ≥ 3 grade shift from baseline (grade 0-3 or 4 and grade 1-4).
Pharmacokinetic Subanalysis
A subanalysis was performed to assess the pharmacokinetic profile of saquinavir-SGC in a subset of randomly selected patients. Intensive pharmacokinetic assessments were performed on patients from 2 preselected study centers after the completion of 4 weeks of therapy. Following a standard breakfast, saquinavir-SGC/ritonavir 1600 mg/100 mg was administered with 2 NRTIs. Samples for analysis of saquinavir-SGC (7 mL) were taken predosing (hour 0) and at 1, 2, 3, 4, 6, 8, 12, and 24 hours post-dose. At week 4, single trough plasma concentrations of saquinavir-SGC were also determined in a larger group of patients. For patients who took their dose in the morning, single samples were taken before their next morning dose (approximately 24 hours later) and for those who took their dose in the evening, samples were taken at least 12 hours after the previous dose.
Pharmacokinetic Analysis
Saquinavir-SGC plasma concentration was assayed using a validated SCIEX API liquid chromatography extraction method with a mass spectrophotometric (LC-MS) detection system. The method was validated for a range of 5-3000 ng/mL. For patients who underwent intensive pharmacokinetic evaluation, the variables assessed were area under the curve over 24 hours (AUC0-24h), maximum plasma concentration (Cmax) and observed trough plasma concentration (Cmin). Cmax and Cmin were read directly from the observed data. The AUC data were obtained by noncompartmental analysis.
In patients with a single trough plasma sample, Cmin at 24 hours (C24h) was the pharmacokinetic variable of interest and saquinavir-SGC C24h was mathematically extrapolated to reflect 24-hour levels, assuming an elimination half-life of 5-6 hours.[18] The equation C1 =C0e(−kel* Δt) was used to calculate the saquinavir-SGC C24h extrapolated levels where: C1 = concentration extrapolated at 24 hours; C0 = observed concentration; kel = the elimination rate constant for the drug (kel = 0.693/t½); Δt = difference in time between C1 and C0; and t½ is defined as the half-life of the drug.
Descriptive statistics for the pharmacokinetic parameters of saquinavir-SGC were summarized to compare them with historical values obtained with the licensed dose of saquinavir-SGC (1200 mg 3 times daily [TID]).
Statistical Analyses
This study was designed to compare the efficacy of once-daily dosing of saquinavir-SGC/ritonavir 1600 mg/100 mg vs efavirenz 600 mg at weeks 24 and 48, using the proportion of patients with HIV-RNA values < 50 copies/mL (response rate). Group differences in the proportion of patients with HIV-RNA levels < 50 copies/mL were analyzed using a noninferiority test. The Farrington-Manning method for testing noninferiority was used to derive P values and to construct 1-sided 95% confidence intervals (CI) of the rate difference.[19] If the limit of the 95% 1-sided CI of the difference rate was greater than −20%, then noninferiority was concluded. Mean changes from baseline in CD4+ cell counts were analyzed using an analysis of variance (ANOVA) model. Safety variables were summarized using descriptive statistics for the entire study period up to the CSC date. Fisher's exact test was used to statistically compare the incidence of adverse events in both treatment groups.
Results
Patients and Study Course
From July 1999 to October 2001, 269 patients were screened and 171 were randomized to treatment (Figure 1). Six patients withdrew from the primary ITT population before receiving study medication, giving a total of 165 patients who received study drug. Of these, 13 patients were excluded because no postrandomization on-drug efficacy evaluation was available. The resulting primary ITT population included 75 patients in the saquinavir-SGC/ritonavir group and 77 patients in the efavirenz group. Of the primary ITT population, 108 patients completed all 48 weeks of the study (51 in the saquinavir-SGC/ritonavir group and 57 in the efavirenz group). At week 48, the OT population consisted of 110 patients: 52 and 58 patients in the saquinavir-SGC/ritonavir and efavirenz arms, respectively. The safety population consisted of 161 randomized patients: 81 and 80 patients in the saquinavir-SGC/ritonavir and efavirenz group, respectively.
Figure 1.
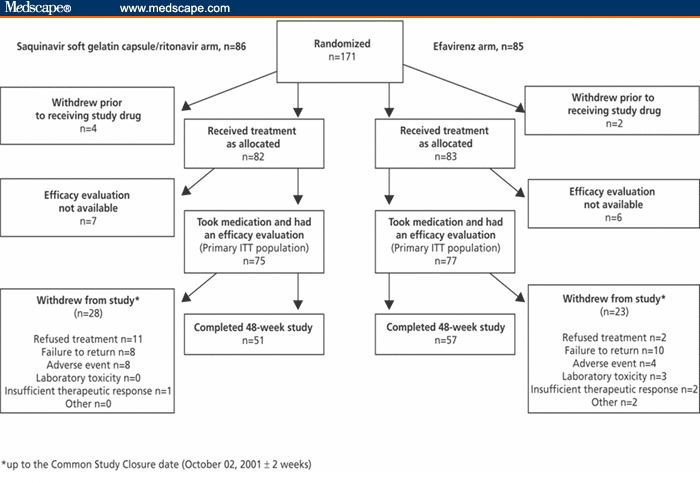
Study design and completion status.
