

Abstract
Field asymmetric waveform ion mobility spectrometry (FAIMS) has emerged as a powerful tool of broad utility for separation and characterization of gas-phase ions, especially in conjunction with mass spectrometry (MS). In FAIMS, ions are filtered by the dependence of mobility on electric field while carried by gas flow through the analytical gap between two electrodes of either planar (p-) or cylindrical (c-) geometry. Most FAIMS/MS systems employ c-FAIMS because of its ease of coupling to MS, yet the merits of two geometries have not been compared in detail. Here, a priori simulations reveal that reducing the FAIMS curvature always improves resolution at equal sensitivity. In particular, the resolving power of p-FAIMS exceeds that of c-FAIMS, typically by a factor of 2 - 4 depending on the ion species and carrier gas. We have constructed a new planar FAIMS incorporating a curtain plate interface for effective operation with an ESI ion source and joined to MS using an ion funnel interface with a novel slit aperture. The resolution increases up to 4-fold over existing c-FAIMS, even though the analysis is ∼2 times faster. This allows separation of species not feasible in previous FAIMS studies, e.g., protonated leucine and isoleucine or new bradykinin isomers. The improvement for protein conformers (of ubiquitin) is less significant, possibly because of multiple unresolved geometries.
Introduction
The need to analyze samples of ever-growing complexity, primarily in biological applications, has been driving the continued innovation in separation technologies.1,2 Separations in condensed media such as liquid chromatography (LC),3 capillary electrophoresis,4 strong cation exchange (SCX),5 and gel electrophoresis6 have been developed extensively and are well-known in the field. These techniques and particularly their combinations are exceptionally powerful, with the peak capacity of two-dimensional methods7-9 reaching ∼103 - 104. However, separations in condensed phases where molecular diffusion is slow inevitably require a long time (many hours for SCX/LC or 2-D gel7-9), limiting the throughput to several samples per day.
The desire for greater throughput has intensified the interest in gas-phase separations based on ion mobility, which are extremely fast (typically taking milliseconds to seconds) thanks to the high speed of ion motion in gases. The technique of ion mobility spectrometry (IMS) established in 1970s sorts ions by mobility (K) at low electric fields.10-13 The other approach of field asymmetric waveform IMS (FAIMS) was known since 1980s,14-18 but became topical in the last few years.19-48 Most recently, FAIMS was coupled to LC49 or IMS, creating first 2-D gas-phase separations.50 In FAIMS, ions are distinguished by the difference between mobilities at high and low electric fields (E), which in general differ as ion mobilities in gases depend on the field:
| (1) |
where N is the gas number density. In a typical FAIMS regime (E/N < 100 Td), the terms up to b(E/N)4 generally represent K(E) with sufficient accuracy. Depending on the relative values of a and b coefficients, K(E) may increase, decrease, or first increase and then decrease, with corresponding ions classified as types A, C, and B, respectively. These designations are not absolute, but depend on the buffer gas: ions exhibiting A-type behavior in one gas may become C-type in another.23 For example,23 the protonated heptadecanoic acid is A-type in CO2 or N2O but C-type in O2 or N2. Most FAIMS studies have used air or N2 gases, but gas mixtures such as He/CO2, N2/CO2, and especially He/N2 are attracting increasing interest. The resolution and sensitivity of FAIMS using binary and ternary mixtures is often better than that with any individual component,25-27,33,48 because of non-Blanc behavior of ion mobilities at high electric fields.48 Addition of volatile vapors to a homomolecular buffer gas produces a similar effect.41
FAIMS is implemented by flowing a gas stream containing ions through the space between two electrodes (the “analytical gap”) carrying a periodic asymmetric waveform VD(t) plus a DC compensation voltage (CV).19,20 The waveform induces ion oscillation in the gap, and the displacements during positive and negative segments of VD(t) slightly differ because of unequal mobilities at high and low E. This would result in ions migrating toward one of the electrodes (depending on the ion type) and neutralizing on impact. For a particular species, a certain CV may produce an offsetting drift, allowing ions to remain in equilibrium inside the gap.19,20 Scanning the CV reveals the spectrum of ionic mixture. Earliest FAIMS devices had a planar analytical gap confined between two parallel electrodes (p-FAIMS).14-17 An alternative configuration with ions separated in the annular space between two coaxial cylinders (c-FAIMS) was demonstrated subsequently.16,18-20 A geometry comprising two concentric spheres was also proposed.21 While the spherical design had not been tested in experiment, a “dome” geometry (cFAIMS plus a hemispherical section) has become widely used.22-35 The advent of micromachined FAIMS “chip” gave raise to miniaturized p-FAIMS devices.38-43
The electric field in a curved analytical gap is inhomogeneous, and thus may focus ions to the gap median. However, VD(t) of either polarity focuses ions of one type (A or C) only, but removes those of the other type from the gap.19,20 Hence, acquiring a full CV spectrum using c-FAIMS requires scanning CV with both VD(t) polarities, which halves the instrumental duty cycle. Further, the focusing strength depends on the slope of K(E) curve over the relevant E range: ions with a near-flat K(E) (often belonging to type B) focus poorly and are depleted in c-FAIMS. This phenomenon distorts the CV spectra measured using c-FAIMS, discriminating against ions with lower absolute CV.51 Homogeneous electric fields cannot focus ions, so the above adverse behaviors are not encountered in p-FAIMS.
Both planar and cylindrical (“dome” and “side-by-side”21,52) geometries have been implemented in commercial FAIMS systems, the DMS41,43 (Sionex, Bedford, MA) and Selectra33,34 (Ionalytics, Ottawa, Canada), with the number and diversity of applications growing swiftly. These span the fields such as detection of drugs, explosives, and chemical warfare agents,31,35,36,40 environmental monitoring,22,24,25,27,29,39 bacterial typing,42 product quality assurance,32 natural resource management,30 and biomedical research.33 However, the utility of FAIMS has been restricted by resolving power46,47 (R) of ∼10 - 20 (compared to R ∼150 in best IMS designs).53-55 This has limited the separation of even simple mixtures, e.g., H34SO4− and 35ClO4− isobars24,27 or three phthalic acid isomers.26 While those species can still be distinguished via buffer gas modifications,26,27 such changes have to be made for each individual case by trial and error, and the overall peak capacity may not increase. The resolution may also be improved by reducing the analytical gap width (g) or the waveform frequency (wc),47 superposing a low-frequency ripple56 onto VD(t), or varying the hemispherical gap width in “dome” design,35 but all those approaches also diminish sensitivity.35,56 Importantly, the resolution cannot be raised beyond a certain limit (depending on CV and the VD(t) amplitude known as the dispersion voltage, Vmax) even as ion transmission through FAIMS approaches zero.56
The remaining instrumental parameter is the gap curvature, which (for c-FAIMS) depends on the internal (rin) and external (rout) electrode radii. Increasing rin (at constant g = 2 mm) from 4 to 12 mm raises R several times,57 because weaker ion focusing at lower curvature narrows the CV range of ions that may pass the gap. Extrapolating that trend to rin ∞, the p-FAIMS with no ion focusing was surmised to have the highest R, but this was not substantiated or quantified in either theory or experiment. An experimental and modeling investigation of p- and c- FAIMS has shown the latter to provide a higher transmission for ions with strong focusing properties (i.e., high |CV|) and lower transmission for other ions.45 However, the compared devices were dissimilar in several key aspects in addition to their curvature. First, p- and c-FAIMS used {g = 0.5 mm; wc = 1.3 MHz} and {g = 2 mm; wc = 200 - 250 kHz}, respectively, and performance is quite sensitive to those parameters.47 Second, the gap length (L) was 15 mm for p- but 90 mm for c-FAIMS and the ion residence times (tres) differed by a factor of >100: ∼2 ms vs. ∼360 ms respectively, which unduly favored the p-FAIMS in a sensitivity comparison.46,47 While the inevitable trade-off between resolution and ion transmission means that sensitivities of different designs should be studied for a fixed R, the resolutions achievable for p- and c-FAIMS have not been compared previously. The model45 has also neglected the space-charge effects and anisotropy of ion diffusion that are important for a realistic description of FAIMS performance,46 and made other approximations needed for a closed-form mathematical solution.