ITT=intent to treat
At the CSC date, 51/152 (34%) patients had withdrawn from the primary ITT population: 28/75 (37%) patients from the saquinavir-SGC/ritonavir group and 23/77 (30%) patients from the efavirenz group (Figure 1). Of these, 18 patients withdrew due to failure to return for reassessment (8 in the saquinavir-SGC/ritonavir arm and 10 in the efavirenz arm), 13 patients refused treatment/withdrew consent (11 in the saquinavir-SGC/ritonavir arm and 2 in the efavirenz arm), and 12 withdrew due to adverse events (8 in the saquinavir-SGC/ritonavir arm and 4 in the efavirenz arm). The demographic and baseline characteristics of the patients in the saquinavir-SGC/ritonavir and efavirenz arms were similar, with respect to gender, age, race, baseline HIV-RNA levels and CD4+ counts (Table 1). The majority (73%) of patients in both arms was male, 47% were black, and the age range was 19-61 years (median, 36 years).
Table 1.
Summary of Patient Characteristics at Baseline (Primary Intent-to-Treat Population)
| Characteristic | Saquinavir-SGC 1600 mg + ritonavir 100 mg, Once Daily (n = 75) | Efavirenz 600 mg Once Daily (n = 77) |
|---|---|---|
| Male n (%) | 53 (71) | 58 (75) |
| Female n (%) | 22 (29) | 19 (25) |
| Race n (%) | ||
| White | 28 (37) | 29 (38) |
| Black | 34 (45) | 38 (49) |
| Hispanic | 6 (8) | 8 (10) |
| Other | 7 (9) | 2 (3) |
| Age (y); mean ± SD (range) | 37.2 ± 9.7 (20-61) | 37.2 ± 9.9 (19-61) |
| Weight (kg); mean ± SD (range) | 78.0 ± 16.5 (52-126) | 75.4 ± 15.0 (44-136) |
| Plasma HIV RNA (log10 copies/mL); mean ± SD (range) | 4.8 ± 0.6 (3.34-6.49) | 4.7 ± 0.5 (3.51-5.66) |
| CD4+ cell count (cells/mcL); mean ± SD (range) | 372 ± 190 (75-1025) | 343 ± 180 (80-900) |
| Nucleoside reverse transcriptase inhibitor combination therapy; n (%) | ||
| 3TC-AZT | 47 (63) | 52 (68) |
| 3TC-d4T | 27 (36) | 22 (29) |
| 3TC-ddI | 0 | 2 (3) |
| AZT-ddI | 1 (1) | 0 |
| d4T-ddI | 0 | 1 (1) |
| Stratification (HIV RNA); n (%) copies/mL | ||
| ≥ 5000-75,000 | 39 (52) | 43 (56) |
| > 75,000 copies/mL | 36 (48) | 34 (44) |
SGC = soft gelatin capsule; SD = standard deviation; 3TC = lamivudine; AZT = zidovudine; d4T = stavudine; ddI = didanosine.
Efficacy
Plasma HIV RNA Response
As shown in Figure 2, in the primary ITT population at week 24, 63% (47/75) and 83% (64/77) of patients in the saquinavir-SGC/ritonavir and efavirenz groups, respectively, had HIV-RNA values < 50 copies/mL (P = .5255, 95% 1-sided CI = −32.0%). At the 48-week time-point, the respective values were 51% (38/75) and 71% (55/77) (P = .5392, 95% 1-sided CI = −33.5%).
Figure 2.
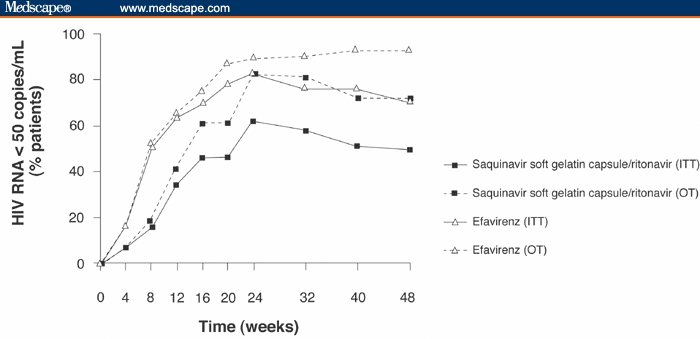
Percentage of patients treated with either saquinavir-soft gelatin capsule/ritonavir or efavirenz both once-daily in combination with 2 nucleoside reverse transcriptase inhibitors, with undetectable HIV-RNA levels (< 50 copies/mL), as detected by the Ultra-Sensitive assay.