The operational regimes of p- and c- FAIMS matched in Ref. [45] are typical for existing devices (e.g., DMS and Selectra): for p-FAIMS, g is 3 - 4 times smaller (0.5 vs. 1.5 - 2 mm), wc is ∼2 - 10 times higher (1 - 2 MHz vs. 80 - 750 kHz), and tres are shorter by two orders of magnitude (∼1 - 5 vs. ∼100 - 500 ms). However, these drastic differences reflect a historical legacy not related to inherent properties of the two geometries.
While FAIMS and IMS may work as stand-alone analyzers with a Faraday cup detector,14-16,19,20,36 much interest involves their coupling to mass-spectrometry (MS). The hemispherical section of “dome” FAIMS that collects ions separated in the cylindrical part provides a natural MS interface, resulting in a broad acceptance of that design.22-35 In a planar analytical gap, ions freely spread laterally to the gas flow and only a minor fraction enters a small orifice leading to MS. This has compelled virtually all FAIMS/MS systems to adopt c-FAIMS, regardless of its intrinsic merits vs. p-FAIMS.
Here we employ first-principles calculations to compare the performance of planar and cylindrical FAIMS a priori, keeping other factors equal. The modeling has revealed a fundamental advantage of p-FAIMS in terms of resolution-sensitivity balance, in addition to the duty cycle gain and lack of discrimination discussed above. Guided by those simulations, we have constructed a new p-FAIMS with resolving power several times that of any known FAIMS. This device has been coupled to MS using a novel “slit-aperture” interface, enabling practical high-resolution FAIMS/MS analyses. The improved peak capacity allowed separating ionic species previously indistinguishable by FAIMS (or IMS).
Theoretical foundation
Computational treatment
The FAIMS model used herein has been described and extensively validated by measurements.46,47,56 The overall concept follows SIMION:58 a representative ensemble of ions is propagated through the device by trajectory integration. The statistics of those passed is aggregated, with ion transmission given by the ratio of the number of exiting and initial trajectories. Evaluation of this ratio as a function of CV produces the peak profile and yields the full width at half-maximum (fwhm) and R, defined as (peak CV)/(fwhm).
The underlying physics incorporates ion diffusion (accounting for the high-field and anisotropic components), Coulomb repulsion of ions within the ensemble,46 and a non-uniform profile of gas flow velocity across the analytical gap.56 This capability was used to probe the dependence of FAIMS response on various design and operational parameters, including g, L, the VD(t) profile, wc, and the identity and flow velocity of buffer gas.46,47,56 Those assessments were restricted to c-FAIMS with “median radius” rcen = (rin + rout)/2 = 8 mm (adopted in most analyzers including Selectra), but the model would work for any geometry as long as E(t) and gas flow are defined at any point. Present simulations are for the bisinusoidal waveform46,47,56
| (2) |
with f = 2 as used in Selectra, DMS, and the new p-FAIMS reported here. This value of f is optimum for FAIMS performance.47 The FAIMS response substantially depends46,47,56 on the ion current intensity, I. Here we model the extremes of (i) low I where the Coulomb repulsion is negligible and (ii) saturation where the FAIMS space-charge capacity is filled. The results turn out similar, thus the conclusions would hold in any real situation. While all calculations below are for homomolecular buffers, they could be extended to heteromolecular media via a formalism treating non-Blanc effects.48
Simulations as a function of FAIMS analyzer curvature and their experimental validation
Since the present computational model was previously corroborated for rcen = 8 mm only,46,47,56 first we test its performance as a function of gap curvature using the data for bromochloroacetate (BCA) anion (m/z = −173 Da) in N2 carrier gas.57 In practice, increasing rcen from 5 to 9 mm and further to 13 mm (at fixed g = 2 mm) narrows the CV spectral peaks at any Vmax in the −(1.80 -3.96) kV range studied,57 as shown for minimum and maximum values (Fig. 1 a-f). This behavior is due to a weaker ion focusing at lower curvature, as described above. However, at low |Vmax| the peaks cease to narrow above certain rin, e.g., at Vmax = −1.80 kV, once fwhm of 0.2 V is reached at rin ∼8 mm (Fig. 1 c, e). This happens because focusing (always weak at low |Vmax|) becomes negligible at low curvature. Further, increasing |Vmax| (at any curvature) has not significantly improved R; while CVs increase, peaks widen nearly in proportion because of stronger ion focusing at higher |Vmax|.57
Fig. 1.

CV peaks for BCA− at Vmax = −1.80 kV (left column) and −3.96 kV (right column), for c-FAIMS geometries with rcen = 5 mm (a, b), 9 mm (c, d), and 13 mm (e, f), and p-FAIMS (g, h); g = 2 mm for all. Shown are measurements (circles) and present simulations (lines) in the limits of zero (dashed) and saturated (solid) ion current. In (c), (e), (g), and (h), the solid and dashed lines coincide within the computational accuracy. The calculated (c) and experimental (e) values of fwhm (V) and the resolving power are listed in each panel.
We have simulated the evolution of CV spectra for BCA− as a function of Vmax and rin (for experimental57 wc = 750 kHz, T = 298 K, a = 7.984 × 10−6 Td−2, b = −3.049 × 10−10 Td−4; assuming57 tres = 0.2 s and K(0) = 1.7 cm2/(V×s) at 298 K), Fig. 1 a - f. As usual, features at saturated I are narrower than those in the limit of I fl 0, because Coulomb repulsion preferentially removes from the analytical gap the ions with CVs near the edges of transmission band that would otherwise still pass FAIMS.46 At high Vmax = −3.96 kV, the measured peaks lie between the two theoretical limits. At lowest Vmax = −1.8 kV, the calculated peaks appear slightly broader than the measured ones (in Fig. 1 c, e), but the latter are clearly narrowed by having just two-three points across the peak. Overall, the agreement with all observed dependences of peak profiles (and hence R) on both Vmax and rin is very good.
We have then extended the model to planar FAIMS geometry (Fig. 1 g, h). When a peak has ceased to sharpen with increasing rcen because of negligible ion focusing at low curvature, it obviously would not narrow further in a p-FAIMS. This is the case at the lowest Vmax = −1.8 kV, where fwhm = 0.2 V (Fig. 1g) as in c-FAIMS with rcen = 9 - 13 mm (Fig. 1 c, e). To the contrary, at high Vmax (−3.96 kV) the computed peak in p-FAIMS (Fig. 1h) is ∼5 times narrower than that in any c-FAIMS considered previously (Fig. 1f) and ∼10 times narrower than that in the Selectra device (Fig. 1d). This is not surprising: absence of ion focusing in planar FAIMS means a constant peak width (here 0.2 V) regardless of Vmax, thus the resolution advantage of p-FAIMS grows with increasing |Vmax|. The most striking result is the potential for reaching R ∼100 (Fig. 1h), which would bring the FAIMS resolving power close to that of IMS.
One critical difference between p- and c- FAIMS is in the dependence of peak widths on the ion residence time. In c-FAIMS, filtering out ions with CVs outside of a finite range allowing equilibrium in the gap requires a certain tres (∼50 ms in Selectra), and further FAIMS action does not improve resolution. 47 In p-FAIMS, there is a single CV permitting ion equilibrium, and ions with even a tiny CV difference will eventually be eliminated given sufficient time. Hence, longer separation times would increase R (in principle) indefinitely. To test the ability of present model to quantify that effect, we have simulated the toluene peaks in micromachined p-FAIMS38 with wc = 2 MHz and Vmax = 1.1 kV (Fig. 2). Knowing the gap dimensions38,61 of L = 15 mm, S = 10 mm (span), and g = 0.5 mm, the gas flow rate (1 - 5 L/min) may be converted into linear velocity and further into tres (0.9 - 4.5 ms). The evolution of features over that timescale was modeled assuming {K(0) = 2.3 cm2/(V×s) at 298 K, a = 1.28 × 10−5 Td−2, b = −8.2 × 10−10 Td−4}, with K(0) derived from IMS62 and {a, b} obtained from FAIMS data.43 Excellent agreement between predicted and measured peak profiles (Fig. 2) supports the applicability of the present model to p-FAIMS over a range of separation times.
Fig. 2.
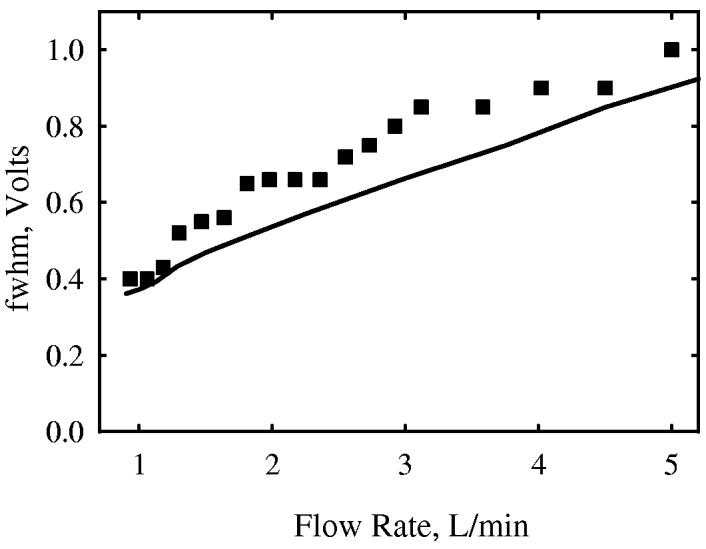
Peak width for toluene ions filtered by micromachined p-FAIMS as a function of gas flow rate: measurements (circles) and present calculation (line).
Relative intrinsic merits of planar and cylindrical FAIMS
With the simulation validated for FAIMS analyzers of any (and no) curvature, we proceed to compare the response of c- and pFAIMS with all other factors equal. Benchmarking will be made for the exemplary system of deprotonated leucine anion in air, for which the performance of c-FAIMS (with rcen = 8 mm) was extensively modeled,46,47,56 in agreement with experiments. As previously, we assume conditions of (Vmax = −3.3 kV, wc = 210 kHz, L = 30 mm, tres = 0.2 s, T = 298 K) and use {K(0) = 2.18 cm2/(V×s) at 298 K, a = 5.43 × 10−6 Td−2, b = −1.85 × 10−10 Td−4}46,47 derived from FAIMS measurements.59,60 Similarly to the behavior in Fig. 1, increasing rin (at constant g = 2 mm, in the I 0 limit) raises R from 10 at rcen = 8 mm to ∼45 at rcen = 73 mm (Fig. 3a). The magnitude of improvement (a factor of 4.5) is less than ∼10 for BCA− (Fig. 1h vs. 1d): the ion focusing for (leucine − H)− is weaker because of (i) lower |Vmax| = 3.3 kV vs. 3.96 kV for BCA− and (ii) lower CV at equal Vmax due to a less steep K(E) curve with the value of a equal to ∼70% of that for BCA−. A weaker ion focusing with increasing rcen also leads to greater losses, reducing ion transmission from 96% at R = 10 to 2% at R ∼ 45 (Fig. 3a). Further curvature reduction to p-FAIMS results in yet lower transmission and has not been modeled.
Fig. 3.
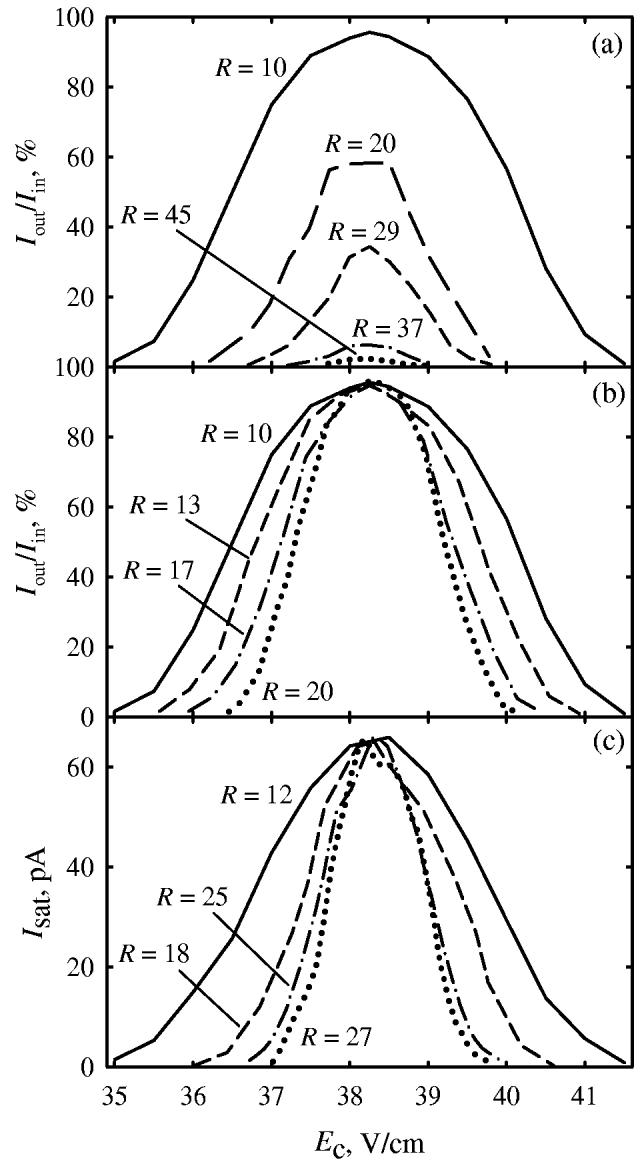
Simulated CV peaks for (leucine – H)− as a function of analytical gap curvature, for the limits of zero ion current (a, b) and saturated current (c). From outside to inside, curves are for rcen = 8, 13, 18, 38, and 73 mm in (a); 8, 18, 38, and ∞ (p-FAIMS) in (b), and 8, 19, 49, and ∞ in (c). The gap width is fixed at g = 2 mm (a) or adjusted to keep constant sensitivity (b, c), with respective g of 2, 2.7, 3.5, and 8.5 mm (b) and 2, 2.4, 2.7, and 2.7 mm (c). To account for a variable g, CV is expressed via compensation field, Ec = (CV)/g. The computed values of resolving power are given in all cases.