ITT=intent to treat; OT=on treatment
In the OT population, at week 24, 82% (46/56) and 90% (61/68) of patients had HIV-RNA levels < 50 copies/mL in the saquinavir-SGC/ritonavir and efavirenz groups, respectively (P = .0374, 95% 1-sided CI = −19.05%). By week 48, 73% (38/52) and 93% (54/58) of patients had HIV-RNA levels < 50 copies/mL, respectively (P = .5015, 95% 1-sided CI = −33.3%).
Changes in CD4+ Cell Counts
At week 24, the mean increase in CD4+ cell counts was 170 cells/mcL in the saquinavir-SGC/ritonavir arm compared with 143 cells/mcL in the efavirenz arm (P = .344; OT population). At the 48-week study endpoint, this had increased to 239 cells/mcL and 204 cells/mcL, in the saquinavir-SGC/ritonavir and efavirenz groups, respectively (P = .058; OT population).
Safety
Overall, adverse events were reported by 98% and 94% of patients in the saquinavir-SGC/ritonavir and efavirenz groups, respectively. The majority of adverse events were mild or moderate in intensity. During the entire study period, 11% (18/161) of patients in the safety population discontinued treatment due to adverse events. In the saquinavir-SGC/ritonavir group 15% (12/81) of patients experienced adverse events that led to withdrawal and 8% (6/80) of patients withdrew from the efavirenz group due to adverse events. The most commonly reported adverse events leading to withdrawal in the saquinavir-SGC/ritonavir group were gastrointestinal in nature, including nausea and vomiting. In contrast, weight loss, peripheral neuropathy and abnormal dreams were the reported adverse events leading to withdrawal in the efavirenz group.
Adverse events of moderate, severe or life-threatening intensity that were considered possibly or probably related to study medication occurring in 2% or more of patients are shown in Table 2. Overall, 42% (34/81) and 29% (23/80) of patients in the saquinavir-SGC/ritonavir and efavirenz groups, respectively, reported at least 1 adverse event of moderate, severe, or life-threatening intensity considered possibly or probably related to study medication. Among adverse events, the rates of nausea differed significantly among treatment groups (Table 2).
Table 2.
Clinical Adverse Events Considered to Be at Least Possibly or Probably Related to Treatment, at Least of Moderate, Severe or Life-Threatening Intensity, and Occurring in ≥ 2% of Patients in the Safety Population
| Adverse Event (ordered by body system) | Saquinavir-SGC 1600 mg + ritonavir 100 mg, Once Daily (n = 81) | Efavirenz 600 mg Once Daily (n = 80) |
|---|---|---|
| n (%) | n (%) | |
| Gastrointestinal disorders | ||
| Nausea* | 18 (22.2) | 3 (3.8) |
| Vomiting NOS | 5 (6.2) | 0 |
| Diarrhea NOS | 4 (4.9) | 4 (5.0) |
| Abdominal pain NOS | 2 (2.5) | 0 |
| Diarrhea aggravated | 2 (2.5) | 0 |
| Esophageal reflux | 2 (2.5) | 0 |
| General disorders | ||
| Fatigue | 2 (2.5) | 4 (5.0) |
| Dizziness (excluding vertigo) | 1 (1.2) | 4 (5.0) |
| Skin and subcutaneous tissue disorders | ||
| Dermatitis NOS | 1 (1.2) | 5 (6.3) |
| Metabolic and nutritional disorders | ||
| Appetite decreased | 3 (3.7) | 1 (1.3) |
| Neurologic disorders | ||
| Headache NOS | 2 (2.5) | 2 (2.5) |
| Insomnia | 1 (1.2) | 2 (2.5) |
| Weakness | 2 (2.5) | 0 |
| Psychiatric disorders | ||
| Depression NOS | 2 (2.5) | 1 (1.3) |
| Anxiety | 1 (1.2) | 2 (2.5) |
| Abnormal dreams | 0 | 2 (2.5) |
| Vascular disorders | ||
| Hypertension NOS | 0 | 2 (2.5) |
SGC = soft gelatin capsule; NOS = not otherwise specified.
P = .0004; Fisher's exact test.
In total, 15 patients (7 [8.6%] and 8 [10.0%] patients in the saquinavir-SGC/ritonavir and efavirenz groups, respectively) reported at least 1 serious adverse event, none of which were considered to be related to study medication. One patient in the saquinavir-SGC/ritonavir group died due to myocardial infarction, which again was not considered to be related to study medication.
Laboratory Variables
Overall, laboratory grade shifts in hematology and chemistry values were similar in both treatment groups. Grade 3/4 total cholesterol laboratory shifts were reported in 3 patients in the saquinavir-SGC/ritonavir group compared with 7 patients in the efavirenz group. Mean triglyceride and total cholesterol levels assessed throughout the 48-week study period are presented in Figure 3. At weeks 24 and 48, mean changes in triglyceride values from baseline were 29.4 mg/dL and 51.2 mg/dL, respectively, in the saquinavir-SGC/ritonavir group, compared with 27.0 mg/dL and 71.4 mg/dL, respectively, in the efavirenz group. The differences between groups were not statistically significant at weeks 24 and 48. Mean changes in total cholesterol levels were comparable between the 2 treatment arms throughout the 48-week study period. Mean changes in high-density lipoprotein cholesterol at weeks 24 and 48 were 7.9 mg/dL and 8.7 mg/dL, respectively, in the saquinavir-SGC/ritonavir group compared with 9.3 mg/dL and 12.1 mg/dL, respectively, in the efavirenz group.
Figure 3.
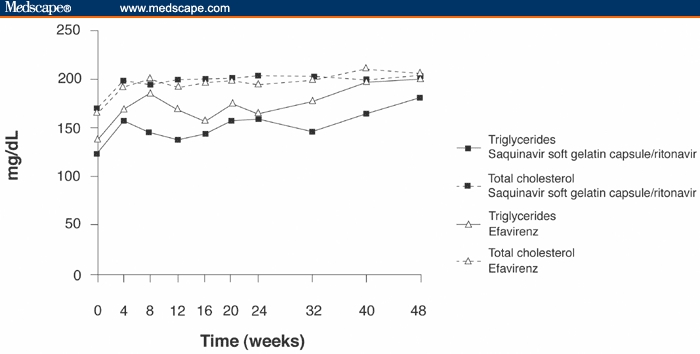
Mean levels of triglycerides and total cholesterol in the safety population of patients treated with either saquinavir-soft gelatin capsule/ritonavir or efavirenz both once daily in combination with 2 nucleoside reverse transcriptase inhibitors.
Pharmacokinetics
Of 72 patients who were enrolled in the pharmacokinetic sub-analysis, 6 underwent intensive pharmacokinetic evaluation. Sixty-five patients had single plasma samples taken at week 4 and data from 44 patients were suitable for evaluation of trough concentrations (data from the remaining 21 could not be extrapolated because the samples from 11 patients were taken less than 12 hours after the previous dose and the remaining 10 patients were noncompliant). Furthermore, of the 6 patients included in the intensive pharmacokinetic analysis, 1 patient was noncompliant and, therefore, not included in the single-trough evaluable cohort of patients. The single-trough evaluable cohort, therefore, consisted of 49 patients (5 patients with intensive pharmacokinetics and 44 with single pharmacokinetic evaluations).
Intensive Pharmacokinetic Analysis at 4 Weeks.
The intensive pharmacokinetic profile of the 6 unselected patients evaluated at 9 time points over 24 hours is shown in Figure 4. The median observed Cmin at 24 hours, Cmax and AUC0-24 for these 6 patients were 429 ng/mL (range, 68-1750 ng/mL), 6435 ng/mL (range, 1720-13,400 ng/mL), and 66,920 ng.h/mL (range, 10,542-137,563 ng.h/mL), respectively.
Figure 4.
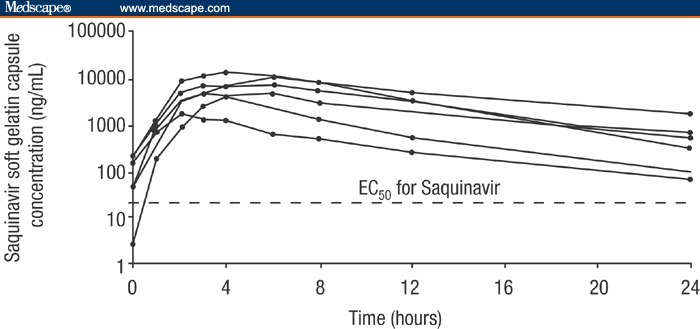
Intensive pharmacokinetic profile of 6 patients at week 4. EC50 = concentration for 50% maximal effect
Saquinavir-SGC Trough Levels at Week 4.
The median saquinavir-SGC C24h extrapolated level at week 4 for all evaluable patients (n = 49) was 376 ng/mL (range, 2.5-5709 ng/mL). Eight patients had saquinavir-SGC C24h extrapolated levels under the in vivo concentration for half-maximal effect (EC50) value of 50 ng/mL (range, 2.5-45.2 ng/mL).[20] Despite this, these patients had sustained HIV viral load suppression up to week 24. There was no significant correlation between saquinavir-SGC Cmin values and the reduction in plasma HIV RNA values from baseline to week 48.
Discussion
This 48-week study compared the efficacy and safety of the once-daily HIV treatment regimens, including saquinavir-SGC/ritonavir 1600 mg/100 mg, to that of efavirenz 600 mg. The primary ITT analysis showed that a higher proportion of patients in the efavirenz-treated group achieved HIV-RNA levels < 50 copies/mL compared with the saquinavir-SGC/ritonavir-treated group. More patients withdrew from randomized treatment due to gastrointestinal disorders in the saquinavir-SGC/ritonavir arm. The lower virologic response rate observed in the saquinavir-SGC/ritonavir arm in the ITT population was predominately due to the high rate of discontinuations, with 37% of patients withdrawing from the saquinavir/ritonavir arm, compared with 30% from the efavirenz arm. These withdrawals were related to gastrointestinal side effects, and possibly higher pill counts (patients taking saquinavir-SGC/ritonavir had to take 9 capsules at 1 time compared with 3 efavirenz capsules, together with the background NRTIs). This study included a high percentage of African-American patients and no specific adherence intervention was provided to overcome the difference in pill burden in the 2 treatment arms.