Hence the question is: how to exploit the resolution advantage of (more) planar FAIMS without sacrificing sensitivity? This may be achieved by widening the gap while reducing its curvature. For example, the ion transmission of ∼95% (at I 0) may be retained by increasing g from 2 to ∼8.5 mm as rcen is raised from 8 mm to infinity (Fig. 3b). Then R improves less dramatically, doubling from 10 for standard c-FAIMS to 20 for p-FAIMS. This also occurs in the other limit of saturated I (Fig. 3c), with R at equal ion current increasing from 12 to ∼27. Again, the effect is greater in cases of higher CV (that correspond to stronger ion focusing in c-FAIMS). Therefore the transition to planar geometry could improve R at any ion intensity twofold or more with no sensitivity impairment. Accordingly, the curve summarizing the trade-off between FAIMS resolution and sensitivity (Fig. 4) moves to the right when the gap curvature is reduced. The greatest gain is at highest sensitivity, moderating at lower sensitivity (and higher R). This happens because higher FAIMS resolution is achieved by constraining the gap, meaning a lower curvature of c-FAIMS. The advantage of p-FAIMS is yet greater when compared vs. c-FAIMS with R controlled using ripple56 (Fig. 4).
Fig. 4.

Calculated resolution-sensitivity trade-off (at I 0) for (leucine – H)− in p-FAIMS (empty circles) and c-FAIMS with Rcen = 8 mm (filled circles) [56], both obtained by varying the gap width. Triangles are for the trade-off obtained in c-FAIMS by scaling a 60-Hz “ripple” harmonic superposed on the asymmetric waveform [56]. Lines are quadratic regressions through the data for each curve.
Implementation of new high-resolution planar FAIMS
While, by theory, p-FAIMS allows a higher resolution, it is not achievable using existing designs with insufficient ion residence times. In fact, the resolution of DMS p-FAIMS was generally worse than that of Selectra c-FAIMS, as the difference of tres by a factor of ∼100 outweighed the intrinsic resolution advantage. Also, the DMS chips do not provide for the desolvation of ions generated by ESI, and thus are less suitable for ESI-MS coupling.
Hence we have constructed a new p-FAIMS analyzer permitting much longer separations (Fig. 5). In this device, the analytical gap is formed by two polished stainless steel plates secured inside a plastic (peek) enclosure and precisely aligned by ceramic spacers. The gap dimensions are g = 2 mm (same as in most c-FAIMS systems including Selectra), L ∼50 mm, and S = 20 mm. Thus, the cross-sectional area is 40 vs. 113 mm2 in Selectra, meaning a proportionately shorter tres at equal gas flow rate, e.g., ∼0.1 s vs. ∼0.2 s typical in Selectra. Still, that exceeds the tres in previous p-FAIMS by ∼20 - 100 times, which should improve resolution.
Fig. 5.

Scheme of the new planar FAIMS analyzer coupled to MS using an ion funnel interface.
The new p-FAIMS also features an effective ESI/FAIMS interface (Fig. 5). Ions enter the gap through a curtain plate chamber resembling that in Selectra,57 with curtain plate and sampling apertures of 2.5 and 1.5 mm, respectively. The carrier gas enters through a side opening in the chamber and splits into two streams: the major (∼70 %) flows out of the curtain plate orifice and desolvates incoming ions while minor carries ions into the sampling orifice and through the gap (Fig. 5). To enable FAIMS/MS analyses, the device was installed in a custom peek holder secured to the inlet of time-of-flight (ToF) mass spectrometer. The FAIMS exit opposes the sampling aperture, leaving a break of ∼0.5 mm for electrical insulation. The gas is supplied to the device by a unit that formulates gas mixtures of up to three components (each pre-dried by hygroscopic filters) and controls the flow rate in the 0.5 - 5 L/min range. Here we employed N2 and He/N2 mixtures with 10 - 50% He (v) at total flow rates of 2 - 3 L/min. Then the flow through the gap is ∼0.5 - 0.8 L/min, approximately matching the MS inlet conductance. The ESI emitter is mounted a few mm away from the inlet on an X-Y translation stage for fine position adjustment (Fig. 5). Samples are infused to the emitter (at 0.4 μL/min) through a metal union using a microsyringe driven by a pump (kd Scientific, Holliston, MA).
The bisinusoidal asymmetric waveform (eq. 2) with wc = 750 kHz and f = 2 is produced by a power supply similar to that in Selectra, that adds the 750 and 1500 kHz harmonics (with 2 : 1 amplitude ratio) output by resonating LC circuits.63 The CV is generated by a programmable DC power supply. The VD(t) and CV are applied to opposite electrodes, simplifying the arrangement in Selectra where they are co-applied to the internal electrode. The Vmax value is adjustable from 2.3 to 4 kV, here we use 3.9 kV. The whole FAIMS unit is biased at 190 V, the curtain plate is at 1 kV, and the ESI emitter is at ∼3 kV (referenced to ground). Spectra are obtained by scanning the desired CV range with a speed of 0.5 - 5 V/min. All instrumental parameters are controlled by custom software resident on a dedicated PC. Since there is no ion focusing, the VD(t) polarity is immaterial and (unlike for c-FAIMS) need not be switched depending on the ion type.
The ToF MS is LC/MSD TOF (Agilent Technologies, Palo Alto, CA) with the capillary-skimmer cone at the front end replaced by electrodynamic ion funnel that significantly raises the ion utilization efficiency at API/MS interfaces.64-66 The present funnel has 100 round electrodes with IDs reducing from 25 mm at the “mouth” to 2 mm at the exit to MS, and features a jet disrupter66 that improves sensitivity. The funnel chamber is evacuated by a mechanical pump (pumping on the inlet-skimmer chamber in the original ToF). The capillary inlet normally leading into the ion funnel is replaced by an aperture cut through a 0.2 mm steel sheet (Fig. 5). We have tested two apertures - a circle of 0.43 mm diameter and a non-contiguous “slit” made of 11 circular 0.13-mm holes disposed uniformly along a 4-mm segment. These apertures have the same cross-sectional area of 0.145 mm2, resulting in equal funnel chamber pressure of 2 Torr. The rationale for slit aperture is to increase the spatial overlap with the ribbon-shaped ion beam exiting p-FAIMS. The funnel focuses and guides broad or divergent ion beams in a pseudopotential well created by combination of DC and RF voltages applied to the electrodes.64-66 Here the voltages were 190 V on the MS aperture, 187 V to 40 V DC along the funnel, and 175 V on the jet disrupter; the peak RF amplitude was 45 V (at 560 kHz). All voltages above are for positive ions, and should be reversed for negative ion analyses.