A previous comparative randomized trial of the boosted PI saquinavir-SGC/ritonavir has also shown lower virologic response rates, which have been attributed to differences in study withdrawal rates as a result of gastrointestinal toxicities in the saquinavir-SGC study arms.[21] In the MaxCmin 2 study, which compared twice-daily saquinavir-SGC/ritonavir with lopinavir/ritonavir, comparable viral suppression was demonstrated in the OT analyses; however, the difference in study withdrawal rates due to gastrointestinal toxicity in the saquinavir-SGC/ritonavir arm led to a lower overall response in the ITT analyses.[21] The gastrointestinal toxicities encountered in the saquinavir-SGC study arm of MaxCmin 2 were attributed to the glyceride capmul component of the SGC formulation of saquinavir.
Saquinavir is available in 2 formulations: the SGC formulation, Fortovase, and the original hard capsule (HC) saquinavir mesylate formulation, Invirase. In the present study, the SGC formulation of saquinavir was used. As a sole PI (unboosted), the oral bioavailability and effectiveness of saquinavir with the SGC formulation is higher than that of the HC formulation. This difference is due to the glyceride excipient component of the SGC formulation, capmul, which allows saquinavir to dissolve and disperse rapidly upon administration. However, the SGC formulation is associated with more gastrointestinal adverse events.[22,23] In a recent study of the saquinavir/ritonavir boosted regimen, the HC formulation of saquinavir mesylate demonstrated similar exposure to the SGC formulation but was better tolerated.[22,24] Consequently, it might be expected that the tolerability issues encountered in this study could be ameliorated if the HC saquinavir mesylate formulation of saquinavir was used. Interestingly, in a further study the boosted saquinavir-HC/ritonavir 1600 mg/100 mg regimen demonstrated potent viral suppression (89% of patients had HIV RNA values < 50 copies/mL) with no patients withdrawing from the study due to adverse events.[25] Of note, the saquinavir mesylate is now available as a 500-mg film-coated tablet, which is similar in size to the saquinavir mesylate 200 mg HC.
Changes in lipid levels have been associated with the use of NRTIs and PIs and, consequently, the impact on lipid levels should be an important consideration when choosing a treatment regimen.[26] With PIs, this risk has been shown to be greatest with ritonavir, intermediate with amprenavir and nelfinavir, and lowest with indinavir and saquinavir.[27,28] The recently approved PI, atazanavir appears to have little effect on lipid concentrations.[16,27] The lower doses of ritonavir (100-200 mg) that are suitable for boosted saquinavir/ritonavir regimens may result in some increases of fasting triglyceride and cholesterol levels.[29] The data from this study confirm a low lipid profile risk associated with a once-daily saquinavir-SGC/ritonavir dose of 1600 mg/100 mg. During this study, data evolved regarding the contribution of NRTIs to hyperlipidemia and a post-hoc analysis was conducted that demonstrated that the predicting factor for hyperlipidemia in this study appeared to be the use of stavudine as the background NRTI.[30]
Efavirenz once daily is a highly effective and commonly prescribed part of ARV therapy; however, it may not be suitable for all patients. Patients taking efavirenz may experience central nervous system adverse events such as dizziness, insomnia, poor concentration, and vivid dreams.[8] With the recent classification of efavirenz as pregnancy category D caution, it is advised in the treatment of women of child-bearing potential. Also, efavirenz use may be a cause for concern in drug users currently taking methadone because efavirenz-mediated reduction of methadone blood concentrations has also been reported.[31,32] No methadone dose adjustments have been found to be required in patients receiving once-daily saquinavir/ritonavir 1600 mg/100 mg therapy.[33]
The results of the pharmacokinetic sub-analysis illustrated that a once-daily regimen including saquinavir-SGC/ritonavir 1600 mg/100 mg provides a median AUC0-24h of 66,920 ng.h/mL. This is significantly higher than might be anticipated following the unboosted saquinavir SGC regimen of 1200 mg 3 times daily, where the AUC0-8h is 7249 ng.h/mL.[34] In addition, the median observed Cmin at 24 hours for saquinavir-SGC of 429 ng/mL was substantially higher than the EC50 value of 50 ng/mL.[20] There is some evidence that failure on PI therapy can be linked to low plasma concentrations,[35] but in this study no relationship was found between HIV RNA concentrations and saquinavir Cmin values.[36] Currently, with antiretroviral therapy, it is unknown which pharmacokinetic parameter correlates best with efficacy.[37] While there is some evidence that failure on PI therapy can be linked to low Cmin plasma concentrations,[20] in this study no relationship was found between HIV RNA concentrations and saquinavir Cmin values.
In conclusion, once-daily efavirenz was statistically superior to the saquinavir-SGC/ritonavir (1600 mg/100 mg) once-daily regimen, as part of combination ARV therapy. Gastrointestinal adverse effects were commonly associated with (ITT) treatment failure in the saquinavir-SGC/ritonavir arm of the study, which may be attributed to the SGC formulation used in this study. Future studies should evaluate the use of the saquinavir mesylate formulation, which has demonstrated similar exposure to the ritonavir-boosted SGC formulation with a better tolerability profile and by virtue of the 500-mg tablet also provides a lower pill burden.
Acknowledgments
We thank Pascal J. de Caprariis, MD, and Huilin Hu for assistance with the trial. We also would like to express our thanks to the study coordinators from all investigational sites.
FOCUS Study Team
Nicholaos Bellos, MD, Southwest Infectious Disease Associates, Dallas, Texas; Alfred F. Burnside Jr, MD, Burnside Clinic, Columbia, South Carolina; Joseph Cervia, MD, Comprehensive HIV Center, New Hyde Park, New York; Ann C. Collier, MD, University of Washington School of Medicine, Seattle, Washington; Paul Cook, MD, ECU School of Medicine, Greenville, North Carolina; Stephen Follansbee, MD, HIV Clinical Trials Unit, San Francisco, California; Joseph C. Gathe Jr, MD, Houston, Texas; Bruce Hathaway, MD, ECU School of Medicine, Greenville, North Carolina; Margaret Hoffman-Terry, MD, Lehigh Valley Hospital AAO, Allentown, Pennsylvania; Dushyantha T. Jayaweera, MD, University of Miami School of Medicine, Miami, Florida; Jazila Alattar Mantis, MD, Queens Hospital Center, Jamaica, New York; Joseph Masci, MD, Elmhurst Medical Center, Elmhurst, New York; Julio S.G. Montaner, MD, St Paul's Hospital, Vancouver, British Columbia, Canada; Mahmoud Mustafa, MD, Physicians' Home Service PC, Washington, DC; Kristin Ries, MD, University of Utah, Salt Lake City, Utah; Robert J. Roland, DO, Infectious Disease Specialists, Union, New Jersey; Danielle Rouleau, MD, CHUM, Montreal, Quebec; Michael S. Saag, MD, AIDS Outpatient Clinic, Birmingham, Alabama; Stefan Schneider, MD, Living Hope Clinical Trials, Long Beach, California; John Schrank, MD, ID Care Center, Greenville, South Carolina; Malte Schutz, MD, Illinois Masonic Medical Center, Chicago, Illinois; Robert Schwartz, MD, Associates in Research, Fort Myers, Florida; Gladys Sepulveda, MD, Anexo Hospital Oncologica, Ponce, Puerto Rico; Leon Smith, MD, St Michael's Medical Center, Newark, New Jersey; Joseph Timpone, MD, Georgetown University Hospital, Washington, DC; Paul Volberding, MD, UCSF AIDS Program, San Francisco, California; Sharon Walmsley, MD, The Toronto Hospital, Toronto, Ontario.
Funding Information
This study was supported by a research grant from Roche Pharmaceuticals.
Contributor Information
Julio S.G. Montaner, St Paul's Hospital, University of British Columbia, Vancouver, BC, Canada. Email: jmontaner@cfenet.ubc.ca.
Malte Schutz, Roche Laboratories, Inc, Nutley, New Jersey.
Robert Schwartz, Associates in Research, Fort Myers, Florida.
Dushyantha T. Jayaweera, University of Miami School of Medicine, Miami, Florida.
Alfred F. Burnside, Burnside Clinic, Columbia, South Carolina.
Sharon Walmsley, Toronto Hospital, Toronto, Canada.
Michael S. Saag, University of Alabama at Birmingham.