For benchmarking purposes, p-FAIMS was replaced by the Selectra c-FAIMS with no other changes to the system. The exit orifice of Selectra is circular, hence the circular MS inlet aperture was used. The VD(t) waveform and gap width in Selectra and present p-FAIMS are identical, allowing a direct performance comparison (Vmax = 4 kV or −4 kV was chosen for A-and C-type ions, respectively). The resolution of Selectra may be tuned by longitudinal translation of the internal electrode that adjusts the axial gap width in hemispherical region (gH) from 1.7 to 2.7 mm in 0.1 mm increments.35 Increasing gH above g = 2 mm improves the FAIMS resolution but lowers sensitivity.35 Here we used the maximum practical35,50 gH = 2.3 - 2.5 mm to provide a stringent benchmark for resolution of new p-FAIMS; the comparison with Selectra operated at more typical gH ∼ 2.1 mm50 would favor p-FAIMS even more.
Increased resolution of new planar FAIMS
First, we compared the merits of two MS inlet apertures described above. The ion signal with the “slit” was consistently >250% of that with the circular aperture (Fig. 6). While this clearly shows the advantage of elongated apertures at the p-FAIMS/MS interface, our present “slit” design can be refined further, and its optimization is in progress.
Fig. 6.
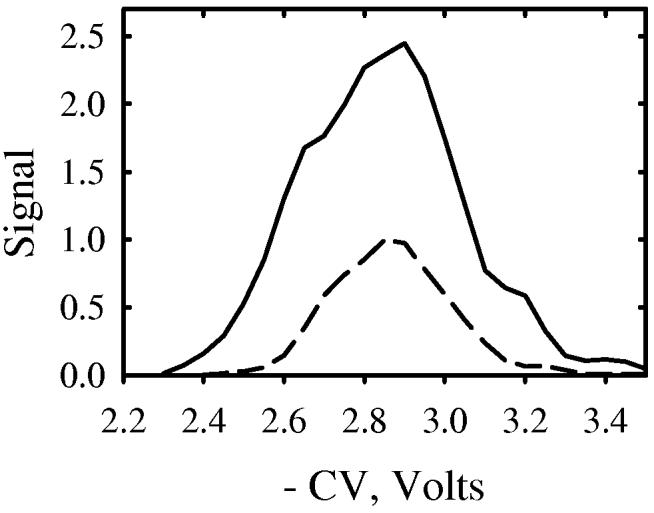
CV spectra of H+reserpine measured using p-FAIMS with the circular (dash) and “slit” (solid line) MS inlet apertures.
The separation performance of new p-FAIMS has been characterized using several representative analytes: 10 μM reserpine, 50 μM 1:1 leucine/isoleucine, 10 μM bradykinin, and 50 μM bovine ubiquitin; all obtained from Sigma Aldrich. For infusion into ESI, those chemicals were dissolved in 50:49:1 methanol:deionized water:acetic acid.
Reserpine is a customary standard for MS system evaluation. With N2 buffer, the CV spectrum of H+reserpine (m/z = 609 Da) comprises a dominant feature at −3 V and two lesser ones at −4 V and ∼ −4.5 V (Fig. 6). To compare the resolution of two FAIMS geometries, we have chosen the major peak. As He content grows from 0 to 50%, the CV in c-FAIMS moves from −3 V to −7 V while the width increases from 0.6 V to 0.9 V (Fig. 7a). The peak broadening at higher |CV| is typical of c-FAIMS; stronger ion focusing at greater CV widens the CV transmission window. This does not happen in p-FAIMS; in fact the features narrow from 0.5 V in N2 to 0.3 V at 50% He (Fig. 7b) because at higher He concentration K(0) increases, effectively constraining the gap. Overall, the resolution advantage of p-FAIMS is by a factor of 1.2 - 2.7, which is close to the above theoretical prediction.
Fig. 7.

CV spectra of H+reserpine (a, b) and H+leucine/H+isoleucine (c, d) acquired using Selectra (a, c) [with Vmax = −4 kV (a) and 4 kV (c)] and p-FAIMS (b, d), in N2 (solid line) and He/N2 with He fraction of 20% (long dash), 30% (short dash), 40% (dash-dot), and 50% (dotted). For clarity, we plot only the major peak for reserpine (at CV = −3 V in Fig. 6a). Spectra in (b, d) were aligned with those in (a, c) to match the CVs at peak apexes. Measured fwhm values are given for all features.
IMS and FAIMS (typically in conjunction with MS) are often employed to characterize isomeric mixtures. For example, a prototypical bioanalytical problem is distinguishing leucine and isoleucine. These amino acids were previously separated by c-FAIMS as negative deprotonated ions,59 but not as positive protonated ions (m/z = 132 Da) for which the forms of K(E) (and thus CVs) differ less.60 We also could not separate H+isoleucine from H+leucine using Selectra (Fig. 7c). In N2, there is a single peak of ∼0.7 V width at CV = −9.6 V, consistent with previous data.59,60 As discussed above, ions in He/N2 tend to exhibit non-Blanc effects that often improve separation.33,48 Here, CV shifts with increasing He fraction to more negative values up to −11.3 V at ∼30% He, then the trend reverses with −9.7 V reached at 50% He (Fig. 7c). Such a maximum of absolute CV (evidencing non-Blanc behavior) is similar to that for many other species48 in He/N2, but the peak width remains at ∼0.7 - 0.8 V and there is no separation improvement. In p-FAIMS, the peaks are consistently sharper (0.25 - 0.55 V wide) at any He fraction (Fig. 7d). This suffices to clearly separate the two isomers at 50% He, though not at lower He concentrations. The increase of R is from 12 - 15 in c-FAIMS (Fig. 7c) to 20 - 42 in p-FAIMS (Fig. 7d), i.e., by a factor of 2.5 on average: again close to theoretical predictions.
One promising FAIMS application is the analysis of proteolytic digests.28,49,50,67 Global proteome digests can consist of 105 - 106 different peptides,68 and the need for their rapid characterization is the major driver for continued innovation in separations science. The speed of gas-phase ion mobility methods makes them extremely attractive for high-throughput proteomics platforms based on multidimensional separations, such as LC/FAIMS/MS49 and FAIMS/IMS/MS.50 Ion mobility approaches may also distinguish isomeric/isobaric peptides, including sequence inversions.69 Since tryptic peptides cover a limited CV range, a modest FAIMS resolution has particularly constrained its utility in proteomics. A classical model peptide in MS and IMS studies is bradykinin (RPPGFSPFR), which under soft ESI conditions produces mainly H2+bradykinin70 (m/z = 531 Da). Two conformers of this ion (minor I and major II at higher |CV|) were distinguished by Selectra,70 the result reproduced here (Fig. 8). However, combined FAIMS and H/D exchange data expose four different species.70 Using p-FAIMS, we resolved six features (a - f) in the CV spectrum for H2+bradykinin (Fig. 8). The isomers b and d match respectively I and II, but we discover new isomers a in the ledge of I, c hidden between I and II, and e and f in the extended tail of II (Fig. 8). Well-defined features b - f are ∼0.3 - 0.4 V wide meaning R ∼25 - 35, which exceeds the R of Selectra by a factor of 3 - 4 (as expected from simulations). The species a, b, c, and d apparently correspond to 4, 3, 2, and 1 distinguished by FAIMS-H/D exchange,70 whereas e and f were not reported previously.
Fig. 8.

CV spectra of H2+bradykinin obtained using Selectra with Vmax = −4 kV (dotted) and p-FAIMS (full spectrum - solid line, smaller features expanded by × 25 - dashed line).
Finally, we have applied the new p-FAIMS to characterize macromolecular conformers. Proteins and other flexible macromolecules may adopt a plurality of 3-D structures in solution that are partly conserved by soft ionization mechanisms such as ESI.71-73 This allows using IMS and FAIMS to explore protein polymorphism and isomeric transitions (denaturation) in solution depending on the temperature, solvent composition, acidity (ionic strength), and presence of specific chemicals, including as a function of time.71-73 The growing focus on misfolded proteins (prions) as the cause of transmissible spongiform encephalopathies, Alzheimer's, and other neurodegenerative diseases has made this research increasingly topical.74 All methods for protein structure elucidation (in either solution or gas phase) face a fundamental challenge: any protein exists as an ensemble of numerous similar but slightly different geometries. This causes blurring of NMR structures for proteins in solution, a diminished resolution of LC or CE separations, and broadening of IMS peaks that limits R to ∼30 - 35, whatever the instrumental performance.71,73,75 The resolution of FAIMS should be affected likewise, but the actual limitation could not be determined because of low instrumental R.
Ubiquitin is a common small protein (76 residues, 8565 Da for bovine) often used as a model for biophysical investigations. ESI of acidified ubiquitin solutions generally yields ions of charge states (z) of 6+ to 14+, with most having several isomers distinguished by IMS71 and FAIMS.72,76-78 Here we focus on z = 10 - 13, where the protein is largely unfolded (by Coulomb repulsion) resulting in simpler CV spectra that allow a direct comparison between c- and p-FAIMS. In agreement with earlier measurements,72,78 the data acquired using Selectra show peaks at CV = −(8 - 9) V (Fig. 9). The peak widths are 0.8 - 1.0 V for 10+ and 13+ and ∼1.5 V for 11+ and 12+ that exhibit poorly resolved features,72,78 also with fwhm of 0.8 - 1.0 V as determined by Gaussian peak fitting. Switching to p-FAIMS reduces the fwhm for 10+ and 13+ to 0.4 - 0.7 V, but reveals no new conformers (Fig. 9). Four and two features are partly resolved for 11+ and 12+, respectively, with fwhm of 0.3 - 0.7 V. These more structured spectra are close to those measured77 using Selectra at Vmax = −4.4 kV, with peaks ∼0.4 - 0.5 V wide. Hence the resolution advantage of p-FAIMS for ubiqutin is only ∼1.5 - 2 times, i.e., less than that for bradykinin with similar CVs (Fig. 8) though comparable to that for leucine/isoleucine (Fig. 7 c, d). These data suggest ∼0.3 - 0.4 V as the minimum peak width for protein ions set by multiplicity of exact geometries making up a “single” conformer; firm clarification of this issue is pending detailed studies involving other proteins.
Fig. 9.
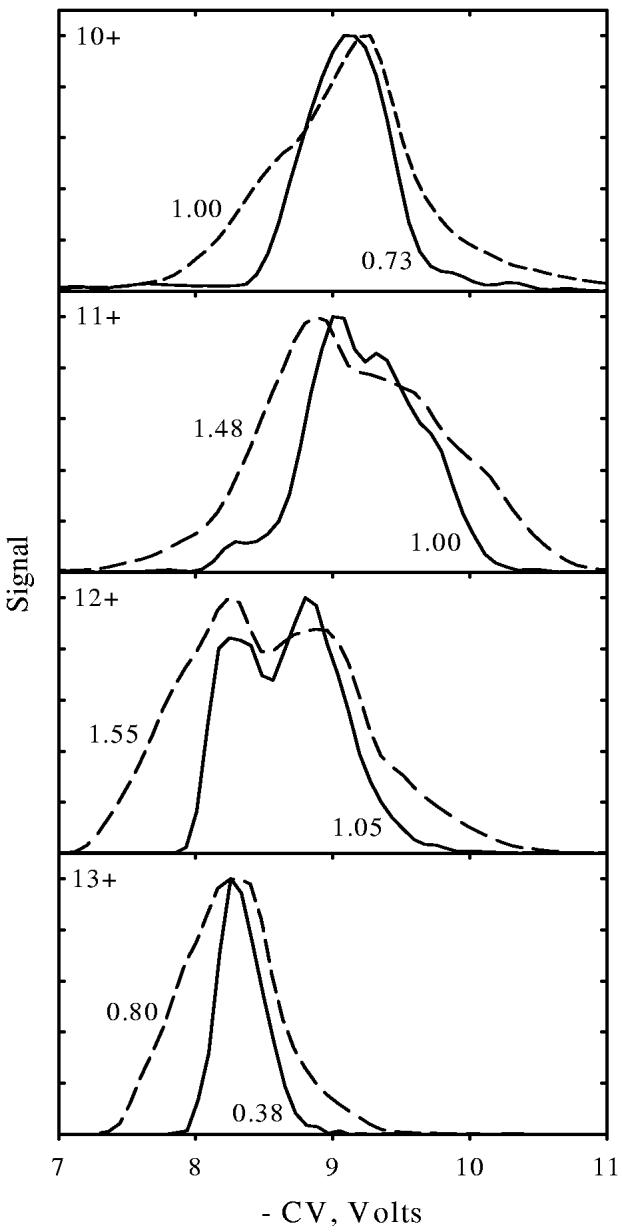
CV spectra for protonated bovine ubiquitin (z = 10 - 13) obtained using Selectra with Vmax = −4 kV (dash) and p-FAIMS (solid line). Measured fwhm are indicated.
Conclusion
A comprehensive modeling of FAIMS separations based on first-principles molecular dynamics has revealed a uniform improvement of performance (in terms of resolution/sensitivity trade-off) with reduction of analytical gap curvature. In particular, planar (p-) FAIMS is superior to cylindrical (c-) FAIMS, always providing a higher resolving power at equal ion utilization efficiency, for any ion current intensity. The resolution advantage increases with increasing absolute CV of an ion, with a gain of ∼2- to 4-fold over commercial c-FAIMS instrumentation for typical analytes and carrier gases. In contrast to c-FAIMS, the resolution of p-FAIMS is highly sensitive to the duration of analysis over any timescale, and, given sufficient time, much higher resolution may be achieved. These results are in a quantitative agreement with FAIMS measurements as a function of curvature and ion residence time.
Guided by simulations, we developed and demonstrated a planar FAIMS with separation timescales approaching that in Selectra c-FAIMS (∼0.1 s), i.e., ∼20 - 100 times longer than in existing miniature p-FAIMS devices. As predicted by theory, the resolution of new system exceeds that of c-FAIMS in all cases. The gain ranges from ∼20% to a factor of ∼4 (generally increasing at higher |CV|), with peaks narrowing to 0.25 V and the resolving power reaching 40. A proportional increase of peak capacity allows p-FAIMS to deal with significantly more complex analytes. In an exemplary isomeric separation, protonated leucine and isoleucine with a CV difference of ∼3.5 % could be distinguished by p-FAIMS but not Selectra. With application to conformational analyses, p-FAIMS has found four new conformers of the bradykinin dication, in addition to the two separated by c-FAIMS. The improvement for proteins (exemplified by ubiquitin) is lesser, likely because the resolution becomes limited by the multiplicity of conformers occupying neighboring minima on the potential energy surface rather than instrumental factors, as previously seen in IMS. Still, p-FAIMS has resolved ubiquitin conformations not found using c-FAIMS under otherwise identical conditions.