References
- 1.Palella FJ, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 2.Detels R, Munoz A, McFarlane G, et al. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. AIDS Cohort Study Investigators. JAMA. 1998;280:1497–503. doi: 10.1001/jama.280.17.1497. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JA, DeMasi R, Quinn J, et al. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 4.Altice FL, Friedland GH. The era of adherence to HIV therapy. Ann Intern Med. 1998;129:503–505. doi: 10.7326/0003-4819-129-6-199809150-00015. [DOI] [PubMed] [Google Scholar]
- 5.Monforte d'Arminio A, Lepri AC, Rezza G, et al. Insights into the reasons for discontinuation of the first highly active antiretroviral therapy (HAART) regimen in a cohort of antiretroviral naive patients. I.CO.N.A. Study Group. Italian Cohort of Antiretroviral-Naive Patients. AIDS. 2000;14:499–507. doi: 10.1097/00002030-200003310-00005. [DOI] [PubMed] [Google Scholar]
- 6.Cohen CJ, Hellinger J, Norris D. Evaluation of simplified protease inhibitor dosing regimens for the treatment of HIV infection. AIDS Read. 2000;10:296–299. 304-307, 311-313. [PubMed] [Google Scholar]
- 7.Staszewski S, Morales-Ramirez J, Tashima KT, et al. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. N Engl J Med. 1999;341:1865–1873. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 8.US Prescribing Information. New Jersey: Bristol-Myers Squibb; Sustiva (efavirenz capsules and tablets) January 2002. [Google Scholar]
- 9.DHHS Guidelines. Guidelines for the use of antiretroviral agents in HIV-infected adults and adolescents. US Dept Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. October 6, 2005. Available at aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
- 10.BHIVA Writing Committee British HIV association (BHIVA) guidelines for the treatment of HIV-infected adults with antiretroviral therapy. HIV Med. 2001;2:276–313. doi: 10.1046/j.1464-2662.2001.00083.x. [DOI] [PubMed] [Google Scholar]
- 11.Kilby JM, Hill A, Buss N. The effect of ritonavir on saquinavir plasma concentration is independent of ritonavir dosage: combined analysis of pharmacokinetic data from 97 subjects. HIV Med. 2002;3:97–104. doi: 10.1046/j.1468-1293.2002.00090.x. [DOI] [PubMed] [Google Scholar]
- 12.Cardiello PG, Van Heeswijk RP, Hassink EA, et al. Simplifying protease inhibitor therapy with once-daily dosing of saquinavir soft-gelatin capsules/ritonavir (1600mg/100mg): HIVNAT 001.3 study. J Acquir Immune Defic Syndr. 2002;29:464–470. doi: 10.1097/00042560-200204150-00006. [DOI] [PubMed] [Google Scholar]
- 13.Arasteh K, Wood R, Teofilo E, et al. Amprenavir (APV) 600 mg/ritonavir (RTV) 100 mg BID or APV 1200 mg/RTV 200 mg QD given in combination with abacavir (ABC) and lamivudine (3TC) maintains efficacy in ART naive HIV-1 infected adults over 24 weeks ( APV20001). Eighth European Conference on Clinical Aspects and Treatment of HIV Infection; October 28-31; Athens, Greece. 2001. Abstract 218. [Google Scholar]
- 14.Gathe JC, Jr, Ive P, Wood R, et al. SOLO: 48-week efficacy and safety comparison of once-daily fosamprenavir/ritonavir versus twice-daily nelfinavir in naive HIV-1-infected patients. AIDS. 2004;8:1529–1537. doi: 10.1097/01.aids.0000131332.30548.92. [DOI] [PubMed] [Google Scholar]
- 15.Eron J, Bernstein B, King M, et al. Once-daily versus twice daily Kaletra (lopinavir/ritonavir) in antiretroviral-naive HIV+ patients: 48 week follow-up. Ninth Conference on Retroviruses and Opportunistic Infections; February 24-28; Seattle, Washington. 2002. Abstract 409. [Google Scholar]
- 16.Piliero PJ. Atazanavir: A novel once-daily protease inhibitor. Drugs Today (Barc) 2004;40(11):901–912. doi: 10.1358/dot.2004.40.11.872579. [DOI] [PubMed] [Google Scholar]
- 17.Peytavin G, Landman R, Lamotte C, et al. Saquinavir (SQV) plasma and intracellular concentrations in a once daily dosing combination Fortovase[R] (SQV-SGC)-low dose ritonavir (RTV) in a prospective study (IMEA 015) in HIV-infected patients. Second International Workshop on Clinical Pharmacology of HIV Therapy 2001; April 2-4; Noordwijk, The Netherlands: Abstract 3.16. [Google Scholar]
- 18.Buss N, Snell P, Bock J, et al. Saquinavir and ritonavir pharmacokinetics following combined ritonavir and saquinavir (soft gelatin capsules) administration. J Clin Pharmacol. 2001;52:255–264. doi: 10.1046/j.0306-5251.2001.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farrington CP, Manning G. Test statistics and sample size formulae for comparative binomial trials with null hypothesis of non-zero risk difference or non-unity relative risk. Stat Med. 1990;9:1447–1454. doi: 10.1002/sim.4780091208. [DOI] [PubMed] [Google Scholar]
- 20.Gieschke R, Fotteler B, Buss N, et al. Relationships between exposure to saquinavir monotherapy and antiviral response in HIV-positive patients. Clin Pharmacokinet. 1999;37:75–86. doi: 10.2165/00003088-199937010-00005. [DOI] [PubMed] [Google Scholar]