Broad use of planar FAIMS has been impeded by the lack of practical ESI and MS interfaces. We have integrated a curtain plate ESI interface into p-FAIMS and developed a new elongated slit-ion funnel MS interface that captures a ribbon-like ion beam delivered by p-FAIMS, converts it into a circular shape, and effectively conveys it to MS analyses. A “slit” aperture substantially increases the sensitivity of FAIMS/MS, and the ongoing optimization of aperture geometry will yield further improvements.
In conclusion, the new planar design enables high-resolution FAIMS with the peak capacity approaching that of modern IMS. Since FAIMS is intrinsically more orthogonal to MS than IMS is, this development makes FAIMS/MS a promising tool for many applications. Unlike in IMS, no a priori means to deduce ionic structures from FAIMS data currently exist. However, FAIMS pre-separation greatly augments the structural elucidation power of IMS,50,75 and the work to couple planar FAIMS to IMS/MS is in progress.
Acknowledgments
The authors thank Dr. Roger Guevremont and Dr. Randy Purves (Ionalytics, Ottawa, Canada) for numerous insightful discussions of FAIMS technology and providing the original experimental data. This work was supported by the NIH National Center for Research Resources (RR 18522). PNNL is a multi-program national laboratory operated by the Battelle Memorial Institute for the U.S. Department of Energy through Contract DE-ACO5-76RLO1830.
References
- 1.Smith RD, Shen YF, Tang KQ. Acc. Chem. Res. 2004;37:269. doi: 10.1021/ar0301330. [DOI] [PubMed] [Google Scholar]
- 2.Evans CR, Jorgenson JW. Anal. Bioanal. Chem. 2004;378:1952. doi: 10.1007/s00216-004-2516-2. [DOI] [PubMed] [Google Scholar]
- 3.Shen Y, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. Anal. Chem. 2002;74:4235. doi: 10.1021/ac0202280. [DOI] [PubMed] [Google Scholar]
- 4.Shen Y, Xiang F, Veenstra TD, Fung EN, Smith RD. Anal. Chem. 1999;71:5348. doi: 10.1021/ac9909305. [DOI] [PubMed] [Google Scholar]
- 5.Washburn MP, Wolters D, Yates JR. Nature Biotechnol. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 6.Shevchenko A, Wilm M, Vorm O, Mann M. Anal. Chem. 1996;68:850. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 7.Wolters DA, Washburn MP, Yates JR. Anal. Chem. 2001;73:5683. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 8.Shen YF, Jacobs JM, Camp DG, Fang RH, Moore RJ, Smith RD, Xiao WZ, Davis RW, Tompkins RG. Anal. Chem. 2004;76:1134. doi: 10.1021/ac034869m. [DOI] [PubMed] [Google Scholar]
- 9.Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. Proc. Natl. Acad. Sci. USA. 2000;97:9390. doi: 10.1073/pnas.160270797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill HH, Siems WF, Louis RH, McMinn DG. Anal. Chem. 1990;62:A1201. doi: 10.1021/ac00222a001. [DOI] [PubMed] [Google Scholar]
- 11.Bowers MT, Kemper PR, von Helden G, van Koppen PAM. Science. 1993;260:1446. doi: 10.1126/science.260.5113.1446. [DOI] [PubMed] [Google Scholar]
- 12.Shvartsburg AA, Hudgins RR, Dugourd P, Jarrold MF. Chem. Soc. Rev. 2001;30:26. [Google Scholar]
- 13.Hoaglund-Hyzer CS, Lee YJ, Counterman AE, Clemmer DE. Anal. Chem. 2002;74:992. doi: 10.1021/ac010837s. [DOI] [PubMed] [Google Scholar]
- 14.Gorshkov MP. 1982 USSR Inventor's Certificate # 966583. [Google Scholar]
- 15.Buryakov IA, Krylov EV, Soldatov VP. 1988 USSR Inventor's Certificate # 1412447. [Google Scholar]
- 16.Buryakov IA, Krylov EV, Soldatov VP. 1989 USSR Inventor's Certificate # 1485808. [Google Scholar]
- 17.Buryakov IA, Krylov EV, Nazarov EG, Rasulev UK. Int. J. Mass Spectrom. Ion Processes. 1993;128:143. [Google Scholar]
- 18.Carnahan BL, Tarassov AS. U.S. Patent # 5420424 1995
- 19.Purves RW, Guevremont R, Day S, Pipich CW, Matyjaszczyk MS. Rev. Sci. Instrum. 1998;69:4094. [Google Scholar]
- 20.Guevremont R, Purves RW. Rev. Sci. Instrum. 1999;70:1370. [Google Scholar]
- 21.Guevremont R, Purves RW, Barnett DA. US Patent # 6,713,758 2003
- 22.Ells B, Froese K, Hrudey SE, Purves RW, Guevremont R, Barnett DA. Rapid Commun. Mass Spectrom. 2000;14:1538. doi: 10.1002/1097-0231(20000830)14:16<1538::AID-RCM61>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 23.Barnett DA, Ells B, Guevremont R, Purves RW, Viehland LA. J. Am. Soc. Mass Spectrom. 2000;11:1125. doi: 10.1016/S1044-0305(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 24.Handy R, Barnett DA, Purves RW, Horlick G, Guevremont R. J. Anal. At. Spectrom. 2000;15:907. [Google Scholar]
- 25.Ells B, Barnett DA, Purves RW, Guevremont R. Anal. Chem. 2000;72:4555. doi: 10.1021/ac000341v. [DOI] [PubMed] [Google Scholar]
- 26.Barnett DA, Purves RW, Ells B, Guevremont R. J. Mass Spectrom. 2000;35:976. doi: 10.1002/1096-9888(200008)35:8<976::AID-JMS25>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 27.Ells B, Barnett DA, Purves RW, Guevremont R. J. Environ. Monit. 2000;2:393. doi: 10.1039/b005601o. [DOI] [PubMed] [Google Scholar]
- 28.Guevremont R, Barnett DA, Purves RW, Vandermey J. Anal. Chem. 2000;72:4577. doi: 10.1021/ac0000271. [DOI] [PubMed] [Google Scholar]
- 29.Gabryelski W, Wu F, Froese KL. Anal. Chem. 2003;75:2478. doi: 10.1021/ac026466c. [DOI] [PubMed] [Google Scholar]
- 30.Gabryelski W, Froese KL. Anal. Chem. 2003;75:4612. doi: 10.1021/ac026439m. [DOI] [PubMed] [Google Scholar]
- 31.McCooeye MA, Mester Z, Ells B, Barnett DA, Purves RW, Guevremont R. Anal. Chem. 2003;74:3071. doi: 10.1021/ac011296+. [DOI] [PubMed] [Google Scholar]
- 32.McCooeye M, Ding L, Gardner GJ, Fraser CA, Lam J, Sturgeon RE, Mester Z. Anal. Chem. 2003;75:2538. doi: 10.1021/ac0342020. [DOI] [PubMed] [Google Scholar]
- 33.Cui M, Ding L, Mester Z. Anal. Chem. 2003;75:5847. doi: 10.1021/ac0344182. [DOI] [PubMed] [Google Scholar]
- 34.Guevremont R. J. Chromatogr. A. 2004;1058:3. [PubMed] [Google Scholar]
- 35.Guevremont R, Thekkadath G, Hilton CK. J. Am. Soc. Mass Spectrom. 2005;16:948. doi: 10.1016/j.jasms.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Buryakov IA. Talanta. 2003;61:369. doi: 10.1016/S0039-9140(03)00305-9. [DOI] [PubMed] [Google Scholar]
- 37.Buryakov IA. J. Chromatogr. B. 2004;800:75. doi: 10.1016/j.jchromb.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 38.Miller RA, Nazarov EG, Eiceman GA, King AT. Sens. Actuators A. 2001;91:301. [Google Scholar]
- 39.Eiceman GA, Tarassov A, Funk PA, Hughs SE, Nazarov EG, Miller RA. J. Sep. Sci. 2003;26:585. [Google Scholar]
- 40.Krylov E, Nazarov EG, Miller RA, Tadjikov B, Eiceman GA. J. Phys. Chem. A. 2002;106:5437. doi: 10.1021/jp020009i. [DOI] [PubMed] [Google Scholar]
- 41.Eiceman GA, Krylov EV, Krylova NS, Nazarov EG, Miller RA. Anal. Chem. 2004;76:4937. doi: 10.1021/ac035502k. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt H, Tadjimukhamedov F, Mohrenz IV, Smith GB, Eiceman GA. Anal. Chem. 2004;76:5208. doi: 10.1021/ac0497611. [DOI] [PubMed] [Google Scholar]
- 43.Veasey CA, Thomas CLP. Analyst. 2004;129:198. doi: 10.1039/b310760d. [DOI] [PubMed] [Google Scholar]
- 44.Krylov EV. Tech. Phys. 1999;44:113. [Google Scholar]
- 45.Krylov EV. Int. J. Mass Spectrom. 2003;225:39. [Google Scholar]
- 46.Shvartsburg AA, Tang K, Smith RD. J. Am. Soc. Mass Spectrom. 2004;15:1487. doi: 10.1016/j.jasms.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 47.Shvartsburg AA, Tang K, Smith RD. J. Am. Soc. Mass Spectrom. 2005;16:2. doi: 10.1016/j.jasms.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 48.Shvartsburg AA, Tang K, Smith RD. Anal. Chem. 2004;76:7366. doi: 10.1021/ac049299k. [DOI] [PubMed] [Google Scholar]
- 49.Venne K, Bonneil E, Eng K, Thibault P. Anal. Chem. 2005;77:2176. doi: 10.1021/ac048410j. [DOI] [PubMed] [Google Scholar]
- 50.Tang K, Li F, Shvartsburg AA, Strittmatter EF, Smith RD. Anal. Chem. 2005;77:6381. doi: 10.1021/ac050871x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnett DA, Guevremont R, Purves RW. Appl. Spectrosc. 1999;53:1367. [Google Scholar]
- 52.Guevremont R, Ding L, Ells B, Barnett DA, Purves RW. J. Am. Soc. Mass Spectrom. 2001;12:1320. doi: 10.1016/S1044-0305(01)00321-X. [DOI] [PubMed] [Google Scholar]
- 53.Asbury GR, Hill HH. J. Microcolumn. Sep. 2000;12:172. [Google Scholar]
- 54.Srebalus CA, Li J, Marshall WS, Clemmer DE. Anal. Chem. 1999;71:3918. doi: 10.1021/ac9903757. [DOI] [PubMed] [Google Scholar]
- 55.Dugourd Ph., Hudgins RR, Clemmer DE, Jarrold MF. Rev. Sci. Instrum. 1997;68:1122. [Google Scholar]
- 56.Shvartsburg AA, Tang K, Smith RD. J. Am. Soc. Mass Spectrom. 2005;16:1447. doi: 10.1016/j.jasms.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 57.Guevremont R, Purves RW. J. Am. Soc. Mass Spectrom. 2005;16:349. doi: 10.1016/j.jasms.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 58.Dahl DA. Int. J. Mass Spectrom. 2000;200:3. [Google Scholar]
- 59.Barnett DA, Ells B, Guevremont R, Purves RW. J. Am. Soc. Mass Spectrom. 1999;10:1279. doi: 10.1016/S1044-0305(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 60.Guevremont R, Barnett DA, Purves RW, Viehland LA. J. Chem. Phys. 2001;114:10270. [Google Scholar]
- 61.Miller RA, Eiceman GA, Nazarov EG, King AT. Sens. Actuators B. 2000;67:300. [Google Scholar]
- 62.Lubman DM. Anal. Chem. 1984;56:1298. [Google Scholar]
- 63.Krylov EV. Instrum. Exp. Tech. 1997;40:628. [Google Scholar]
- 64.Kim T, Tolmachev AV, Harkewicz R, Prior DC, Anderson GA, Udseth HR, Smith RD, Bailey TH, Rakov S, Futrell JH. Anal. Chem. 2000;72:2247. doi: 10.1021/ac991412x. [DOI] [PubMed] [Google Scholar]
- 65.Tang K, Shvartsburg AA, Lee HN, Prior DC, Buschbach MA, Li F, Tolmachev AV, Anderson GA, Smith RD. Anal. Chem. 2005;77:3330. doi: 10.1021/ac048315a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang K, Tolmachev AV, Nikolaev E, Zhang R, Belov ME, Udseth HR, Smith RD. Anal. Chem. 2002;74:5431. doi: 10.1021/ac0202583. [DOI] [PubMed] [Google Scholar]
- 67.Barnett DA, Ells B, Guevremont R, Purves RW. J. Am. Soc. Mass Spectrom. 2002;13:1282. doi: 10.1016/S1044-0305(02)00527-5. [DOI] [PubMed] [Google Scholar]
- 68.Aebersold R, Mann M. Nature. 2003;422:198. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 69.Wu C, Siems WF, Klasmeier J, Hill HH. Anal. Chem. 2000;72:391. doi: 10.1021/ac990601c. [DOI] [PubMed] [Google Scholar]
- 70.Purves RW, Barnett DA, Ells B, Guevremont R. Rapid Commun. Mass Spectrom. 2001;15:1453. doi: 10.1002/rcm.390. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Taraszka JA, Counterman AE, Clemmer DE. Int. J. Mass Spectrom. 1999;185/186/187:37. [Google Scholar]
- 72.Purves RW, Barnett DA, Guevremont R. Int. J. Mass Spectrom. 2000;197:163. doi: 10.1002/1096-9888(200008)35:8<976::AID-JMS25>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 73.Hudgins RR, Woenckhaus J, Jarrold MF. Int. J. Mass Spectrom. Ion Processes. 1997;165/166:497. [Google Scholar]
- 74.Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, Bitan G, Teplow DB, Bowers MT. J. Am. Chem. Soc. 2005;127:2075. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 75.Shvartsburg AA, Li F, Tang K, Smith RD. Anal. Chem. in press. [Google Scholar]
- 76.Purves RW, Barnett DA, Ells B, Guevremont R. J. Am. Soc. Mass Spectrom. 2000;11:738. doi: 10.1016/S1044-0305(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 77.Purves RW, Barnett DA, Ells B, Guevremont R. J. Am. Soc. Mass Spectrom. 2001;11:894. doi: 10.1016/S1044-0305(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 78.Robinson EW, Williams ER. J. Am. Soc. Mass Spectrom. 2005;16:1427. doi: 10.1016/j.jasms.2005.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]