- 21.Youle M, Gerstoft J, Fox Z, et al. The final 48 week analysis of a phase IV, randomized, open-label, multicentre trial to evaluate safety and efficacy of lopinavir/ritonavir (400/100 mg BID) versus saquinavir/ritonavir (1000/100 mg BID): the MaxCmin 2 trial. Program and abstracts of the 9th European AIDS Conference (EACS); October, 2003; Warsaw: Abstract F11/3. [Google Scholar]
- 22.Kurowski M, Sternfeld T, Sawyer A, et al. Pharmacokinetic and tolerability profile of twice-daily saquinavir hard gelatin capsules and saquinavir soft gelatin capsules boosted with ritonavir in healthy volunteers. HIV Med. 2003;4:94–100. doi: 10.1046/j.1468-1293.2003.00143.x. [DOI] [PubMed] [Google Scholar]
- 23.Mitsuyasu RT, Skolnik PR, Cohen SR, et al. Activity of the soft gelatin formulation of saquinavir in combination therapy in antiretroviral-naive patients. AIDS. 1998;12:F103–109. doi: 10.1097/00002030-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Cardiello P, Monhaphol T, Mahanontharit A, et al. Pharmacokinetics (PK) of once daily saquinavir-hard gels caps (SQV-HGC) and saquinavir-soft gel caps (SQV-SGC) boosted with ritonavir (RTV) in HIV-1+ Thai patients: HIV NAT001.4 substudy. Program and abstracts of the 3rd International Workshop on Clinical Pharmacology of HIV Therapy; April 2002; Washington, DC: Abstract 1.2. [Google Scholar]
- 25.Ananworanich J, Hill A, Siangphoe U, Ruxrungtham K, et al. Staccato Study Group A prospective study of efficacy and safety of once-daily saquinavir/ritonavir plus two nucleoside reverse transcriptase inhibitors in treatment-naive Thai patients. Antivir Ther. 2005;10:761–767. [PubMed] [Google Scholar]
- 26.Dale E, Manuney J, Uffelman K, et al. Incidence of lipodystrophy in START (Selection of Thymidine Analog Regimen Therapy) Studies. Program and abstracts of the 1st International Workshop on Adverse Drug Reactions and Lipodystrophy in HIV; June 26-28; San Diego, California: 1999. Abstract 30. [Google Scholar]
- 27.Dube MP, Stein JH, Aberg JA, et al. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group Adult AIDS Clinical Trials Group Cardiovascular Subcommittee; HIV Medical Association of the Infectious Disease Society of America. Clin Infect Dis. 2003;37:613–627. doi: 10.1086/378131. [DOI] [PubMed] [Google Scholar]
- 28.Moyle GJ, Baldwin C. Lipid abnormalities during saquinavir soft gel-based highly active antiretroviral therapy. Program and abstracts of the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy; September 26-29; San Francisco, California: 1999. Abstract 1292. [PubMed] [Google Scholar]
- 29.O'Brien WA, Rojo D, Acosta E, et al. Switch of Saquinavir 400mg/ritonavir 400mg to saquinavir 1000mg/ritonavir 100mg during BID four drug antiretroviral therapy in patients with viral load less than 200 copies/mL. Program and abstracts of the 3rd International Workshop on Clinical Pharmacology of HIV Therapy; April 11-13; Washington, DC: 2002. Abstract 2.1. [Google Scholar]
- 30.Walmsley S, Montaner J, Hill A, et al. Nucleoside reverse transcriptase inhibitor treatment as a risk factor for hyperlipidemia: results from the FOCUS trial. Antiviral Ther. 2002;7 [Google Scholar]
- 31.Marzolini C, Troillet N, Telenti A, et al. Efavirenz decreases methadone blood concentrations. AIDS. 2000;14:1291–1292. doi: 10.1097/00002030-200006160-00036. [DOI] [PubMed] [Google Scholar]
- 32.Clarke SM, Mulcahy FM, Tija J, et al. The pharmacokinetics of methadone in HIV-positive patients receiving the non-nucleoside reverse transcriptase inhibitor efavirenz. Br J Clin Pharmacol. 2001;51:213–217. doi: 10.1046/j.1365-2125.2001.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munsiff AV, Einstein Coll A, Patel J. Regimens with once daily ritonavir + Fortovase are highly effective in PI-experienced HIV-HCV co-infected patients on Methadone. Program and abstracts of the 39th Annual Meeting of the Infectious Diseases Society of America (IDSA); October 25-28; San Francisco, California: 2001. Abstract 684. [Google Scholar]
- 34. Fortovase. EU Summary of Product Characteristics. Available at: www.emea.eu.int/humandocs/PDFs/EPAR/Fortovase/H-178-PI-en.pdf Accessed April 21, 2006.
- 35.Durant J, Clevenbergh P, Garraffo R, et al. Importance of protease inhibitor plasma levels in HIV-infected patients treated with genotypic-guided therapy: pharmacological data from the Viradapt study. AIDS. 2000;14:1333–1339. doi: 10.1097/00002030-200007070-00005. [DOI] [PubMed] [Google Scholar]
- 36.Schutz M, Goodly J, Chen K, et al. Lack of correlation between saquinavir trough levels and adverse events or clinical outcomes in saquinavir cohort of focus study. Program and abstracts of the 41st Annual Meeting of the Infectious Diseases Society of America (IDSA); October 9-12; San Diego: 2003. Poster 1059. [Google Scholar]
- 37.Back DJ, Khoo SH, Gibbons SE, et al. The role of therapeutic drug monitoring in treatment of HIV infection. Br J Clin Pharmacol. 2001;51:301–308. doi: 10.1046/j.1365-2125.2001.01380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]